JOURNALOFVIROLOGY,JUlY 1991, p.3504-3513 Vol.65, No.7 0022-538X/91/073504-10$02.00/0
CopyrightC)1991,American Society forMicrobiology
Role
of
cx-Transinducing
Factor
(VP16)
in the Induction of
ca
Genes
within the Context of Viral
Genomes
DAVID SPECTOR, FRANCES PURVES, AND BERNARD ROIZMAN*
The
Marjorie
B. Kovler ViralOncologyLaboratories, The University ofChicago, 910 East 58th Street,Chicago, Illinois 60637
Received 12February1991/Accepted 27March1991
In herpes simplex virus 1, thefive agenesareinducedby a-transinducing factor(aTIF; VP16), a virion protein, acting in concertwithOct-1 and other cellularproteinson acis-actingsite in thepromoterdomainof agenes. BecauseaTIF is an essential virionprotein,its function as an inducer can best beevaluatedonlyby mutating thecis-acting site. Earlierwereportedon aseries of17mutationsin andaround the-cis-actingsite ofa275-bpa27promoterfused to areporter geneandrecombinedinto the viralgenome.Theserecombinant viruses were testedin Vero cells in thepresenceofcycloheximide,and we demonstrated that mutations inthe sequence required for Oct-I binding abolished transactivation whereas mutations in the aTIF-dependent GARAT sequence decreased but did not abolish transactivation. We now report that (i) in limited-passage human embryonic lung cells, a gene expression from promotersmutated in the GARAT sequences is often higher and more variable thaninVerocells, (ii)inthe absenceofcycloheximide,the mutantvirusesshow less significant impairment ofreporter gene expression, (iii) Oct-1 can bind either to the overlapping octamer element or tovarious TAATGARAT sequences with differingdegrees ofbindingstrength andtheserelative bindinglevelscorrelate wellwithlevels ofgeneexpression observedin infectedcells, (iv) in thecis-actingsite upstream of the a4 gene,nodegenerateoverlapping Oct-i sequenceexists,andtherefore in thisinstance Oct-1 must bebindingdirectly to theTAATGARAT sequence, (v) extension ofthea27 promoterbyan-additional 1,334 bp resultsinmuch higherexpression ofthereporter gene as aresult ofadditional upstreamcis-acting sites, and (vi) obliteration ofthe mostproximalOct-1binding element withinthe275-bppromoterdramatically reducesgeneexpression evenin thepresence oftheadditional upstreamcis-acting sites.
The herpes simplex virus 1 (HSV-1) genes form three
majorgroups, a*, ,,and -y, whoseexpressioniscoordinately
regulated and sequentially orderedinacascadefashion (25, 26).Several years ago, in studies designed to determine why at genes areexpressed first, thislaboratory discovered that
(i) ax genes are induced by a structural component of the
virus contained in the tegument and transported into the nucleus independentlyofthe viral DNA (4, 44) and (ii) the
specific cis-actingsitewith the consensus 5'-GyATGnTAAT
GARATTCyTTGnGGG (noncoding) is present in one to severalcopies in the 5' transcribed noncoding domains of all
ax
genes (39). The gene encoding the transactivator was mapped, and its sequence was determined (11, 13, 43). The geneproduct, designatedby thislaboratory as theox-transin-ducing factor
(oxTIF),
had been previously identified as thevirion protein 16 (VP16) (23, 50). It soon emerged that the cis-acting site binds a cellular host protein designated by this
laboratoryasotHl (32, 33). This protein has proven to be the
ubiquitousoctamer-binding protein variously referred to as
Oct-1, NFIII, OBP-100, NF-A1, and OTF-1 (5, 16, 35, 46,
55). Subsequent studies revealed that aTIF forms stable
complexes with its cis-acting site, but only in concert with
Oct-1and other cellular proteins (19-22, 30, 40, 42, 45, 53,
58,60).
Therole ofcxTIFin natural infections is a puzzle inasmuch as the protein is an essential structural component of the virus, and as such its gene cannot be deleted from the viral genome. Inan attempt to define the precise contribution of aTIF to the expression of ot genes in infected cells, we constructed a series of 17 mutations in and around the
*Correspondingauthor.
proximal cis-acting site of the a27 gene promoter. A275-bp fragment containing a single cis-acting site of either the wild-type or a mutated sequence was fused to a reporter gene, the viral thymidine kinase (tk) gene, and recombined into the viral genome. The first series of
studies,
reported elsewhere, were performed in Vero cells, a continuous African greenmonkey cell line, in the presence of cyclohex-imide (51). These studies demonstrated that the reporter gene mRNAaccumulation was mostreduced by mutations in the ATGnTAAT sequence whichabolished Oct-i binding. Mutations in the GARAT sequence, which abolished the formation ofDNA-proteincomplexes containing acTIF, also reduced the synthesis of reporter gene mRNA, but to a significantly lesser degree.In the studiesreported here, we extend our investigation of the a27 cis-acting site within the context of the viral genome.We have favored thisapproachbecause the results obtained in transientexpression systems frequently do not
reflecttheexpression ofgenesembedded in the viral genome and expressed during infection (7, 15, 49, 51). While the
previous report focused on identifying those sequences involved in the induction of a genes, the present studies
investigateseveraldifferentaspectsofageneregulation: (i)
differentiation of the role of the overlapping octamer se-quenceand the TAATGARAT in mediating agene
induc-tion, (ii) comparison of the effects ofmutations within the
cis-acting site in the primary limited-life-span and contact-inhibited humanembryoniclung (HEL) cell strain with those obtainedpreviouslyin the continuous transformed Vero cell
line, (iii) the effect of extending the 275-bp a27 promoter used inprevious studies to 1,609bp by restoring additional native sequences to both the wild-type promoter and the most impaired mutant promoters, and (iv) the level of 3504
on November 10, 2019 by guest
http://jvi.asm.org/
MUTATIONAL ANALYSIS OF THE HSV-1 a GENE PROMOTER 3505
-
-
er~~~~~~27
___+55-219
..
.
u
1-19____
aTIF REPONSIVE ELET
COSESNSIIUS
SEECE GYATNTMT TTC y6T
DfILD
TYPESEGUENCE iNa27
PRmmTCCCCCATlT-C-C-C-CC
CCCGTCC
C -C66AACCGTTAT
GTATATOCT AATTAAATACATGCCAC6TA
^TTAGIGT
CTTSTCTSSm
ICCGAGTC
NuTmr
SEQUENICES
IN0a27
PRoM210 200 190 180 170 160 1SO 140 130 120 110 100
1. CCAOCATCT Cup TICC GCG C AACGTCATATGTATATST AATTAMTAC ATCCACSTACTTATGCTST CTGA1TSSTC CIGTCTgTgCCSGAGT
2. 6CCACATAT cCACCcCCTcccMTTccc
rCCtACAC
mSG6TCTAT
GTATATGCTAATTAATAC ATCCTA ITTGCTT CTGATTGSTC CITBTCTCSi CCSGASGTB3. CCACATAT CCA CGC Cc=TCCC WM= AcCGTGTAT GATATCT AATTAAATAC TGGT CTTGTCTT CCSGAeST
4. GCCACCATATcCAccCCGc
ccCCTcccc
cACTcTT cCCTA TCTATCTTATATGCTAATTAAATAC ATGcCACtT TCTTATTCTSATTGTC CTGTCTGIsCCCUCCTS0009000
5.
CCASCATAT
ccAccccCcC
CCCtiCTccC
mCCCmcC
ATATTCTS
TTTTATT
nTTTC
ATCCTACTA
TATTT CTSAITGGTC CTTGTCT CCGAGGT6.
CCACCATAT
CCACCCCcCCCCCSTCCCC
CCtC
CGTTAT
GCATCACT
CTATAC
ATICCUACTA
1COTCTAICCGTC
CTTgTCTGt CCtCACCTC9. SCCACATAT
CCACCCCCGC CCCCCTCCCC C0WUCCCC
AACCTCTAT
TGTTTmTTAtrAC
ATCCACTCItATUT
CT1TGGTC CTTTCTCTS CCGGCMT80. GCCAGCATAT CCACCCCCGCCCCCCTCCCC GCGGACGCCC MCGGTGTAT GIGTATOCT AATAATAC ATCC&<TI CiAOBU CTATtGGTC mCTTCTGTGCCGGTC
19.
GCCAGCATATCCACCCCCCC CCCCtTCCCC GcGGAMcGG MmCGTGTAT GTATATGCT ATTAATAC ATrACA6TA TC1TT TTT CTGATTGGTC CTTmTITCII CCSGAGGTG10.
GCCACCATATCCACCCCCGC CCCCCTCCCC GcGGD.McGG AAcunTGTAT GTATAnTCT AATTAAATAC ATGCCA6TA rCATTGT CT?&T??TCmCTITCT6T CMCGGTT1. GCCACATAT C>CCCCCCCC CTCGTCCC GCGACCG AACGTGTT STITATCT AATTAATAC ATCCCTA CIAcTIE CTGATGTCmCTrCTGrsCCGGAGGTG
12. oCCACATATCCCCCCCC CCGCUTCCCC mC C ACTnTAT aTCT Tc ATGCCACTA CTATST CITTTTC CrGTCT CCTTCTGIM
13. rGccACATATccACCCCCGc ccGGTcccc ACGG AAcuGTTAT rGTTC6CGTCT ATTAAATAc ATGccACTA CIGATOBT CTGAT1 TCCTTGTCTGTi CCGGAGBT
14. GCCAGCATATCCCCCCCC CCGGTCCCC GccaAmcumcAGTGTAT CTATATGCTMATT&&& ATACOAT CITATiGT CTATTGTC CTTgTCTGi CCGGAGTG
15. OCCACCATAT CCCCCTCCCC
MCCCTAT^TGTATAnTCT
AATTAAATAC CTACAITITSTCTGATTGGTCCTGTCTTi CCCGGAGTG16. CCAGCATAT cucccccrGC ccGGTCCCC GwGGAGc AACGGTTAT GTGATaT ATTAAATAC
ALMZAAGGA
Isi'sTGT CCGGMG17. CCACATAT CCACCCCCGCCCCCCTCCCC
CCeAAGCC
CGTTAT GTGTATGCt ATFATAC TCCCGTT cUAT GT CTGATTAGTCCTTGTCTCTe CCGGAGGT 18. GCCACATATCCACCCCCUCCCC6TCCCC
CCGGACACG ACGGTTAT GT> MATTAAATACATGCCAMTA
C1TAT CTUTGTGTC CTiGTCTGTi CcGGGTG19.
rCCCuATAT
CCACCCCCCGC
CCCCTCCCC
GcGGCC
C cmCGGTGTAT
CTTC6C6TCT
AAT6JAT& ATCCACGTA
mCTATGT
CTGATrGGTC CT1TCTGCCUCCTG
FIG. 1. HSV-1 genome sequence arrangement and sequence of the mutagenized domain of the a27 gene promoter. (A) Sequencearrangementofthe HSV-1genome. Filled rectanglesrepresentthe invertedrepeatsflankingtheuniquesequences(represented bythinlines) of the longand shortcomponents. (B) Expansionof the domainshowingthe1,609-bpa27promoterandassociatedleadersequence fusedto
the tkreportergeneinmutants20 and21. Circlesrepresenthomologsof the aTIF cis-actingsite.(C) The275-bpportion ofthe a27promoter and associated leadersequencefusedtothetkreportergeneinmutants1through 19.This fragmentcontainsonlyoneand themostproximal aTIF cis-acting site. (D) Expansion of the region subjectedtomutagenesis. The replaced nucleotidesareidentified byuppercase lettersand
superimposedcircles. Recombinant virus 20 contained thesamemutationswithin themostproximalelementasdid virus6. Intheengineered recombinant viruses, the chimeric a27-tkgenereplaced thetkgeneatthe natural locationofthetkgene. WT, wildtype.
expression observedfor themutant virusesin theabsence of
protein synthesis inhibitors such as cycloheximide.
MATERIALS AND METHODS
Cells and viruses. The parent and mutant viruses were
grownandtitered in Verocells. Transcriptionof the reporter
genes linked to mutant and wild-type promoters was
mea-sured in infected Vero or HEL cells, as stated in Results.
The mutant viruses were derived from HSV-1(F)A305, a
virus derivedfrom the wild-typestrain HSV-1(F)bydeletion
of 500 bp from the tkgene as previouslydescribed (44).
Construction of thea27-tk mutant viruses. The structures of the chimeric reporter genes inserted into the HSV-1(F) genomesandthemutationsin thetestpromoterare shownin
Fig. 1. Construction of mutant viruses 1 to 17 and their
parentwild-type a27-tkvirus hasbeen previously described (51). Mutantviruses 18 and 19wereconstructed in thesame
way except that site-directed mutagenesis was done onthe
M13mpl9
counterpart of pRB8032, previously used in theconstruction ofmutant virus 13. As before, EcoRI-HindIII
fragmentscontainingthemutanta27-tkgenes wererecloned
into
pRB4050
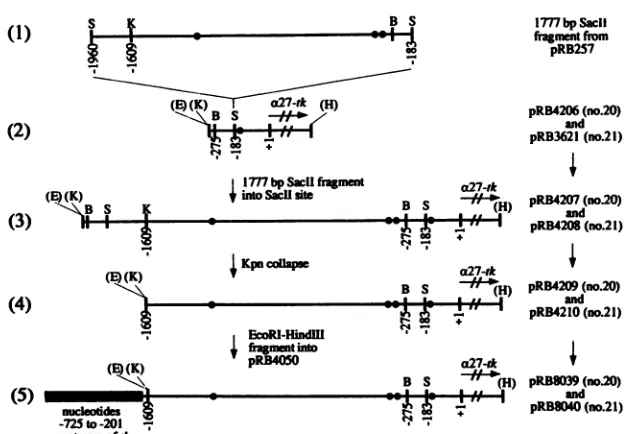
for recombination into the viral genome, creatingthe constructspRB8037 andpRB8038, correspond-ingto mutant recombinant viruses 18 and 19, respectively.Construction oftheplasmids used to construct viruses 20 and 21, which harbor the extended a27 promoter, is illus-trated inFig.2. Asshown, the 1,777-bp SacII fragmentfrom
pRB257 containing EcoRV-G
(Fig.
2, line1)
was cloned inthe correct orientation into the SacII sites ofpRB4206 and pRB3621(Fig. 2, line 2)to constructpRB4207andpRB4208,
respectively (Fig. 2, line 3). pRB3621 contains the parent a27-tk gene used throughout these studies. pRB4206 was
constructedbycloningtheEcoRI-HindIII fragment contain-ing themutanta27-tkgenefrompRB8006previouslyused in
the construction ofmutant virus 6 into pUC18. To remove those sequencesduplicatedbetween theBamHI site at -275
and the SacII site at -187 from plasmids pRB4207 and pRB4208, a KpnI collapse was done on each plasmid
be-A
B
C VOL. 65,1991
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.93.534.76.436.2]3506 SPECTOR ET AL.
(1) I s1Bi
o0s~~~~~~~~~~~~~0
(E)(K) r o27-rk (H)
(2)
ifI _
1777bpS;1t fragment
+intoSacxl site
B S
-4
;Kpnoollapoe
B S
Ia*
~
I,EoR-HinU
-f_into
B s
q
a27-k
_>H) pRB4207(no.20)
pRB4208(no.21)
a274k
//
(-H)
pRB4209(no.20)*SH| and
' " pRB42lO (no.21)
a27-d t
(H) pRB8039 (no.20)
Iis,aand
.+-I pRB8040(no.21)
FIG. 2. Construction ofplasmidsusedto constructthe chimerica27-tkgenesfor recombinant viruses20 and 21. All intermediateandfinal plasmids usedin the construction of recombinant virus 20 containedamutantaxTIF-responsiveelement located betweennucleotides -156 and -141, whereas those used in the construction of recombinant virus 21 were wild type. B, BamHI; E, EcoRI; K, KpnI; S, SacII. Restriction sites indicatedinparentheseswerederived frompolylinkersequences.
tween the KpnI site in the polylinker and the KpnI site located at position -1609 of the a27 promoter, creating plasmids pRB4209 andpRB4210 (Fig. 2, line4). The result-ing x27-tk genes containing the extended a27 promoter
sequences were then recloned asEcoRI-HindIII fragments from both pRB4209 andpRB4210 intopRB4050 for
recom-bination into the viral genome, creating plasmids pRB8039
andpRB8040 (Fig. 2,line5), corresponding torecombinant viruses 20 and21, respectively. While both viruses contain identical extended promoter sequences, mutant virus 20 is mutated in its firstoctamerbinding site in the same fashion as mutantvirus6, while virus 21 harborsnomutations. All
plasmidconstructswereverifiedby sequencing using Seque-nase (UnitedStatesBiochemical). The recombinant viruses
wereplaque purifiedatleast twice and checked for purity by hybridization oftheelectrophoretically separated fragments. The increase in size of the viral EcoRI N fragment was
readily detectable, and this band hybridized with both a27-and tk-specific probes.
Gelretardation assay. HeLa cells were infected with 10
PFU of HSV-1(F) percell and harvested at18 h postinfec-tion. Nuclear extracts were prepared as described by
Dig-nametal. (14). Protein concentrations weredetermined by
themethod of Bradford (9). 32P-end-labeled DNA probe(10 fmol) was incubated with 5 ,g of nuclear extract in the
presence of 10 ,g of poly(dI)-poly(dC) (Pharmacia P-L
Biochemicals) in 20 mM Tris-HCl (pH 7.6)-S50 mM KCl-1 mM EDTA-0.05% Nonidet P-40-5% (vol/vol) glycerol-5 mM 1-mercaptoethanol-100 ,g of bovine serum albumin
(Sigma)per ml ina reaction volume of 20
RIu
for 30 minat25°C. The reaction mixtures were electrophoretically
sepa-rated in nondenaturing 4.5% polyacrylamide gels at 4°C,
dried, and exposedto Kodak X-Omat film for 12 h. Preparationof RNA probes.The probeswere constructed asfollows.pRB4051wasmade by cloning the 675-bp
EcoRI-NruIfragment from pRB8000 containing nucleotides -272to
+400 of theparentot27-tkgeneinto the EcoRI-HinclI sites of
pGEM-3Z. pRB4086 is a SacII-MluI collapse ofpRB4051.
Cleavage ofpRB4051 withEcoRI andtranscription by SP6
polymerase (Promega Biotec) generated a 700-bp RNA probe (TK1)thatprotected400bpof ot27-tkmRNA,whereas pRB4086 treated similarly generated a 350-bp RNA probe (TK2) that protected 264 bp of cx27-tk mRNA. pRB4052 contains the 510-bp HindIII-AccI fragment (nucleotides -330to +180)of theao4genefrompRB4011 clonedintothe HindIII-AccI sites ofpGEM-4Z. Cleavage with PvuII and transcription with the SP6 polymerase generated a 700-bp
RNA probe thatprotected 180 bp ofat4 mRNA. Template DNA was removed by RQ1 RNase-free DNase(Promega),
and unincorporated nucleotides were removed on Quick-SpinRNase-free spincolumns(Boehringer Mannheim).
Conditions of infection. In theprevious studies and in the initial studies described in this report, replicate Vero cell cultures in 150-cm2 flasks were preincubated for 1 h in mediumcontaining cycloheximideat 100 ,g/ml, exposedto 20 PFU of recombinant virus per cell for 1 h, and then overlayed with fresh medium containing the drug. In all studies,extensiveprecautionsweretakentoensurethat the
replicate cultures infected with the different mutants were
exposedtothesameratio of 20 PFU of viruspercell. After 4 hat37°C,testRNAswerepreparedasdescribed below.In the second set ofexperiments, conditions remainidentical exceptthat HEL cellsweresubstitutedfor Verocells. In the
finalset ofexperiments, Vero cellswereinfectedunder the conditions described aboveexcept that cycloheximide was
notused andcellswereharvested either 3or7 h afterthe end
of the adsorptioninterval.
PreparationoftestRNAs.Cellswerewashed,scraped into
ice-cold isotonicphosphate-buffered saline,spundown, and
resuspended in 3 ,lI of 10 mMTris-HCl (pH 7.6)-150 mM NaCI-1.5 mM MgCl2-0.2% Nonidet P-40. Nuclei were
re-movedby centrifugation, andthe supernatantfluid contain-ing totalcytoplasmic RNAwastreated with
phenol-chloro-form (24:1) three times, treated withchloroform once, and ethanolprecipitated.
RNaseprotectionassays.Totalinfected-cellRNA(40to50
1777bp SacIl fragmentfrom
pRB257
pRB4206(no.20)
and pRB3621 (no.21)
(E)(K)
(3)
(4)
(5)
-(E)(K)
(E)(K)
nucletdes § -725to-201
-upsuem of tk
2
N-CR 0 - -.L K
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.147.460.72.289.2],ug)
wascoprecipitated
with anexcessofa4andeitherTK1 or TK2 probe and hybridized overnight at 50°C in 30 ,ul of 80% (vol/vol) formamide-40 mMpiperazine-N,N'-bis(2-ethanesulfonic acid) (PIPES; pH
6.4)-i
mM EDTA-0.4 M NaCI. RNase digestion buffer (300 ,ul) (10 mM Tris-HCl [pH7.6], 5.0 mM EDTA, 0.3 M NaCl, RNase A [40 ,ug/ml],
RNase T1 [2
jug/ml])
was added, and incubation continued for 1 h at 30°C. Sodium dodecyl sulfate and proteinase K(Sigma)wereadded, andincubation continued for 30 min at
37°C. The RNAs were phenol-chloroform extracted once andethanol precipitated, using glycogen as a carrier. After resuspension in 10 mM EDTA-0.1% xylene cyanol-80%
formamide, the samples were electrophoretically separated onan 8 Murea-6% polyacrylamide gel. The dried gels were exposedfor approximately 1 h to Kodak X-Omat S film.
Analysis of results. Tables 1 to 3, which summarize the results of our studies, each display the results of two to five
experimentsandtheir means. Each value was determined by
assaying independently derived mRNA from a separate
infection of cells with the indicated virus. Radioactivity in protectedbandsfor the a4 anda27-tkmRNAs from both the parent and the indicated recombinant virus plus
repre-sentative background bands was measured three times on a Betagen Betascope model 603 blot analyzer, and the per-centageof mutanta27-tkmRNA wascalculated after theoA levels were scaled to be equal. The standard error was generated by the three quantitations on the Betagen. The meanvalue listed represents the mean + standard deviation of the two tofiveindependentdeterminations for each virus under theconditions tested.
RESULTS
Mutations. The 19 sets of substitutions in the wild-type nucleotide sequences of the 110-bp domain of the a27 promoter are identified by dots and large capital letters in
Fig. 1. The viruses are designated by the mutant sequence
numbers shownin thatfigure.
Analysis of recombinant viruses mutated in the proximal
aTIFcis-acting site in the promoter of the a27 gene. In the
previous report, wedemonstrated that transversions in the TAAT sequence of the aTIF cis-acting site of the a27 promoter (mutant 6) abolished both the ability of that se-quence toformOct-1 andaTIF protein-DNA complexes and the capacity of the corresponding reporter gene to be
in-duced (51). Transversions in the sequences immediately
upstreamofthe TAATnucleotides, which destroythe
over-lapping octamer consensus site to which Oct-1 was
foot-printed, alsodestroyed theability of that sequenceto form
Oct-1 and aTIF protein-DNA complexes except at vastly increased amounts ofprobe. The levels ofa27-tk mRNA accumulated after infection with the virus containing this
mutation (mutant 13)was significantly reduced in
compari-son with that of the wild-type a27-tk mRNA but also
significantly higherthan thelevels observed in cellsinfected with mutant virus 6. In light of these results and the observation that many of the aTIF-responsive elements upstreamofother a genes,particularly those present in the promoter of the a4 gene, either donotpossess an overlap-ping octamer element or possess a
highly
degenerate se-quence, wewishedtodetermine whetherOct-1binds totheoverlapping octamer element or to the more
highly
con-served TAATGARAT sequence during induction of the a genes.Althoughthereduced levelsofa27-tk mRNA accumulated after infection with mutant virus 13 provided evidence for
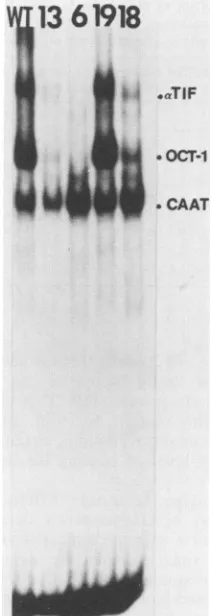
V
13 61918
CA
aTIF
.0
OCT-1
[image:4.612.392.498.81.389.2]4
.~~l
CAATFIG. 3. AutoradiographoflabeledDNA-protein complexesfrom infected-cellextractselectrophoretically separatedon a nondenatur-ing gel.ThemutantDNAprobetestedforbindingisindicated above each lane and consists of the 167-bp ApaI-NcoI fragment located betweennucleotides -86 and -253of thea27promoter. WT,wild type.
the importance of the overlapping octamer element within thecontext of thea27 promoter, thepossibility existed that theparticular transversions that weintroduced might have destabilized the binding of Oct-1 to the TAATTAAAT sequence. To test this possibility, we constructed a virus (mutant18) whichwasmutated in its a27 promoter such that thesequences upstream of the TAAT nucleotides mimicked the sequences immediately upstream of the TAATGAGA Tgc located at position -253 of the a4 gene. Such mutations should recreate anydegenerateoverlapping octamer element thatmight exist upstream of this a4 TAATGARAT. Another a27-tk virus (mutant 19) was also constructed in which the overlapping octamer element was destroyed by transver-sions exactly as in mutant virus 13, but in addition, the TAATTAAATac sequence was mutated to mimic the TAATGAGATgc present upstream of the a4 gene.
The results of the DNA binding assays are presented in Fig. 3. Mutant 18 could not be differentiated from mutant13, being unable to bind Oct-1 except at vastly increased amounts of probe. This result suggests that no degenerate
overlapping octamer element exists in theaTIF-responsive element upstream of the a4 gene, and thereforeOct-1 must bind directly to the TAATGAGATgc sequences in that promoter.Theweakbinding of Oct-1 to thecis-acting sites in mutants 13 and 18 appears to be due toavery low level of binding to the TAATTAAATac sequence. Perhaps more
on November 10, 2019 by guest
http://jvi.asm.org/
3508 SPECTOR ET AL.
TABLE 1. Activities ofmutated a27 promotersexpressedin terms ofaccumulation ofRNArelativetothat of theparent promoter inVerocells in the presence ofcycloheximide(4hp.i.)
(mRNAinitiated frommutanta27promoter/mRNA Parentor initiated fromparent promoter control) x 100
mutantvirus
1 2 3 Mean
WT 100 ± 3 100 4 100 2 100
13 36 2 37 2 38 2 37 ± 1
18 39 2 38 2 44 1 40±3
6 7±1 7±1 9±1 8±1
19 62 2 68 3 54 2 61 7
14 27 3 18 1 17 ±0 21 6
20 182 4 156 6 168±3 169 13
21 639 14 376 13 545 ±11 520 133
interestingwastheeffect ofthe threeguanines introducedin mutant 19, which vastly increased the capacity of this
sequence toformOct-1 and aTIF DNA-protein complexes
in comparison with mutant 13. Still, the wild-type a27 promoterwith theintactoverlappingoctamerelement
main-tained the highest level ofbinding for the specific DNA-protein complexes.
Table 1 summarizes the results ofRNase protection as-says on three sets of independently derived infected-cell
RNAs afterinfection with the indicated virusesunder
con-ditions in which only a genes are expressed. The data suggest thatexpression ofthea27-tk mRNAby themutant promoters correlated well withtheir respective Oct-1 bind-ing levels. The key observation is thattherelative levelof Oct-1bindingandthe level of a27-tk induction decreasedin the orderATGCTAAT> TAATGAGATgc >TAATTAAA Tac> nobinding site. Weconcludethat therelative levelof binding of Oct-1 is morecritical in determining the levelof inductionthan whether theprotein isbindingtothe
overlap-pingoctamerelement or the TAATGARAT sequence itself. Analysis of the expression of a27-tk chimeric genesin HEL cells. Inthe studies reportedearlier, Verocellswereinfected with the panel of mutant viruses to determine the effect of the specific mutations on a gene induction (51). Several factorsledusto retestselectedmutantsin another cellline,
HEL. First, the mutations introduced in mutants 3 and 4,
modifyingthehighlyconservedGA-richregionoftenfound upstreamofagenes, hadvirtuallynoeffectonthe
accumu-lationofa27-tkmRNA in Vero cells, suggesting a possible
species or cell type specificity for this element. Second,
inspectionof the results obtained in Vero cells revealed that
viruses mutated within the TAATGARAT sequences fell
intothree distinct groups: (i) those with no octamer binding
site(mutant 6), which expressed a27-tk mRNA at the lowest level (6 to 7% of the wild-type level), (ii) those with strong octamer binding sites but mutated within the GARAT se-quences (mutants 7 and 14), which formed Oct-1 but not aTIFDNA-proteincomplexes and expressed
a27-tk
mRNA at an intermediate level (15 to 30% of the wild-type level), and(iii)those which formed complexes with both Oct-1 andaTIFand expressed a27-tk mRNA at or close to wild-type
levels. The question was whether these results reflected the
properties of the promoter sequences or the cell lines in
whichthe promoters functioned. HEL cells differ markedly
from Vero cells in that they are a limited-passage primary
human cell strain which is not transformed.
The results of RNase protection assays on three to five
setsof
independently
derived infectedHEL cell RNAs afterTABLE 2. Activities of mutateda27promotersexpressedin termsofaccumulation of RNA relativetothat of theparent promoterin HELcells in the presence ofcycloheximide (4 hp.i.) Parent or (mRNAinitiated from mutant a27promoter/mRNAinitiated
mutant fromparentpromoter control) x 100
virus 1 2 3 4 5 Mean
WT 100 2 100 2 100 1 100 2 100
2 102 3 83 7 156 1 114± 38
3 74 1 88 4 97 4 95 2 89± 10
4 79 1 100 5 104 3 122 2 101 ±22
13 31 2 36 3 34 2 39 1 46±4 37±6
18 37 1 42 5 45 2 49 5 43 ±5
6 8± 0 19 3 19 1 15 2 14±2 15 ±5
19 57±2 79 5 58 2 101 7 74± 21
7 63 3 76 3 59 2 66 9
14 20 1 79 4 28 1 27 1 77±6 46 29
15 65 2 95 4 80 1 80 15
17 79 1 91 4 87 2 86 6
10 25±1 30±1 22±1 26±4
12 55 1 25 ± 1 55 2 76 3 24±4 47 22
20 131 10 119 ±7 125 8
21 787 66 775 ±29 781 8
infection with the indicated viruses under conditions in which only a genes are expressed are presented in Table 2. Itis of interest that the ratio of a4 to a27-tk mRNA observed was greater in Vero cells than in HEL cells. For ease of comparison, Fig. 4 shows a bar graphdisplaying the means and standarddeviations of the percentage of a27-tk mRNA accumulated relative to wild-type virus after infection with eachindicatedmutantvirus in both Vero and HEL cells. It is apparent that most of the mutant viruses express the a27-tk mRNA at similar levels in both cell lines. One difference ofpossible significance, however, is the higher expression detected in HEL cells than in Vero cells of the reporter gene after infection with viruses containing muta-tions in the GARAT sequences (mutants 7 and 14). While mutantvirus 7consistently expresseda27-tk mRNAat66% ± 9%of thewild-type level in HELcells versus25% + 7% in Vero cells, mutant virus 14 expressed the gene with greater variability in HEL cells, sometimes expressing at levels 77 to79% of thewild-typeandatother times
express-ingatlevelsof20to28% ofthewild-type level, similartothe levels of 17to27% consistentlyseen in Vero cells.
The variability that we have observed may arise in part from the unusual properties ofthe HEL cells themselves. These cells tend to be more contact inhibited than Vero
cells, and after forming a confluent monolayer they can remain inastateofarrestedgrowth for many weekswithout significant cell deathordeterioration. It isconceivable that once the cells reach confluency, the levels of the various
transcription factors change according to the needs of the cell. Because of thesecharacteristics,itisdifficultto ensure fromoneexperimenttothe next the exactphysiologic state ofthese cells, evenif in all experiments the cultures reach apparently identical levels of confluency.
Expressionofwild-type and mutanta27-tkgenescontaining an extended promoter with multiple aTIF cis-acting sites. Mutantviruses 1to 19 and theparentalwild-typevirus each contain a chimeric a27-tk gene consisting of a 275-bp a27
promoter-regulatory region fused to the tk gene. This pro-moterwaschosen because it contains only one copy of each of the elementsidentified todateasbeingpotentially impor-tant for the regulation of a genes (26a). Moreover, unlike J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.51.293.111.225.2]a- 160%
E 140% 2 VERO CELLS
0
140%/
a.
[20%.
. x2HEL
CELLS7E 120%
a)
(0 a.1000/0
> 80o/
60%i
a: 40% ~
E
c,
20%/
1 2 3 4 5 13 6 7 14 8 15 16 17 9 10 11 12 WT7
Parent (WT) or Mutant Virus
FIG. 4. Comparison of activities of mutated a27 promoters in terms of accumulation of mRNA relative to that of the parent (wild-type) promoterin Vero and HEL cells. Cells were infected with the indicated recombinant virus under conditions in which onlyagenes were expressed. Vero cell results, shown for purposes of comparison, were those previously derived (51); HEL cell results are those listed in Table 2. Thegraph depicts the mean and standard deviation of a minimum of three and a maximum of five analyses on independently derived RNA from cellsinfected with the various mutants. Recombinant viruses with mutations which had no demonstrable effects in Vero cells and which were not in conservedelements ofagene promoters, orwhich contain mutations similar to those of another tested virus, were not tested in HEL cells.
other a genes, the a27 promoter does not contain binding
sites for,or appear tobe down-regulated by, the products of thea4gene. Inasmuch as sequences upstream of nucleotide -275 of the a27 promoter contain homologs of aTIF-responsive elements, we investigated the effect of longer promoter sequencesin our recombinant virus constructs.
Inrecombinant viruses 20 and 21, thea27-tkchimeric gene containedan a27 promoter extended toposition -1609, as shown diagrammatically in Fig. 1. The locations of the homologs of the consensus sequence of the aTIF-responsive elements are indicated by the circles. The differences be-tweenrecombinant viruses 20 and 21 are in the Oct-1 binding siteproximaltothe cap site of the tk gene. In virus 20, this siteis mutated and is identical to that of mutant 6, whereas in virus 21, the a27 promoter sequences fused to the tk gene arewild type. Theresultsof RNase protection assays on two to three sets of independently derived infected Vero and HEL cell RNAsafter infection with either mutant virus 20 or 21 underconditions in which onlyagenes areexpressed are
presentedin Tables 1 and 2,respectively. Forconvenience, the percentaccumulationofa27-tk mRNAforeach virusis
shown relative to that accumulated in cells infected with the virus containingthe 275-bpa27promoter-tk chimeric gene. In cells infected with recombinant virus 21, the levels of a27-tk mRNA were four- to eight-fold higher than those detected in cells infected with the viruscarryingthe275-bp
a27 promoter-tk chimeric gene. The longer promoter in-creased the mRNAaccumulationto aslightly greaterextent
in HELcellsthan in Vero cells. We concludethat the level
of
expression
ofan ageneisinlargepartdeterminedbythenumber ofaTIF-responsive elements located upstream of that gene, concordant with similar observations made
re-gardingtheexpressionof chimeric a4-tk and aO-tk chimeric genes by MackemandRoizman(37).
Comparison ofthe levelof a27-tk mRNA accumulatedby
mutant viruses 20 and 21 revealed the importance of the most proximal aTIF-responsive element located between nucleotides -156 and -141. Even though several other
aTIF-responsive elements are present, including those lo-cated between nucleotides -315 and -300 and between
nucleotides -366 and -351, the level of a27-tk mRNA is reduced approximately threefold in Vero cells and over sixfold in HEL cells when the proximal octamer element is
abolished. These results indicate that the aTIF-responsive element located nearest the TATA box exerts a greater influence on the level ofa gene expression than do those elementslocatedfarther away. If themostproximalelement is mutated, the distal aTIF response elements can still mediatesignificant induction albeit to a lesser degree.
Expressionofwild-type andmutant a27-tk chimeric genes in cells infected and maintained in theabsence of cyclohexi-mide. In all earlier studies, the expression of a-reporter genes within the context of the viral genome was studied underconditions which permittedtheexpression ofa genes
only (51).Theobjective ofthoseexperiments wasto deter-mine the effect of aTIF introduced into the cell during infection on the expressionofagenes,withoutinterference
from changes in the physiology of the cell due to gene
products expressed later in infection. To determine what actually happens in untreated infected cells, a series of
experiments was done on cells infected and maintained in theabsenceofcycloheximide. Table 3 shows the results of RNase protection assays on three sets of independently
derived infected Vero cell RNAsharvested 3 h after infec-tion with the indicated viruses. Thestrikingobservationwas that in the absenceofcycloheximide there was little or no difference between expression of wild-type and mutant a27-tk chimeric genes. Even mutant virus 6, which barely expresseda27-tk mRNA ininfected cells treatedwith cyclo-heximide, expressedatlevels 68to76%thatofthe wild type in the absence of thedrug.
Experiments were also done using RNA extracted from cells infected and maintained for 7 h in the absence of
cycloheximide.
We should stress, however, that theassaysonthe RNAharvestedfrom cells infectedand maintained for 7 h were less reproducible. The reason for this lack of
reproducibility is not known. One
possible
explanation is that the a.4 transcription which serves to normalize the a27-tk mRNA isnegatively regulated
at this time and the extentofaccumulationof this RNA may vary.Nevertheless,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.155.473.78.243.2]3510 SPECTOR ET AL.
TABLE 3. Activities ofmutated a27 promotersexpressed in termsof accumulationof RNA relative to that of the parent promoter in Verocells in theabsence of cycloheximide(3 hp.i.)
(mRNA initiated from mutant a27 promoter/mRNA Parent or initiated from parent promoter control) x 100 mutant virus
1 2 3 Mean
WT 100 4 100 ±3 100 ±5 100
1 100 4 89± 1 92± 3 94±6
2 65 2 69± 1 74 ±3 69 ±5
3 68 3 91 ± 1 98 ±4 86± 16
4 97 3 88 ± 1 115 ±4 100 ± 14
5 52 2 67 ± 1 61 ±2 60 ±8
13 66±2 84 ±2 70 ±2 73 ±9
6 68 ±2 73 ± 1 75 ±2 72 ±4
14 121 ± 15 111 ±5 128 ±5 120±9
8 131 ± 6 131 ±5 131 ±0
15 115 ±5 777 3 80 ±3 91± 21
i6 123 ±5 113 ±5 114 ±4 117±6 17 112 ±5 111 ±5 116 ±3 113 ±3
9 153 ±6 161 ± 7 147 ±4 154 ±7
10 89 ±4 92 ±5 107 ±3 96± 10
11 142 ±6 129 ±5 147 ±4 139 ±9 12 106 ±5 107 ±5 199 ±4 111 ± 7
in the experiment showing thelowest accumulation of
mu-tant 6 a27-tk mRNA at 7 h postinfection, the level of this mRNA was 40% of the wild-type mRNA level (data not shown).
DISCUSSION
A complete understanding of HSV-1 a gene regulation
requiresknowledgeof theproteinfactorsinvolved,the DNA
sequences upon which they act, and the complex
environ-ment of the infected cell in which these elements function.
Much progress has been made in recent years to elucidate thecomponentsof the system,but little is knownof howthe
system actually operates. Because aTIF is an essential
structuralproteinof thevirion,theeffects of mutations in the protein cannot be readily and unambiguously assigned to eithertransactivatingor structuralfunctions. Therefore,we
havechosen toinvestigatethetransinduction role of aTIF in
the infected cell by mutating its cis-acting site in the
pro-moter domains of chimeric a27-tk genes embedded in the
viralgenome.Thesalientfeaturesof the resultsconcernthe
interactionofOct-1with theaTIF-responsiveelement inthe
agenesand theroleof aTIF in theexpressionofagenes in
infected cells.
Interaction ofOct-1withHSV-1 DNA. aTIFcis-actingsites
consistof anoctamermotifoverlappingwiththemorehighly
conserved TAATGARAT sequence (3, 5, 33, 41). The
se-quenceATGCTAATTAAATACcontained inthemost
prox-imal otTIFcis-acting siteof the a27 promoterused inthese
studies differsinonlyoneofeightresiduesfrom theoctamer
consensus ofATGCAAAT. Footprinting studies identified
theoctamermotifas theOct-1binding site inthispromoter (33).InotheraoTIFcis-acting sites,mostnotablythoseof the
a4gene, no overlapping octamermotif is apparent, yetthe possibilityremained that a degenerate site mightexist. To
testthis hypothesisweconstructed themutant18, inwhich
thesequencesimmediately upstreamof theTAATsequence
resemblethoseof the a4gene aTIFcis-actingsite atposition
-253. This mutation failed to restore Oct-1 binding or
increase a27-tk induction above the levels obtained with
mutant 13,in which the samesequenceswere simply
trans-versed to destroy the octamer motif. By destroying the overlapping octamer motif by transversions and alsoaltering the TAATTAAATAC sequence to TAATGAGATGC
(mu-tant 19), we observed greater Oct-1 binding and greater oa27-tk expression than with mutant 13. The simplest inter-pretation of these results is that Oct-1 can bindtoeither the overlapping octamer motif or the TAATGARAT sequence and does so with highly variable binding strengths. A key
observation is that thestrength with which Oct-1 bindstoits site in vitro correlates well withthe level of a gene induction observed in the infected cell.
The results reported herearecongruent with thefindings
of other laboratories investigating the DNAbinding domains of the Oct-1 protein. Sturm et al. identified Oct-1 as a member of a new class of proteins referred to as POU proteins, which share a 150- to 160-amino-acid-long region
known asthe POU domain (24, 55). The POU domain is a bipartite DNA-binding structure which can be subdivided into POU-homeo andPOU-specificsubdomains (56). Studies with another POU protein, Pit-i, have revealed that the POU-homeo subdomain isabsolutely required and sufficient for low-affinity DNA binding, whereas the POU-specific subdomain is necessary forhigh-affinity and sequence-spe-cific binding (27). Verrijzer et al. reported that whereas the entire POU domain has a 600-fold-higher affinity than the POU-homeo subdomain for the octamer motif, the two regions have nearly equal affinity for the a4 TAATGARAT sequence(59). Mutations in the RAT part of this sequence hadnoeffectonbindingof thePOU domain. Similar studies have suggested that the POU-homeo subdomain of Oct-1 contactstheTAATsequenceof the oLTIFcis-acting site, the
POU-specific subdomain contacts the ATGC sequence im-mediately upstream of the TAAT sequence, and aTIF may even contact the GARAT sequences (34). Importantly, re-centcrystallographicevidence shows that the homeo domain of the engrailed protein contacts an at TAATsubsite in the DNA (29). With respect to these findings, we suggest that mutants13,18, and 19 bind Oct-1weakly by interactionwith thePOU-homeo subdomain and that the wild type sequence is capable ofestablishing high-affinity binding through in-creased interactions with the POU-specific subdomain.
Al-though Oct-1 exhibits weaker relative binding toward the a(27 promoter TAATTAAATac sequence than toward the a4 promoter TAATGAGATgc sequence, the presence of the
high-affinity overlapping octamermotif in the (x27 promoter more than compensates.
Role ofaTIFin theinduction of a genes. Just as an Oct-1
binding site is a critical component of the aTIF cis-acting
site, so is the GARAT sequence which mediates specific aTIFprotein-DNA complex formation. We were intrigued
by ourprevious results obtained in Vero cells, which dem-onstrated that viruses mutated within the TAATGARAT sequences fell into three distinct groups: (i) those with no octamer binding site (mutant 6), which expressed ot27-tk mRNAatthe lowest level (6 to7%of the wild-type level), (ii) those with strong octamerbinding sites but mutated within the GARAT sequences (mutants 7 and 14), which formed Oct-1 butnotaTIFDNA-protein complexes and expressed ot27-tk mRNA at an intermediate level (15 to 30% of the
wild-type level), and (iii) those which formed complexes withboth Oct-1 andaTIFandexpressedot27-tk mRNA at or closetowild-typelevels. Ourexpectation had been that cells infected by viruses mutated in the GARAT sequence would demonstrate thesamereduction of reporter gene expression ascould be observed in cells infected with viruses mutated in theOct-1bindingsite.Clearly, in Vero cells, the presence of J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.57.297.107.302.2]anOct-1 binding site allowed for increased expression of the reporter gene, even in the absence of the GARAT se-quences, although these levels were significantly below wild-type levels. By retesting these viruses in another cell type, limited-passage HEL cells, we have demonstrated an even lesser dependence on the GARAT sequence for signif-icant levels of a gene expression. Indeed, a gene expression from promoters mutated in the GARAT sequences was often as high as 77 to 79% that of wild-type promoters in HEL cells, although these high levels were not regularly repro-ducible. As the growth characteristics of HEL cells are notablydifferent from those of Vero cells, suchvariability in gene expression is not completely surprising. We postulate that (i) an unknown cellularfactor is capable of interacting with Oct-1 to induce a gene expression, albeit to a lesser degree than when Oct-1 is complexed with aTIF, (ii) this cellularfactor has no dependence on the GARAT sequence as doesaTIF, and (iii) HEL cells possess more of this factor than Vero cells, but only in certain physiologic states. The presence of such afactor could explain the high levels of a gene expression observed in transformed cell lines, evenin the absence of aTIF (38).
Role ofmultipleaTIF-responsive elementsin promoters of a genes. By restoring anadditional 1,334 bp ofcontiguous upstream sequences to the 275-bp a27 promoter (virus 21), we were able to increase the level of a gene expression observed four- toeightfold over the level observedwith the 275-bppromoter construct. The increased expression can be attributed to the homologs of the aTIF-responsive element contained within this sequence at thelocations indicated in Fig. 1.Mutation of the proximalOct-I binding site decreased a gene expression three- to sixfold even in the presence of these additional sequences (virus 20), indicating that the aTIF-responsive element located nearest the TATA box exerts agreater influence on the level of a geneexpression than do those elements located farther away. Increasing
interest has focused on the possible role of aTIF as a coactivator, which along with other proteins may form a bridgebetweenOct-i bound to the aTIFcis-acting site in the DNAand the transcriptionalmachinery located at the TATA box (6,28). In thisregard, the acidic activatingdomain of the aTIF molecule has been reported to specificallyretain both the general transcription factor TFIIB and the TATA box factorTFIID on anaffinity column, while specificmutations withinthis domain prevent suchinteractions (12, 36,54). We envisage the aTIF-hostfactor complex bound to the DNA scanning the region for such transcriptional proteins at TATAboxes. The greater the local concentration ofbound
aTIF protein-DNA complexes and the nearertheyare tothe TATAbox, the morefrequently theymight interact with the transcriptional machinery to induce a gene expression. In summary, the organization ofa gene promoters and
avail-able data on mutantsin theaTIFsites suggestthat thelevel of mRNA induction observed for a given a gene is deter-mined by (i) the number of Oct-1 binding sites located
upstreamof the gene,(ii) theproximity of thesesitesto the TATAbox,(iii) theaffinity withwhichthese sequencesbind Oct-1, and (iv) theability of theseelements, mediatedbythe GARAT sequence, to form protein-DNA complexes with aTIF.
Role of aTIF-responsive elements in the expression of a genes in untreated infected cells. As one objective of these studieswas toexamine theroleofthe targetsequences inan infected cell in the genomic environment in which these sequences were intended tofunction, whether
they
should function in induction, shutoff, or maintenance of a geneexpression, we
performed
severalexperiments
in the ab-senceofcycloheximide.Although
wedidnotdetermineany sites involved in the shutoff or maintenance of a geneexpression, we point out that if these sequences were the same asthoseinvolved in
induction,
wecouldnotdetermine themthroughthisapproach.Weweremoresurprised
by
thehighlevels of a27-tk mRNA observed in cellsinfectedwith allofthe mutant viruses in the absence of
cycloheximide,
despitethedrastic mutations introducedin severalofthem. Weconclude thatagene promoters possess morethanone element
capable
ofstimulating significant
levels ofexpres-sion in the infected cell and that loss ofany one of these elements isnotsufficientto
severely
impair
expression
of the gene. Sucharesult isunexpected
in viewofthestrength
ofaTIF-mediated
transinduction,
which isreadily
demonstra-blein the presence ofcycloheximide
in the infected cell andin various transient expression systems
(10, 31,
37,
48,51,
57).
Significantly,
Ace et al.(1,
2)
have constructed anHSV-1mutant
possessing
a12-bp
insertionintheaTIF gene,whichreportedly rendersthemoleculeunabletotransinduce a genes. The virus establishes latent infection in vivo and
reactivates
efficiently
fromexplanted
trigeminal
ganglia
(52).
Whereas the levelsofmost agene mRNAsaccumulated in
cells infected with this virus were reduced
only
four- tofivefold,
the a4 mRNAlevelswerenot redudedatall(2).
Theobservationsthatmutations introducedeither into the
aTIF
protein
orwithin the aTIFcis-acting
site donothavesignificant
effectsontransinduction ofagenes in the infectedcell suggest that similar results will emerge from studies of the effects of mutations in promoter domains ofgenes in other kinetic classes.
Indeed,
all studiesperformed
todateinvolving
specific
mutationalanalysis
ofpromoter-regula-toryelementsofHSV-1geneswithinthecontextof the viral genomehavedemonstrated minimalor noeffectonobserved levels of gene
expression
as a result of the introducedmutations,
even in those instances in which similarmuta-tions have
produced
readily
documented effects whenana-lyzed
in transientexpression
systems. Forinstance,
EverettandOrr
(15)
recently
reported
thatmutationsintroduced intothe a4
binding
site ofthe aO gene hadnoeffectonaOprotein
levels when
analyzed
in the context oftheviral genome, in contrast totheincreasedlevelsofaO mRNAobservedwhen thesamesite
is mutated andanalyzed
inatransient expres-sion system(15,
17, 18, 47).
Boniand Coen(8)
reported
that mutations introduced in both SP1 sites or in an octamermotifof the
P
tk gene promoter at mostmodestly
impaired
viral tk geneexpression.
Experiments
in which a4binding
sites present in the promoters and nontranscribed leader sequences offy genes have been
specifically
mutated havedemonstratedminimaleffects when examined in thecontext
of
the
viral genome(47a).
Ourinterpretation
of these results is that many if not most HSV genes contain numerous,functionally
redundantnonhomologous
domains to secure aspecific
levelofexpression
and thatablationofone sitemay not be sufficient togrossly
affect theexpression
of the mutated gene.ACKNOWLEDGMENTS
ThesestudieswereaidedbyPublic Health Servicegrantsfrom the National CancerInstitute
(CA47451)
and the NationalInstitute for AllergyandInfectiousDiseases(AI124009andA11588-11).D.S. isapredoctoraltrainee under USPHS
training
grantGM7281. REFERENCES1. Ace,C.I., M.A.Dalrymple, F.H.Ramsay, V.G.Preston,and C.M. Preston. 1988. Mutational
analysis
oftheherpes
simplex
on November 10, 2019 by guest
http://jvi.asm.org/
3512 SPECTOR ET AL.
virus type 1 trans-inducing factorVmw65. J. Gen. Virol. 69: 2595-2605.
2. Ace,C. I., T.A.McKee,M. Ryan,J.M.Cameron, andC. M. Preston. 1989. Construction and characterization of a herpes simplex virustype 1 mutant unable totransinduce immediate-earlygeneexpression.J. Virol. 63:2260-2269.
3. ApRhys,C. M.J.,D. M.Ciufo,E. A. O'Neill,T.J. Kelly,and G. S.Hayward. 1989. Overlappingoctamerand TAATGARAT motifs in theVF65-responsiveelements inherpessimplexvirus immediate-earlypromoters representindependent bindingsites for cellular nuclear factor III. J. Virol. 63:2798-2812.
4. Batterson, W.,and B. Roizman. 1983. Characterization ofthe herpes simplex virion-associated factor responsible for the induction ofotgenes. J. Virol.46:371-377.
5. Baumruker, T., R. Sturm, and W. Herr. 1988. OBP100binds remarkably degenerateoctamermotifsthroughspecific interac-tionswithflankingsequences.Genes Dev. 2:1400-1413. 6. Berger,S.L., W. D.Cress,A.Cress,S.J. Triezenberg,and L.
Guarente. 1990.Selective inhibition of activated but not basal transcription bytheacidic activation domain ofVP16:evidence fortranscriptionaladapters. Cell 61:1199-1208.
7. Blair,E.D.,and E. K.Wagner.1986. Asingleregulatory region modulates cis activation and trans activation of the herpes
simplex virus VP5 promoterintransient-expression assays. J. Virol. 60:460-469.
8. Boni, J., and D. M. Coen. 1989. Examination ofthe roles of
transcriptionfactor Spl-binding sites andan octamer motif in
trans induction ofthe herpes simplex virus thymidine kinase gene. J. Virol. 63:4088-4092.
9. Bradford, M. M. 1976. A rapid and sensitive method for the
quantitation of microgram quantities of protein utilizing the
principleofprotein-dyebinding. Anal.Biochem. 72:248-254. 10. Bzik, D. J., and C. M. Preston. 1986. Analysis of DNA
se-quenceswhichregulatethetranscriptionofherpes simplexvirus immediateearlygene 3: DNA sequencesrequiredfor enhancer-like activity andresponse totrans-activationbyavirion
poly-peptide. Nucleic Acids Res. 14:929-943.
11. Campbell, M. E. M., J. W. Palfreyman, and C. M. Preston. 1984. Identification of herpes simplex virus DNA sequences which encode atrans-acting polypeptide responsiblefor stimu-lationofimmediate early transcription.J. Mol. Biol. 180:1-19. 12. Cress, W. D., and S.J. Triezenberg. 1991. Critical structural elements of theVP16transcriptionalactivationdomain.Science 251:87-90.
13. Dalrymple, M. A., D. J. McGeoch, A. J. Davison, and C. M. Preston. 1985. DNA sequence ofthe herpes simplex virus type 1gene whoseproductis responsible fortranscriptional
activa-tionofimmediateearlypromoters.Nucleic AcidsRes. 13:7865-7879.
14. Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from mammalian nuclei. Nucleic Acids Res. 11:1475-1489.
15. Everett,R. D., and A. Orr. 1991. The Vmwl75bindingsite in the IE-1 promoter has no apparent role in the expression of VmwllOduring herpes simplexvirustype 1infection.Virology 180:509-517.
16. Fletcher, C.,N.Heintz,and R.G. Roeder.1987.Purification and characterizationofOTF-1,atranscriptionfactorregulatingcell
cycle expressionofahumanhistoneH2b gene.Cell51:773-781. 17. Gelman, I. H., andS. Silverstein. 1987. Herpes simplex virus
immediate-early promoters are responsive to virus and cell trans-actingfactors. J. Virol.61:2286-2296.
18. Gelman,I.H.,andS.Silverstein. 1987. Dissection of
immediate-earlygene promotersfromherpessimplexvirus: sequences that
respond to the virus transcriptional activators. J. Virol. 61: 3167-3172.
19. Gerster, T., and R. G. Roeder. 1988. A herpesvirus
trans-activatingprotein interactswithtranscriptionfactorOTF-1 and other cellularproteins. Proc. Natl. Acad. Sci. USA 85:6347-6351.
20. Greaves, R., and P. O'Hare. 1989. Separationofrequirements for protein-DNA complex assemblyfrom those forfunctional
activity in theherpes simplex virusregulatory protein Vmw65. J. Virol. 63:1641-1650.
21. Greaves, R., and P. O'Hare. 1990. Structural requirements in the herpes simplex virus type 1 transactivator Vmw65 for interaction with the cellularoctamer-bindingprotein and target TAATGARAT sequences. J. Virol.64:2716-2724.
22. Haigh, A., R. Greaves, and P. O'Hare. 1990. Interference with the assembly of a virus-hosttranscription complexbypeptide competition. Nature(London) 344:257-259.
23. Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol. 14:640-651.
24. Herr, W., R. A. Sturm, R. G. Clerc, L. M. Corcoran, D. Baltimore, P. A. Sharp, H. A.Ingraham, M. G.Rosenfeld, M. Finney, G. Ruvkun, and H. R. Horvitz. 1988. The POU domain: alarge conserved region in the mammaian pit-1, oct-1,oct-2, andCaenorhabditiselegansunc-86 gene products. Genes Dev. 2:1513-1516.
25. Honess, R. W., and B. Roizman. 1974. Regulation ofherpesvirus macromolecularsynthesis. I. Cascade regulation of the synthe-sis of threegroups of viral proteins. J. Virol. 14:8-19. 26. Honess,R.W., and B. Roizman. 1975. Regulation of herpesvirus
macromolecularsynthesis: sequentialtranscription of polypep-tidesynthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 72:1276-1280.
26a.Hubenthal-Voss,J., and B. Roizman. Unpublisheddata. 27. Ingraham, H. A., S. E. Flynn, J. W. Voss, V. R. Albert, M. S.
Kapiloff, L. Wilson, and M. G. Rosenfeld. 1990. The POU-specific domain of Pit-1 is essential for sequence-POU-specific high affinity DNA binding and DNA-dependent Pit-i-Pit-1 interac-tions. Cell 61:1021-1033.
28. Kelleher, R. J., III, P. M. Flanagan, and R. D. Kornberg. 1990. A novel mediator between activator proteins and the RNA polymeraseIItranscription apparatus. Cell 61:1209-1215. 29. Kissinger, C. R., B.Liu,E.Martin-Blanco, T. B. Kornberg, and
C. 0. Pabo. 1990. Crystal structure of an engrailed homeo-domain-DNA complex at 2.8 A resolution: a framework for understanding homeodomain-DNA interactions. Cell 63:579-590.
30. Kristie, T. M., J. H. LeBowitz, and P. A. Sharp. 1989. The octamer-binding proteins form multi-protein-DNA complexes withtheHSVaTIFregulatory protein. EMBOJ. 8:4229-4238. 31. Kristie, T. M., and B. Roizman. 1984. Separationof sequences definingbasalexpressionfrom thoseconferringagene recogni-tion withintheregulatorydomains of herpes simplex virus1 a genes.Proc. Natl. Acad. Sci. USA 81:4065-4069.
32. Kristie, T. M., and B. Roizman.1987.Hostcellproteinsbind to the cis-acting site required for virion-mediated induction of herpes simplex virus 1 agenes. Proc. Natl. Acad. Sci. USA 84:71-75.
33. Kristie, T. M., and B. Roizman. 1988. Differentiation and DNA contact points of host proteins binding at the cis site for virion-mediatedinduction ofagenesof herpes simplex virus 1. J. Virol. 62:1145-1157.
34. Kristie,T.M., and P.A.Sharp. 1990. Interactions of the Oct-1 POU subdomains with specific DNA sequences and with the HSVa-trans-activatorprotein. Genes Dev. 4:2383-2396. 35. LeBowitz, J. H., T. Kobayashi, L. Staudt, D. Baltimore, and
P. A.Sharp. 1988. Octamer-bindingproteins from B or HeLa cells stimulate transcriptionofthe immunoglobulinheavy-chain promoterinvitro.GenesDev. 2:1227-1237.
36. Lin, Y., and M. R. Green. 1991. Mechanism ofaction of an acidictranscriptionalactivator in vitro.Cell 64:971-981. 37. Mackem, S., andB. Roizman. 1982. Differentiation between a
promoterandregulator regions of herpes simplex virus 1: the functional domains and sequence ofa moveable a regulator. Proc.Natl. Acad.Sci. USA79:4917-4921.
38. Mackem, S., and B. Roizman. 1982. Regulation of a genes of herpes simplexvirus: the a27 gene promoter-thymidine kinase chimera is positively regulated in converted L cells. J. Virol. 43:1015-1023.
39. Mackem, S.,and B.Roizman. 1982.Structuralfeatures of the a gene4, 0, and 27 promoter-regulatory sequence which confer a J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
regulationonchimericthymidine kinasegenes. J. Virol. 44:939-949.
40. McKnight, J. L. C., T. M. Kristie, and B. Roizman. 1987. Binding of the virion protein mediating a gene induction in herpes simplex virus 1-infected cells to its cis-site requires cellular proteins.Proc. Natl. Acad. Sci. USA 84:7061-7065. 41. O'Hare, P., and C. R. Goding. 1988. Herpes simplex virus
regulatory elements and the immunoglobulin octamer domain bindacommonfactorand are both targetsfor virion transacti-vation. Cell 52:435-445.
42. O'Hare, P., C. R. Goding, and A. Haigh. 1988. Direct combina-torial interaction between a herpes simplex virus regulatory protein and a cellular octamer-binding factor mediates specific induction of virus immediate-early gene expression. EMBO J. 7:4231-4238.
43. Pellet,P.E., J. L. C. McKnight, F.J.Jenkins, and B. Roizman. 1985. Nucleotidesequenceand predicted amino acid sequence ofa protein encoded in a small herpes simplex virus DNA fragment capable of trans-inducingagenes. Proc. Natl. Acad. Sci.USA 82:5870-5874.
44. Post, L. E.,S. Mackem, and B. Roizman. 1981. Regulation of a genes of herpes simplex virus: expression of chimeric genes produced by fusionof thymidine kinasewith a gene promoters. Cell24:555-565.
45. Preston, C. M., M. C. Frame, and M. E. M. Campbell. 1988. A complexformed between cell components and anHSV struc-turalpolypeptidebinds to aviral immediateearly gene regula-tory DNAsequence.Cell52:425-434.
46. PruUn, G. J., W. van Driel, and P. C. van der Viiet. 1986. Nuclear factor III, a novel sequence-specific DNA-binding protein from HeLa cells stimulating adenovirus DNA replica-tion. Nature(London)322:656-659.
47. Resnick, J., B. A. Boyd, M. L. Haffey. 1989. DNAbinding bythe herpes simplex virus type 1 ICP4 is necessary for efficient down-regulation oftheICPOpromoter. J. Virol. 63:2494-2503. 47a.Romanelli,M. G., and B. Roizman.Unpublished data. 48. Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne. 1988.
GAL4-VP16 is an unusually potent transcriptional activator. Nature(London) 355:563-564.
49. Silver, S., and B. Roizman.1985. -y2-thymidine kinase chimeras are identically transcribed but regulated as -y genes inherpes simplex virus genomes and as
P
genesin cell genomes. Mol.Cell. Biol.5:518-528.
50. Spear, P. G., and B.Roizman. 1972. Proteins specified by herpes simplex virus. V. Purification and structural proteins of the herpesvirion. J. Virol. 9:143-159.
51. Spector, D., F. Purves, and B. Roizman. 1990. Mutational analysis of the promoter region of the a27 gene ofherpes simplex virus 1 within the context oftheviral genome. Proc. Natl. Acad.Sci. USA 87:5268-5272.
52. Steiner, I., J. G. Spivack, S. L. Deshmane, C. I. Ace, C. M. Preston, and N. W. Fraser. 1990. Aherpessimplexvirus type 1 mutant containing a nontransinducing Vmw65 protein estab-lishes latent infection in vivointhe absence ofviralreplication and reactivatesefficiently from explanted trigeminalganglia.J. Virol. 64:1630-1638.
53. Stern, S., M. Tanaka, and W. Herr. 1989. The Oct-1 homeo-domain directs formation ofamultiprotein-DNA complex with theHSV transactivatorVP16. Nature(London)341:624-630. 54. Stringer, K. F., C. J. Ingles, and J. Greenblatt. 1990. Direct and
selective binding ofanacidictranscriptional activationdomain to theTATA-boxfactorTFIID. Nature(London) 345:783-786. 55. Sturm,R., G. Das, and W. Herr. 1988. Theubiquitousoctamer bindingprotein Oct-1 containsaPOU domainwith ahomeobox subdomain.Genes Dev. 2:1582-1599.
56. Sturm, R. A., and W. Herr. 1988. ThePOUdomain isabipartite DNAbindingstructure. Nature(London)336:601-604. 57. Triezenberg,S. J., K. L. LaMarco, and S. L. McKnight. 1988.
Evidence of DNA:protein interactions that mediate HSV-1 immediateearly gene activation by VP16.Genes Dev. 2:730-742.
58. Triezenberg, S. J., R. C.Kingsbury,andS. L.McKnight. 1988. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 2:718-729.
59. Verrizer,C.P., A. J.Kal,and P. C. van der Vliet. 1990. The oct-1homeodomaincontactsonlypartoftheoctamersequence and fulloct-1 DNA-binding activity requiresthe POU-specific domain.GenesDev. 4:1964-1974.
60. Xiao, P., and P. Capone. 1990. Acellular factor binds to the herpes simplex virus type 1 transactivator Vmw65 and is required forVmw65-dependent protein-DNA complex assem-blywithOct-1. Mol. Cell.Biol. 10:4974-4977.