Phylogenetic analysis of dengue virus reveals the high relatedness between imported and local strains during the 2013 dengue outbreak in Yunnan, China: a retrospective analysis
Full text
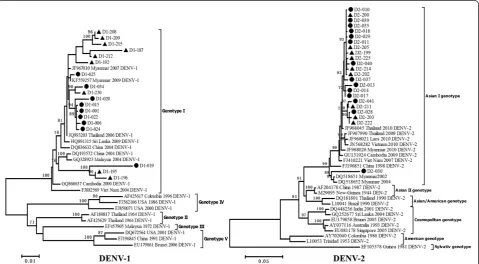
Figure


Related documents
AO: Arbeitsgemeinschaft für Osteosynthesefragen (German for “ Association for the Study of Internal Fixation ” ); AP: Anteroposterior; CT: Computed tomography; DASH: Disabilities of
Cite this article as: Gerritse et al .: Advanced medical life support procedures in vitally compromised children by a helicopter emergency medical service. BMC Emergency Medicine
Also, both diabetic groups there were a positive immunoreactivity of the photoreceptor inner segment, and this was also seen among control ani- mals treated with a
CT scan showing right opacified maxillary sinus with medial bulging causing expansion of the sinus and obstruction of the right nasal
The aim of the I N CA (Impact of NOD2 genotype-guided antibiotic prevention on survival in patients with liver Cirrhosis and Ascites) trial is to investigate whether survival of
Considering all components, the average annual working time per employee is calculated which is strongly deter- mined by the relationship between full-time and part-time employees
Our results confirm (cf. Parisien and Stevenson, 2010) that a higher-level knowl- edge of the verb classes is required to replicate the observed patterns of generalization, such as
Also a time-divided transmit by switch array to detect moving targets causes a phase distortion of echo signals and generates considerably high and periodic side lobes of the
