0022-538X/93/020876-10$02.00/0
Copyright© 1993, American Society for Microbiology
Intragenic Complementation
of
Herpes Simplex
Virus
ICP8
DNA-Binding
Protein
Mutants
MIN GAOtANDDAVID M. KNIPE*
Department ofMicrobiologyandMolecularGenetics, Harvard MedicalSchool, 200
Longwood
Avenue, Boston,
Massachusetts 02115Received 13July1992/Accepted30 October1992
ThemajorDNA-binding protein, orinfected-cell protein 8 (ICP8),ofherpes simplexvirus isrequiredfor viralDNAsynthesisand normalregulationofviralgeneexpression. Previous geneticanalysishasindicatedthat the carboxyl-terminal 28 residues aretheonlyportion of ICP8capableofactingindependentlyas a nuclear
localization signal.In this study,weconstructeda mutantvirus (nllSV) inwhich thecarboxyl-terminal28 residuesofICP8werereplaced bythesimian virus 40large-T-antigennuclearlocalization signal.ThenllSV ICP8 localized intothe nucleus and boundtosingle-strandedDNAinvitroastightlyaswild-typeICP8 didbut
wasdefectiveforviral DNAsynthesisand viralgrowthinVero cells. TwomutantICP8proteins(TL4andTLS) containing amino-terminal alterations could complement the nlSV mutant but not ICP8 gene deletion mutants.Celllinesexpressing TILAandTL5ICP8wereisolated,and inthesecells, complementationofnllSV wasobservedatthelevels of both viral DNAreplicationand viralgrowth.Therefore, complementationbetween nllSVICP8 andTL4orTLM ICP8 reconstitutedwild-typeICP8 functions. Our results demonstrate that(i)the carboxyl-terminal28 residues of ICP8arerequiredforafunction(s)involved in viral DNAreplication, (ii)this
function canbe supplied intrans byanother mutant ICP8, and (iii) ICP8 has multiple domains possessing differentfunctions, and atleastsomeofthesefunctions cancomplementintrans.
The major DNA-binding protein of herpes simplexvirus (HSV), infected-cellpolypeptide 8,orICP8,isoneofseven
virus-encoded proteins required forreplication ofthe HSV
genome(3, 4, 20, 24, 39, 40). ICP8, a 130-kDapolypeptide,
is expressed as a
0i
ordelayed-early-gene product (20, 24).From studies of thephenotypes ofvirus strains containing temperature-sensitive (4, 39), nonsense, deletion, or site-specific mutations (5, 8-10, 12, 23) in the ICP8gene, ICP8 has beenshowntohavetheabilityto(i)bindtoDNA invitro and invivo(1, 16,18, 25, 31), (ii)localizetothe cell nucleus (10), (iii) down-regulate the expression ofviral genes from parental genomes (12), (iv) stimulate late-gene expression fromprogenytemplates (9), and(v)promoteorganizationof nuclearstructures involved in viral and cellular DNA repli-cationproteins (5).
ICP8 bindspreferentiallytosingle-strandedDNA(ssDNA) invitro (1, 16, 18, 25, 32, 33), and the interaction ofICP8 with ssDNA is cooperative (18, 22, 31). On the basis of studies ofICP8 moleculesexpressedbymutantviruses,of in vitrotranscription-translation products ofICP8, and of
par-tialproteasedigestionofpurified ICP8, the ICP8sequences
required for ssDNA-binding activity have been mapped between residues 564 and 849(8, 19, 38). Genetic evidence indicates that ICP8 specifies other nuclear functions in additiontoDNAbinding (8, 9).
Theintranuclearlocalization of ICP8tospecificstructures
appears to be required for ICP8 to exert its nuclear func-tions. In theabsence ofviral DNAreplication, ICP8 local-izes to nuclearframework-associated structures called
pre-replicative sites (5, 26, 27). These structures have been hypothesized to be the sites of viral and cellular protein
*Correspondingauthor.
tPresent address:DepartmentofVirology, Bristol-MyersSquibb Co.,Princeton, NJ08543.
complexes poised to initiate viral DNA synthesis. As viral DNA replication occurs, progeny viral DNA-ICP8
com-plexes and additional viral and cellular proteins migrate to large globular replication compartments(5, 26). HSV
poly-merase colocalizes with ICP8 in both replicative sites and replication compartments. However, localization of HSV polymerase to these structures is dependent on functional
ICP8 molecules. Therefore, ICP8 is required for the
assem-bly of prereplicativesites (5).
Genetic analysis of ICP8genemutantsandICP8-pyruvate kinase fusion proteins demonstrated that the carboxyl-ter-minal 28 residues of ICP8 constitutetheonly portion ofICP8 thatcanfunction aloneas anuclearlocalization signal(NLS) (10). The carboxyl-terminal 28 residues of ICP8 are not needed forssDNA-binding activity(10).
Tostudy the functional domains of ICP8 further,wehave
constructed amutant virus(nllSV)inwhich the carboxyl-terminal 28 residues of ICP8 were replaced by the nuclear
localization signal of simian virus 40 (SV40) T antigen. Despite the fact that nllSV was unable to promote viral DNAsynthesis, nllSV ICP8 still retainssomephysical and
functional properties of wild-type (wt) ICP8. This paper
reports that the carboxyl-terminal 28 residues of ICP8 are
required forafunction(s) involved in viral DNA replication
and that thefunction(s) missinginnllSVcanbesupplied in
trans by anothermutant ICP8 molecule. MATERUILSANDMETHODS
Cellsand viruses. Vero cellswere grownandmaintainedas
described previously (14). The growth medium forthe
neo-mycin-resistant S-2 cell line included 200 ,ug of the anti-bioticG418perml duringthefirstpassageof the cells after
thawing or 500 ,ug of G418 per ml of medium every five passages.
876
on November 9, 2019 by guest
http://jvi.asm.org/
COMPLEMENTATION OF HSV ICP8 MUTANTS 877
L S
-l II -l 4II
Bamn s
0.41 0A0 039 038
wt ICP8
n1l
nllSV
n9
d301
p8 B-S
TL4
TLS
TLI 6
1169
832
264 932
204
207
732
pSVdlOl1
17 564
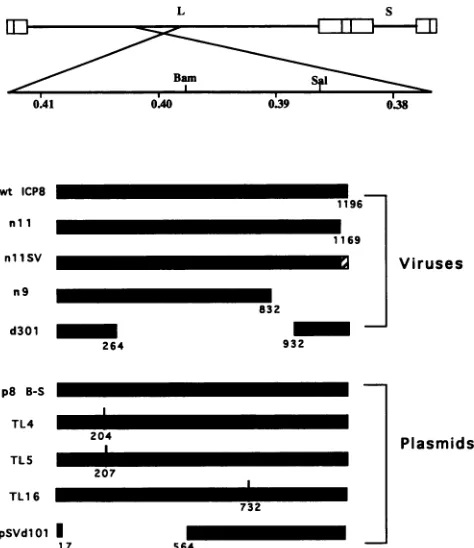
FIG. 1. Locations ofICP8genemutations used in this study. The location of theICP8 coding regiononthe HSVgenomeis shownat thetop.Restriction sites shown areBamHI andSalI. The hatched
barinmutantvirusnllSVrepresentstheSV40 T-antigen NLS (see Materials andMethods). L, longcomponent;S, shortcomponentof the viralgenome.
TL4 and TL5 cell lines were isolated by procedures
described previously (6, 8). Vero cells were transformed
with the plasmid TL4 or TL5 and pSVneo (Fig. 1) in the
presenceof theantibiotic G418 (36).Drug-resistant colonies
wereisolated,grownintocultures,andscreened byindirect
immunofluorescence (27) with anti-ICP8 monoclonal anti-body lOE-3 (30) for the ability to express mutant forms of ICP8 upon infection by nlO mutant virus. The lOE-3 anti-body recognizes TL4ICP8 and TL5 ICP8 butnotnlOICP8 (10).
HSVtype1wtstrainKOS1.1waspropagatedandassayed
asdescribedpreviously (16, 17).Mutant virusesweregrown
inICP8-expressingS-2 cells (8).
Plasmids. Plasmids p8B-S, pICP8, pSV8, pSV8.2, and pSVdlOlaswellasthenucleotide-numberingsystemfor the ICP8 gene were described previously (7-9). The plasmid p8B-S contains the ICP8gene, includingits ownpromoter.
Theplasmid pICP8contains the ICP8genecodingsequences
without its promoter. The plasmid pn9 was generated by
linearization of theplasmid pICP8 (whichwas achievedby partial digestion with SmaI) and subsequent insertion of a
14-nucleotide XbaI linker (New England BioLabs, Inc., Beverly, Mass.) containing stop codons in all threereading frames.Thus, pn9encodesthe first 833 amino acid residues ofICP8. Theplasmid pSV8 wasconstructed by inserting a
5.5-kbp SmaI-SacI fragment (map units 0.374 to 0.409) downstream of the SV40 early promoter. The plasmid pSV8.2 was derived from pSV8 by deleting the polylinker region and the SacI-BglII fragment of thepyruvate kinase
gene. The plasmid pSVdlO1 was generated as described previously (8); it lacks codons for residues 17 to 563 but has an insertion of one Arg codon encoded by the BglII linker sequence. The plasmids p8S/3583 and p8E/3583 were de-rived from pICP8 by deleting the NaeI (nucleotide 3583)-SacI(nucleotide 6075) and the SmaI (nucleotide 436)-NaeI (nucleotide 3583) fragments, respectively. The plasmids
TL4, TL5, and TL16 were constructed as described previ-ously (37).Theycontain eight, six, or two codon insertions afterresidues204, 207, or 732, respectively, of ICP8.
The plasmid p8N/12B-1 was constructed by inserting a 12-nucleotide BglIIlinker at an NaeI site (nucleotide 4111). The plasmid pSVnll was generated by linearization of the plasmid p8N/12B-1 and subsequent insertion of an XbaI
linker containing stop codons in all three reading frames. Thus, pSVnll encodes the first 1,169 amino acid residues of ICP8 aswell as 4 additional amino acids (Thr-Ser-Leu-Asp)
encoded by the XbaI linker sequence. The plasmid pSVnllSV was generated by linearization of the plasmid pSVnll withXbaI and subsequent insertion of a 27-nucle-otide linker (5'-CTAGCACCAAAAAAGAAGAGAAAGG
TA-3')encoding the SV40 large-T-antigen NLS. The correct orientation of the insertion was confirmed by DNA sequenc-ing. Thus, pSVnllSV encodes the first 1,169 amino acid residues of ICP8 as well as the additional amino acids
Thr-Ser-Leu-Ala-Pw-L-L
-Ly-Arg-L
s-Yal-Leu-Asp.
The underlined amino acid sequences represent the SV40 NLS (13).
Isolation of nll and nllSV mutantviruses.The strategy for
isolationof nll and nllSVmutantviruses wasasdescribed
previously(8).The HD-2mutantviruscontainsanin-frame
insertion of thelacZgenein the ICP8 coding sequence and forms blue plaques in the presence of
5-bromo-4-chloro-3-indolyl-p-D-galactopyranoside
(X-Gal). This virus served as a recipient in marker transferexperiments tointroduce then9, nll, and nllSV mutations in theICP8geneinto theviral
genome.
Indirect immunofluorescence. Indirect
immunofluores-cence was performed as described previously (27) with anti-ICP8 monoclonal antibody lOE-3or793 (1:50dilution;
30) and rhodamine-conjugated goat anti-mouse antibody (1:100 dilution).
Marker rescue and complementation experiments. Marker rescueexperimentswereperformed by cotransfection of S-2 cells with 1.0 ,ug each ofinfectiousmutant virus DNAand linearizedwtICP8 geneplasmid DNA(Fig. 1)aspreviously
described (15). Transfected cultures wereincubated at 37°C
for 2 to3 days, and progeny viruswas assayed inS-2 and Verocells.
Complementation of thegrowth ofn9,
d301,
andnllSVmutant viruses by prior transfection of the
plasmid
TLA,
TL5,
TL16, orpSVdlOl
wascarriedoutasdescribedprevi-ously(28).
Analysis
ofviral proteins and viral DNAreplication. Verocell monolayer cultures were infected with
KOS1.1,
nll,
nllR, nllSV, ornllSVR and labeled with
[35S]methionine
for 30 min before harvest at9 h
postinfection (hpi).
Sodiumdodecyl
sulfate-polyacrylamide gel
electrophoresis
(SDS-PAGE) of infected-cell lysates was
performed
asdescribedpreviously (17). After
electrophoresis,
thegels
werefixed,
dried, andexposed toKodak SB5 film.
Analysis ofviral DNA
amplification during
thecourse of infectionwasperformedasdescribedpreviously (9,
29).
Theprobeusedwasthe
plasmid
pBR3441
(21).
VOL. 67,1993
Li
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.64.301.70.344.2]¢ zr,:<X~~EOW.... _
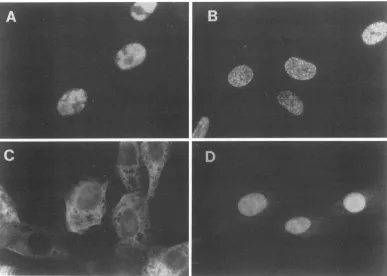
FIG. 2. Subcellular localization of ICP8 encodedbymutantvirusesnll and nllSV.Verocellswereinfected withKOSl.iorICP8mutant
viruses.At 5hpi,the cellswerefixed, permeabilized, and incubatedwith anti-ICP8monoclonalantibody793 andrhodamine-conjugatedgoat anti-mouse immunoglobulin. Immunofluorescencemicrographs areofKOSl.i-infectedcells (A), KOSi.l-infected cells in thepresenceof PAA(B), nll-infected cells(C),andnllSV-infected cells(D).
ssDNA cellulose chromatography. ssDNA cellulose
chro-matography ofinfected-cell extracts was performed as de-scribed elsewhere (8, 14).
RESULTS
Phenotypeof mutant viruses nll andnIlSV.Ourprevious genetic analysis demonstrated that the mutant nlO ICP8, which lacks thecarboxyl-terminal36 residues and localizes to the cytoplasm, does not support viral growth in Vero
cells even though it binds ssDNA in vitro as tightly as wt ICP8 does (8). Further study indicated that the
carboxyl-terminal 28 residues are the onlyportion of ICP8 capable
of acting independently as an NLS (10). Thus, the failure of mutant nlO ICP8 to support viral DNA synthesis may have been solely a consequence of its inability to localize to the cell nucleus. If this were the case, replacement of ICP8 NLS with another known NLS should restore the full functionality of ICP8. Therefore, we constructed two ICP8 mutant viruses. One, nll, lacks the carboxyl-ter-minal 28 residues of ICP8, and the other (nllSV) has the SV40T-antigen NLS (NH2-Pro-Lys-Lys-Lys-Arg-Lys-Val-COOH) replacingthe carboxyl-terminal 28 residues of ICP8
(Fig.
1).
(i) Subcellular locations of nll ICP8 and
nl1SV
ICP8. We first used immunofluorescence microscopy to examine the intracellular localization of the nll and nllSV ICP8poly-peptideswhentheywereexpressed from plasmids in
trans-fectedVero cells.The
nil
ICP8 polypeptide remained within thecytoplasm, while the nllSV ICP8 polypeptide localized in the nucleus (11). We next determined the intracellulardistribution of mutant ICP8 molecules when they were
expressed from recombinant viruses (Fig. 2). At 5 hpi,
infected Vero cellswerefixed andprocessedfor immunoflu-orescence. Cells infected with wt virus showed that ICP8 localized in the nucleus in characteristicreplication compart-ments in the absence ofphosphonoaceticacid (PAA) (Fig. 2A) andatprereplicativesites in the presenceof PAA(Fig.
2B; 26, 27). As expected, nll ICP8 localized in the
cyto-plasm(Fig. 2C)because of lack of the carboxyl-terminal28 residues. Inmostcells,thenllSV ICP8 localizedthroughout
the cell nucleus (Fig. 2D) and did not appear to localize
specificallyeither inreplication compartmentsor at prerep-licative sites. Therefore, replacement of the ICP8 NLS with the SV40 T-antigen NLS restored the capacity of mutant ICP8tolocalize into the nucleus butnot tothesamesitesas wtICP8.
(ii) Growth properties of nll and nllSV. Plaque assays were performed to determine whether the mutantviruses
wereabletogrowin Vero cells. Both mutant viruses failed togrow in Vero cells but grewtotitersnearthoseofwt on the ICP8-expressing S-2 cellline (Table 1). Thus, although
replacementof the ICP8 NLS with the SV40T-antigen NLS restored the ability ofICP8 tolocalize to the nucleus, the substitution didnot restorecomplete functionality of ICP8. (iii) Marker rescueofmutantviruses nll and nllSV. To verify that the phenotypes of mutant viruses nll and nllSV were due to mutations in the ICP8 gene, we performed marker rescue experiments (Fig. 3). Homologous recombi-nation between nll DNA or nllSV DNAand a fragment containingawt ICP8 DNA sequence(p8S/3583) resulted in viral progeny possessing the ability togrow in Vero cells,
thereby rescuingthegrowth deficiencies of nll and nllSV.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.117.504.75.351.2]COMPLEMENTATION OF HSV ICP8 MUTANTS 879 TABLE 1. Growth ofnllandnllSVa
Titer(PFU/ml)
Virus
S-2cells Vero cells
KOSl.1 1.7 x 109 1.8 x 109
nll 2.3x109 2.0x103
nllSV 2.8x 108 2.0 x 103
a Cultures of ICP8-expressingcells(S-2) and Verocellswereinfected with each virus and incubated at 37°C. Plaqueswerecountedat 3days.
TABLE 2. Growthproperties of nllRandnllSVRa
Titer(10' PFU/ml)
virus
S-2cells Verocells
nllR 3.8 2.8
nllSVR 1.3 2.0
a Cultures ofICP8-expressingcells(S-2)and Vero cellswereinfected with each virus andincubatedat37°C. Plaqueswerecountedat3days.
The high percentage of rescue (1.5 to 8.3%)
rescue was likely due to a recombinational el
the transfectedplasmidsandnotadditionaleve ofrescuedviruses nllR and nllSVRwere
ap
orders of magnitude greater than those of viruses nll and nllSV on Vero cells and roi their titers in S-2 cells (compare Tables ] conclude that the phenotype of mutant Vir nllSVwas aresult of mutations within the IC likely the engineered mutations.(iv) DNA-binding
properties
of mutant viinllSV. To attempt to define the nature of functions of the mutant ICP8 molecules, we
ssDNA-binding properties of mutant nll IC]
ICP8. Vero cells were infected with nll c extract from infected cells was passed ov
cellulosecolumn, andICP8was elutedstepw
containing increasing salt concentrations. P thevarious fractionswere analyzed bySDS-F The saltelution patterns fornll ICP8 (Fig.4
BamHI Sall
0.41 0.40 0.39 0.38
wt ICP8
607 4195
n1l
607 4111
nil SV
607
p8S/3583
p8E/3
583p8S/3583
pSV8.2
p8E/3583
p8S/3583
pSV8.2
Viru.
=
% wt recc
<O.C 1 .! 3.,
<0.1
1.
FIG. 3. Markerrescueof mutant virusesnllar
bars indicate coding regions of ICP8 genes. Rel
positions of the recombinant plasmids are as fol nucleotides 432 to 3583; p8S/3583, nucleotides 3 mately 5900; andpSV8.2, nucleotides432 to appi
Percentageofwtrecombinants=(virustiterin Ver
inS-2 cells) x 100%.
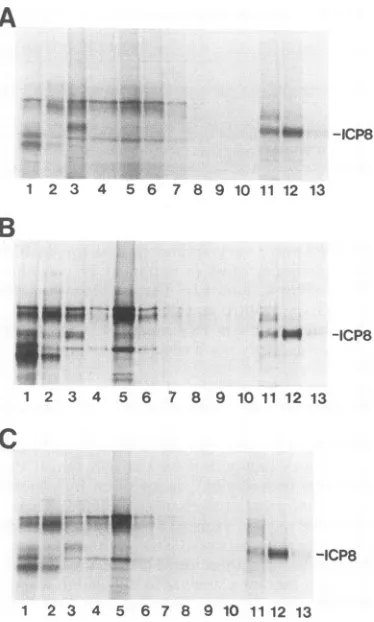
indicated that
(Fig. 4C)
ICP8 werevery similar to that for wtICP8
(Fig.
ventinvolving
4A),
and themajority
of wt ormutant ICP8waseluted with nts Thetivtergs
0.5 M NaCl(Fig.
4, lane12).
Thus, nll andnllSV ICP8 iproximately. bound to ssDNA in vitro astightly
aswt ICP8 did.There-)proximately
5 fore, the inability of thenllSV
mutant to replicate its DNAf the
parental
in Vero cellswaslikely
not aresult ofaninability
to bindtoughly equal
to ssDNAL and 2). We Intragenic complementation of ICP8 mutants. Despite its
ups8es
nil
aonsd
failureto promote viral DNAsynthesis,
the mutantnllSV
ICP8 retained several of thephysical
andfunctional proper-ties ofwtICP8: itlocalizedtothenucleus,
boundtossDNAruses
nil
and invitro,
and was recognized by the 39S anti-ICP8mono-examined the
clonal
antibody
(not shown),
recognition
by
which is
very P8andnllSVsensitive
toconformational
changes
in ICP8. These results
8
nllSVdthn
suggest
that the
mutantvirus
nllSV
maybe altered in
only
er
an1SV
the asingle functional domain of ICP8 which is involved in viraler
an ssDNA DNA replication. We therefore screened a series of ICP8lse
with
buffer
linker insertion
mutantplasmids
todetermine whether they
olypep(Fidegs
4n
couldcomplement the nllSV defect by providing certain
RAGE (Fig.
[B)
andniiSV
4)S
functions(i) Intragenic complementationof ICP8 intrans.between
mutant virusnllSV and mutant
plasmid
TL4 or TL5. Weperformed
complementation
experiments
todeterminewhetherexpres-Sadc
sion of mutant ICP8polypeptides
from linker insertion mutantplasmids
couldcomplement
thegrowth
of nllSV mutantvirusin Vero cells. All of the linker insertionmutantformsofICP8used inthis
study
wereunabletocomplement
the ICP8 gene deletion mutantvirus d301 andappeared
toses localize into the nucleus in transfected cells (37). The
plas-mid
pSVdlOl
containing
alarge
deletion in the ICP8 genewasused as a
negative control,
andTL16
wasusedalinkerinsertionmutantcontrol
(Table 3).
Neitherof thesemutantscomplemented
thegrowth
of either d301 or nllSV. InDNA fragments contrast, cotransfection of the wt ICP8 plasmid (p8B-S)
increased the
yields
ofmutantvirusd3014,400-
to 10,000-fold andofnllSV
1,500-
to3,000-fold.
Two linkerinsertionmutants, TL4and
TL5,
which couldcomplement
growth
of themutantvirus nllSV in Verocells,
wereidentified(Table
3).
Neitherof thesemutantplasmids
wasabletocomplement
imbinants
d301,
butbothplasmids
complemented
nllSV
significantly.
)12
Forplasmid
TIA,
complementation
ranged
from 7to23%of 5 the level ofwt ICP8plasmid
(Table
3).
Theplasmids
TL4
.4
and TL5 contain 8- and 6-amino-acid insertions after resi-dues 204 and 207ofICP8,
respectively (Fig.
1).
Because the0029
insertions inTL4
ICP8 and TL5 ICP8 are only three residues 9 apart, the same functionaldomain(s)
of ICP8 may beaf-.3 fected.
By
looking
for theability
ofmutants to formplaques
onid
nllSV.
Black
Vero cells (Table4),
we also examined infected-celllysateslative
nucleotide'
[lows: p8E/3583, from the
complementation
experiments
shownin Table 3 for3583
to approxi- wt recombinants. The plasmidpSVdlOl
was used as a roximately5900.
negative
control because deletions in the ICP8 genes of ocells/virustiter mutantspSVdlOl
and d301areoverlapping;
thus,norecom-binationbetween these twomutants should occur.
Recom-p8E/3583pSV8.2
Cotransfected Viral DNA fragments
nll nil nil
nl1SV nil SV nl1SV VOL. 67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.60.302.86.148.2] [image:4.612.63.298.412.659.2]880 GAO AND KNIPE
*owN -ICP8
7 8 9 10 11 12 13
B
# 10,** # '60 -ICP8
_f.,
:-; ..;-i
1 2 3 4 5 6 7 8 9 10 11 12 13
C
,,
_i
s. ..w:
r ": 'WR
-1 2 3 4 5 6 7 8 9 10 1112 13
FIG. 4. ICP8 binding to ssDNA-cellulose. Vero cells were in-fected with KOS1.1(A), nll (B),ornllSV(C) in thepresenceof PAA and labeled with [35S]methionine from 4 to 6 hpi. Various protein fractions resolvedon ssDNAcellulose columnswere sub-jectedtoSDS-PAGE.Lanes: 1,pellet from high-saltDNase
extrac-tion; 2, pellet after dialysis; 3, extractput on ssDNA column; 4 through 10,flowthrough and wash; 11, 0.3MNaCleluate; 12,0.5 M NaCleluate; 13,1.0 MNaCl eluate. PositionsofICP8areindicated
attheright.
bination between the transfected wt ICP8 plasmid and in-fectedmutantviruses didoccur atlowfrequency.However, wtvirusfrequencies were almost thesameford301 (where
[image:5.612.86.273.72.383.2]no complementation occurred) and for TL4 or TL5 (where i I00 -ICP8
TABLE 4. Recombinationbetween ICP8 mutantsa
Plasmid Virus Titer (PFU/ml)
transfected superinfected Expt1 Expt2
pSVdlOl d301 ND ND
pSVdlOl nllSV 10 70
TL16 d301 ND ND
TL16 nllSV ND 50
p8B-S (wt) d301 590 300
p8B-S (wt) nllSV 1,300 800
TIA d301 40 ND
TM4 nllSV 10 60
TL5 d301 10 ND
TL5 nllSV 40 80
[image:5.612.316.555.86.211.2]a Infected-cell lysates from theexperimentswhoseresults areshown in
Table 3 wereassayedfor wtrecombinantsbyassayingfortheabilitytoform plaqueson Verocells. ND =titerof<10PFU/ml.
complementation occurred). Thus, the enhancement of growth ofmutantvirusnllSVbyTMAorTL5was notdueto recombination. We conclude thatthe
function(s) required
for themutantvirusnllSVDNAreplicationcould be suppliedin transbypartiallyfunctional TL4orTL5ICP8.
(ii) Phenotype of TL4 and TL5 mutants. To study the mechanism ofintrageniccomplementation byICP8 mutants, we examined the properties of the TML and TL5 ICP8 molecules. Our results indicated that both TLM and TL5
mutantgeneproductsexhibitedatrans-dominantphenotype
which prevented the introduction of these mutations into virus. We therefore isolatedtwocelllines,namedTL4.6 and TL5.2, which expressedTL4 ICP8 and TL5 ICP8, respec-tively, upon HSV infection (11).
Mutant ICP4 moleculescontainingalterations in the trans-activation domain can form a heterodimer with another mutant ICP4 moleculecontainingan alteration in the
DNA-bindingdomain andcancomplementeach other intrans(34, 35). To test whether this interpretation could apply to the mutant ICP8also,weexaminedthe DNA-binding abilityof TL4 ICP8 expressed from the cell line. We concluded that TL4ICP8,like nllSVICP8,binds ssDNA in vitroastightly
as wt ICP8 does (11). These results suggested that the mechanism of intragenic complementation between nllSV and
TMA
ICP8 mutants is different from that of the ICP4 mutants, because both mutant ICP8 molecules can bind DNA.TABLE 3. Intragenic complementation between ICP8mutantsa
Plasmid Virus Virus yield
(PFU/ml)p
Complementationindexctransfected superinfected
Expt
1 Expt2 Expt1 Expt2pSVdlOl d301 9.0 x 101 3.4 x 102 1.0 1.0
pSVdlOl nllSV 3.0x 102 1.1 x 103 1.0 1.0
TL16 d301 1.3 x 102 6.0 x 102 1.4 2.0
TL16 nllSV 2.3 x 102 1.1 X 103 0.8 1.0
p8B-S (wt) d301 9.2 x 105 1.5 x 106 10,000 4,412
p8B-S (wt) nllSV 9.0 x 105 1.6 x 106 3,000 1,455
TMA d301 1.1 x 102 2.8 x 103 1.2 8
TL4 nllSV 6.4 x 104 3.6 x 105 210 327
TL5 d301 6.2 x 102 3.3 x 103 6.9 10
TL5 nllSV 1.2 x 104 1.8 x 105 40 164
aVerocellsweretransfected withthe plasmids indicated. At 20 h posttransfection, cells were superinfected with 3 PFU of either d301 ornllSVper cell and
incubated forafurther24h at37'Cbefore being harvested.
bDeterminedby plaqueassay onS-2cells.
c Expressedasvirus yield relativetoyieldafter transfection with
pSVdlOl
DNA.A
*-,
t0 .1^...
:.3 is*''v -."I .. j :.
1 2 3 4 5 6
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.58.562.564.687.2]COMPLEMENTATION OF HSV ICP8 MUTANTS 881
Vero
n9
n11SV
KOS
TL4.6
h
FIG. 5. Formation ofreplication compartments in nllSV-infectedTILA.6cells. Vero andTL4.6cells,respectively, were infected with n9 (AandB),nllSV(C andD),orKOSl.1(E and F). At 5 hpi, the cells were fixed, permeabilized, and incubated with monoclonal antibody 10E-3andrhodamine-conjugated goatanti-mouseimmunoglobulin.
(iii) Formation of replication compartments in nllSV-in-fectedTLAandTL5 cells. Because theplasmid TI4resulted inmore-significant complementationof thegrowthof nllSV in Verocells thanTL5did(Table 3),weused TIA.6 cells for most of our further studies. To examine the localization
properties ofTL4 ICP8, we infected Vero and TILA.6 cells
with n9, nllSV,or wtKOS1.1for 5h and thenprocessedthe cells forimmunofluorescencebyusingmonoclonalantibody 10E-3astheprimary antibody (Fig. 5).Thisantibody recog-nizes the carboxyl-terminal 28 residues of ICP8 (10) and therefore recognizes only the ICP8 expressed from TIA.6
cells(Fig. 5B andD)andnotthatexpressed byn9
(Fig.
5A)
ornllSV(Fig. SC).Theantibody,of course, alsorecognizes
wtICP8. KOS1.1-infectedVero cellswereused for controls of theantibody andformation ofreplication compartments.
Replicationcompartmentswereobserved innllSV-infected
(Fig.
SD)
butnotn9-infected(Fig.SB)
TIA.6 cells.Thus,we conclude that TLA ICP8 could localize into replicationcompartmentsin the presence ofnllSV ICP8.
(iv) DNA replication in nllSV-infected TL4.6 cells. To determine whether the mutant virus nllSV replicates its DNAin TL4.6cells,we examined viral DNAaccumulation
in these cells. Total viral DNAwas isolated from
mock-,
wt-virus-,
d301-,
or nllSV-infected Vero cells,wt-ICP8-expressing S-2 cell line, and TIA.6 cells at 1 or 16 hpi.
wt-virus-infected Vero cells in the presence or absence of PAA were used as positive and negative controls of viral DNAsynthesis, respectively (Fig. 6). As
expected,
mutantd301-infected TL4.6 cells, like d301-infected Vero
cells,
showedno
amplification
of viral DNAduringthecourse ofinfection, indicating that the
TL4
ICP8was unable to pro-mote synthesis of mutant viral DNA. In contrast,n11SV-VOL. 67,1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.92.523.71.508.2]S-2
oa
o)0
Xi
CiTL4.6
en
or
80
o
csc
v c, c,
Vero
en o
i- 2e at
'r-
DNA(ng)
1000
200 40
8
I6hr
4m 411 -.4
.4 : *m
1000
200
40
[image:7.612.80.537.72.278.2]E:
8FIG. 6. Mutant nllSV DNA replicationin TL4.6 cells.Vero,wt-ICP8-expressing S-2,andTLA.6cellswereinfected withKOS1.1,d301,
ornllSV. Total cellular DNAwasprepared immediatelyafter viralabsorption (1 h) or nearthe endoftheinfection cycle (16 h). Equal
amountsofeach DNAweresubjectedtofivefold serialdilutions,and the DNAswereboundtoanitrocellulosefilter,whichwasprobed with 32P-labeled DNA specific for the VP16gene. Anautoradiographof the blot isshown.
infected TL4cells showed substantial amplificationofviral DNA during the infection. To quantitate these results, the amount of radioactive probe hybridized to each slot was
measured,and the relative amountsofreplicated HSVtype 1 DNA in each sample were determined. In thisparticular
experiment,theamountofnllSVviral DNAsynthesizedin TL4cellswasabout42%of the level of DNAsynthesizedin wt virus-infected Vero cells. We conclude that the func-tion(s) whichisrequired fornllSVtoreplicateits DNAcan
besuppliedintransbythemutant TIAICP8.
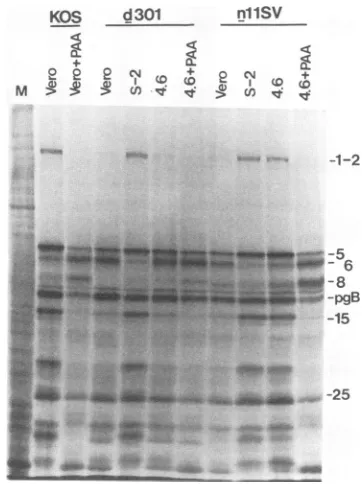
(v) ViralgeneexpressioninnllSV-infected TILAcells. ICP8 is not only required for viral DNA replication but is also involved in stimulation of late-gene expression (9). We therefore examined viral late-gene expression in n11SV-infected TL4.6 cells. Vero cells, wt-ICP8-expressing S-2 cells, and TL4.6 cells were infected with KOS, d301, or
nllSVandlabeledwith [35S]methionine from 9.5to 10hpi, andlabeled proteinswereanalyzed by SDS-PAGE (Fig. 7).
Vero cellsinfectedwithwtvirus inthepresenceorabsence
of PAAwereusedaspositive andnegative controls for the
expression of-y2genes. Whenexamined in Vero cells, the
nllSV mutation resulted inaphenotype similartothat of the negative control with respect to late-gene expression. De-tectable levelsof y2 viralpolypeptides (such asICP1-2 and
ICP15)werenotexpressed. Similar resultswereobservedin
d301-infected Vero cells. These results demonstrated that bothnllSV ICP8 and TM4 ICP8areunableto exert wtICP8 functionsforexpression of lategenes.However,thedefects formutantnllSV virallate-proteinsynthesiswerecorrected
by expressionoftheTL4 ICP8 inTL4.6cells. Thepatternof viralpolypeptides synthesizedinnllSV-infectedTL4.6cells
wasindistinguishable from thatobserved innllSV-infected wt-ICP8-expressing S-2 cells or in wt-infected Vero cells
(Fig. 6).
Tofurther define the level atwhich late-geneexpression
wasrestored,weperformed Northern (RNA)blot analysisto measure thesteady-state level ofy2 glycoproteinC mRNA. These resultsindicated that the intragenic complementation
between nllSV ICP8 and TML ICP8 completely restored viral latemRNA accumulation(resultsnotshown),
although
thecomplementationrestored viralDNAreplicationto
only
42% of the normal level(Fig.
6).
KOS d301
+ 11
M :2?2 uz x t
nilSV
<.
".-..: -1-2
-- 5
Mw 6
-8
_ww -pgB
-15
-25
FIG. 7. Protein synthesis innllSV-infected TLA.6 cells. Vero, wt-ICP8-expressingS-2,orTL4.6 cellswereinfected withKOS1.1, d301,ornllSV. At 8.5 hpi, cellswerelabeled for30min with[35S] methionineand then harvested.Equal fractions of each celllysate
weresubjectedtoSDS-PAGE. An autoradiogram of the resultinggel isshown. At the rightof eachpanelarethemigration positions of several HSVtype1 proteins. Lane M, mock-infected cell lysate.
I
hr
-J
0
E
c0
Cl
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.348.529.413.655.2]COMPLEMENTATION OF HSV ICP8 MUTANTS 883
TABLE 5. Growth of wt andmutant viruses indifferent celllinesa
Virus and Yield (PFU/cell) at MOI of:
cell type 1.0 10.0
KOS1.1
Vero 137 393
S-2 110 306
TL4.6 39 245
TL5.2 4 NT
d301
Vero 0.0006 0.0079
S-2 275 361
TL4.6 0.02 0.33
TL5.2 0.008 NT
nllSV
Vero 0.0003 0.0071
S-2 225 119
TL4.6 44 50
TL5.2 0.9 NT
aCellswereinfected with different viruses, incubated at 37°C for 24 h, and harvested.Titers of progeny viruses were determined in S-2 cells. NT, not tested.
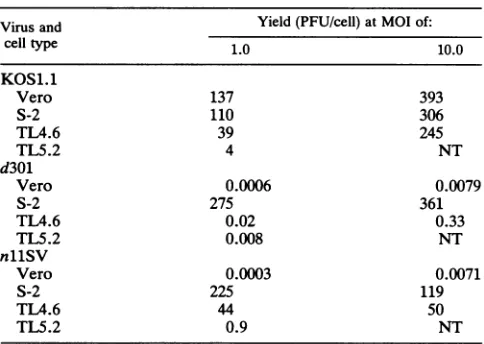
(vi) Growth of nllSVinTL4.6 and TL5.2 cells. Because the plaques of nllSV in TL4.6 or TL5.2 cells were very
small, we performed single-cycle growth experiments to examine thegrowthof nllSV in TL4.6 and TL5.2 cells. The
yields ofwtvirus in TL4.6 andTL5.2cells were 3- to 30-fold lower than those in Vero cells or in the wt-ICP8-expressing
S-2 cellsat amultiplicityofinfection(MOI)of1.0(Table 5), suggestingthattheTL4and TL5mutantgeneproductshave sometrans-dominantnegative phenotype. Theinhibition of
growth ofwtvirusinTL4.6 cellswasovercome at anMOI of 10. Thus, the trans-dominant effect appeared to be due to
competitionbetweenwtICP8 and
TM4
ICP8. Theyields of nllSV in TL4.6 cellswere 7to32%of those ofwtvirus in Verocellsat anMOI of 1 and10,respectively.Becauseonly mutantvirus nllSVbutnotd301could grow inTL4.6 and TL5.2cells,weconcludethat thecomplementationbetween nllSV ICP8 and TL4 or TL5 ICP8 could reconstitute wt ICP8 activities.DISCUSSION
Previous genetic andbiochemical evidence suggests that the functions of the HSV DNA replication protein require
different portions of the molecule: the carboxyl-terminal portion of the molecule is
required
forssDNA-binding,
nuclearlocalization,andstimulation oflate-geneexpression,
and the amino-terminal portion of the molecule has an additional nuclear function(s)
(8).
In this study, we have demonstrated that these functionsarediscrete,becausetwopartially functional ICP8 molecules can complement each other intrans.
Replacement of the ICP8 NLS with the SV40
T-antigen
NLS restoredthe
ability
of ICP8tolocalize intothenucleus,
but themutantvirusnllSV exhibitedadefect in viral DNA
replication and late-gene
expression,
suggesting
that theprimary defect of the nllSV ICP8 is a failure to promote viral DNA synthesis or earlier events such as assembly of viral DNAreplication complexes. The nllSV ICP8 defect can be overcome by
supplying
TLM or TL5 ICP8 intrans. The functions defective innllSVICP8 may be carried outby the carboxyl terminus of ICP8 or other parts of the
molecule; however, extensive conformational
changes
arenot apparent in nllSV ICP8, because it still reacts with anti-ICP8 monoclonal antibody 39S, which is very sensitive to conformational changes in ICP8 (7).
Because monoclonal antibodylOE-3 detectsTM4andTL5
ICP8but notnllSV mutant ICP8, we do not know whether nllSV ICP8 localized alone or formed a complex with
TML
or TL5 ICP8 in thenucleus. However, both
TML
ICP8 and TL5ICP8 localized to replication compartments after super-infection with the mutant virus nllSV but not after super-infection with n9, suggesting that these ICP8molecules can localize to these sites only in the presence of nllSV ICP8. Moreover, because the monoclonal antibody recognized both wt andTM4
ICP8, we know that both ICP8 molecules localized into replication compartments after TL4.6 cells were infected with wt virus. These results suggest thatnllSV ICP8 may alsolocalize toreplication compartments. Because the alter-ation inTML
or TL5 ICP8, as innllSV ICP8, is outside of the region required for ssDNA-binding, both mutant ICP8 molecules, as expected, bind to DNA. It is conceivable thatthe complementing polypeptides of TIA or TL5 ICP8 and nllSV ICP8 do notinteract with each other directly but that both bind DNA and that each molecule contributes some activities from different domains to achieve wt ICP8 func-tions.
Analternativemechanism for intragenic complementation is theformation of a dimer or multimers of nllSV ICP8 and TL4 or TL5 ICP8. Ithas been reported that ICP8 sediments in aglycerol gradient as a monomer (22), but the cooperative nature of the binding of ICP8 tossDNA suggests that ICP8 mayform polymers along ssDNA (22). In multimeric forms, the monomers may be weakly associated and assemble only uponinteractionwith DNA.Therefore, if multimerization is involved in theintragenic complementation of nllSV ICP8 and TL4 or TL5 ICP8, several models can be considered. First, the fact that nllSV ICP8 did not localize to prerepli-cative sites or replication compartments mayindicate that the SV40 T-antigen NLS did not allow proper targeting of ICP8 to specific sites within the nucleus, where viral DNA replication is initiated. This suggests that the
carboxy!-terminal 28 residues of ICP8 may also be involved in determining intranuclear localization and may interactwitha component of the cell nucleus toinitiate viral DNA replica-tion. Therefore, it ispossiblethat thenllSV ICP8 needs
TML
or TL5ICP8 to form the multimers fortargetingto
replica-tionmachinery.
Second, we (8) and others (19) noted that the amino-terminal portion of ICP8 also contributes to the
DNA-binding activity of the intact protein, although the
DNA-binding region has been identified at the carboxyl-terminal
halfof ICP8 molecule(8, 19,38). This contributioncould be due to the intra- or intermolecular interactions of ICP8. Intermolecularinteractions of this type couldexplain
intra-geniccomplementation. Asecondmodel is thatTL4orTL5 ICP8 and nllSV ICP8 bind DNA but that they can bind DNAcooperatively onlywhentheyinteract with each other. Ifthis is true, the domain which isrequiredforformation of
polymers must be disturbed in at least one mutant ICP8 molecule and the TIAorTL5 ICP8canform aheterodimer with nllSV ICP8 but cannot form polymers. This may
explain whytheefficiencyof
intragenic
complementation
of TL4and nllSV ICP8 reachedonly7to20% of that ofwt:the two mutantICP8 moleculescanformonly
adimer instead ofpolymersalongwith DNA. Further
experiments
toexaminecooperative binding ofnll ICP8 is needed to address this
possibility.
Athird model involves theinteraction of ICP8 with other VOL.67,1993
on November 9, 2019 by guest
http://jvi.asm.org/
viral and cellular proteins during viral DNA replication. Recently,Capson etal. demonstrated stepwise
ATP-depen-dentassembly of bacteriophage T4 DNAreplicationproteins by usingprotein-DNAcross-linking(2). Inthissystem, gene 32 ssDNA-binding protein but not DNA polymerase is required for assembly of DNA polymerase accessory pro-teins to form a complex on the primer-template molecule. After ATP hydrolysis, DNApolymerase binds to the com-plex to initiate DNA synthesis. It is possible that the alterations in both nllSV ICP8 and TIA or TL5 ICP8 inactivate domains required for interactions with different DNA replication proteins. However, the complementing ICP8 molecules can interactwith these replication proteins after they form multimers along DNA, resulting in the reconstitution of wt ICP8 activity.
In summary,ourresultsdemonstratethat ICP8consists of separate domainswhich possessdifferentfunctions and that somefunctionscancomplementeach other intrans. Further analysisofintrageniccomplementation of ICP8mutantsmay help definetheinteractions betweenICP8and otherviral and cellular proteins which are involved in viral DNA replica-tion.
ACKNOWLEDGMENTS
We thank L.Pereira for anti-ICP8 monoclonal antibody 793 and E. Villarreal for providing the TL mutant plasmids. We thank SumanShirodkarforassistance with isolation of the nllSV mutant virus andSteve Rice forcomments on the manuscript.
This work wassupported by Public HealthService grant CA26345 from theNational Cancer Institute.
REFERENCES
1. Bayliss, G. J., H. S.Marsden, and J. Hay. 1975. Herpes simplex virusprotein: DNA-binding proteins in infected cells and in the virusstructure. Virology68:124-134.
2. Capson, T. D., S. J.Benkovic, and N. G. Nossal. 1991. Protein-DNAcross-linking demonstrates stepwise ATP-dependent as-sembly of T4 DNApolymerase and its accessory proteins on the primer-template. Cell 65:249-258.
3. Challberg,M. D. 1986.A method for identifying the viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA83:9094-9098.
4. Conley, A. J., D. M. Knipe,P. C. Jones, and B. Roizman. 1981. Moleculargenetics ofherpes simplex virus. VII. Characteriza-tion of a temperature-sensitive mutant produced by in vitro mutagenesisanddefective in DNA synthesis and accumulation ofTpolypeptides. J.Virol. 37:191-206.
5. de Bruyn Kops, A., andD. M. Knipe. 1988. Formation of DNA replication structures in herpes virus-infected cells required a viral DNAbindingprotein. Cell 55:857-868.
6. Deluca, N. A., A.McCarthy, and P. A. Schaffer. 1985. Isolation andcharacterization ofdeletion mutants of HSV-1 in the gene encodingtheimmediate-early regulatory protein ICP4. J. Virol. 56:558-570.
7. Gao, M., J.Bouchey,K. Curtin, and D. M. Knipe. 1988. Genetic identification of a portion of the herpes simplex virus ICP8 proteinrequiredforDNA-binding. Virology 163:319-329. 8. Gao, M., and D. M.Knipe. 1989. Genetic evidence for multiple
nuclearfunctionoftheherpes simplex virus ICP8 DNA-binding protein. J. Virol.63:5258-5267.
9. Gao, M., and D. M. Knipe. 1991. Potential role for herpes simplex virus DNA replication protein in stimulation of late geneexpression. J.Virol. 65:2666-2675.
10. Gao, M., and D.M.Knipe. 1992. Distal protein sequences can affect the function of a nuclear localization signal. Mol. Cell. Biol. 12:1330-1339.
11. Gao, M., and D. M. Knipe.Unpublished results.
12. Godowski,P. J.,and D. M.Knipe. 1986. Transcriptional control of herpesvirus gene expression: gene functions required for positiveand negative regulation. Proc. NatI. Acad. Sci. USA
83:256-260.
13. Kalderon,D., B. L. Roberts, W. D. Richardson, andA. E. Smith. 1984. A short amino acid sequence able to specify nuclear location. Cell 39:499-509.
14. Knipe, D. M., M. P. Quinlan, and A. E. Spang. 1982. Charac-terization of two conformational forms of the major DNA-binding protein encoded by herpes simplex virus 1. J. Virol. 44:736-741.
15. Knipe, D. M., W. T. Ruyechan, and B. Roizman. 1979. Molec-ular genetics of herpes simplex virus. III. Fine mapping of a genetic locus determining resistance to phosphonoacetate by two methods of marker transfer. J. Virol. 29:698-704.
16. Knipe, D. M., and A. E. Spang. 1982. Definition of a series of stages in the association of two herpesvirus proteins with the cell nucleus. J. Virol. 43:314-324.
17.Lee, C. K., and D. M. Knipe. 1983. Thermolabile in vivo DNA-binding activity associated with a protein encoded by mutants of herpes simplex virus type 1. J. Virol. 46:909-919. 18. Lee, C. K., and D. M. Knipe. 1985. An immunoassay for the
study of DNA-binding activitiesof herpessimplex virusprotein ICP8. J. Virol. 54:731-738.
19.Leinbach, S. S., and L. S. Heath. 1988. A carboxyl-terminal peptide of the DNA-binding protein ICP8 of herpes simplex virus contains a single-stranded DNA-binding site. Virology 166:10-16.
20. Littler, E., D. Purifoy, A. Minson, and K. L. Powell. 1983. Herpes simplex virus nonstructural proteins. III. Function of the major DNA-binding protein. J. Gen. Virol.
64:983-995.
21. McKnight, J. L. C., T. M. Kristie, S. Silver, P. E.
Pellett,
P. Mavromara-Nazos, G. Campadelli-Fiume, M. Arsenakis, and B. Roizman. 1986. Regulation of herpes simplex virus 1 gene expression: the effect of genomic environments and its implica-tions for model systems. Cancer Cells 4:163-173.22. O'Donnell, M. E., P. Elias, B. E. Funnell, and I. R. Lehman. 1987. Interaction between the DNA polymerase and single-stranded DNA-binding protein (infected cell protein 8) of herpes simplex virus 1. J. Biol. Chem.262:4260-4266.
23. Orberg, P. K., and P. A. Schaffer. 1987. Expression of herpes simplex virus type 1 major DNA-binding protein, ICP8, in transformed cell lines: complementation of deletion mutants and inhibition of wild-type virus. J. Virol. 61:1136-1146.
24. Powell, K. L., E.Littler,and D. J. M. Purifoy. 1981. Nonstruc-tural proteins of herpes simplex virus. II. Major virus-specific DNA-binding protein. J. Virol. 39:894-902.
25. Powell, K. L., and D. J. M. Purifoy. 1976. DNA-binding proteins of cells infected by herpes simplex virus type 1 and type 2. Intervirology 7:225-239.
26. Quinlan, M. P., L. B. Chen, and D. M. Knipe. 1984. The intranuclear location of a herpes simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:657-868.
27. Quinlan, M. P., and D. M. Knipe. 1983. Nuclear localization of herpesvirus protein: potential role for the cellular framework. Mol. Cell. Biol. 3:315-324.
28. Quinlan, M. P., and D. M. Knipe. 1985. A genetic test for expression of a functional herpes simplex virus DNA-binding protein from a transfected plasmid. J. Virol. 54:619-622. 29. Rice, S. A., and D. M. Knipe. 1990. Genetic evidence for two
distinct transactivation functions of the herpes simplex virus a proteinICP27. J. Virol.64:1704-1715.
30. Rose, D. S. C., K. Shriver, D. S. Latchman, and N. B. Lathangue. 1986. A filamentous distribution for the herpes simplex virus type 2-encoded major DNA binding protein. J. Gen. Virol. 67:1315-1325.
31. Ruyechan, W. T. 1983. The major herpes simplex virus DNA-binding protein holds single-stranded DNA in an extended conformation. J. Virol. 46:661-666.
32. Ruyechan, W. T., A. Chytil, and C. M. Fisher. 1986. In vitro characterization of thermolabile herpes simplex virus DNA-binding protein. J. Virol. 59:31-36.
33. Ruyechan, W. T., and A. C. Weir. 1984. Interaction with nuclear acids and stimulation of the viral DNA polymerase by the herpes simplex virus type 1 major DNA-binding protein. J.
on November 9, 2019 by guest
http://jvi.asm.org/
COMPLEMENTATION OF HSV ICP8 MUTANTS 885 Virol. 52:727-733.
34. Shepard, A. A., and N. A. Deluca. 1991. Activities of het-erodimerscomposed ofDNA-binding- and transactivation-defi-cient subunits of the herpes simplex virus regulatory protein ICP4. J. Virol. 65:299-307.
35. Shepard, A. A., and N. A. Deluca. 1991. A second-siterevertant
ofadefectiveherpes simplex virus ICP4 protein with restored regulatory activities and impaired DNA-binding properties. J. Virol. 65:787-795.
36. Southern, P. J., and P. Berg. 1982. Transformation of mamma-lian cells to antibiotic resistance with a bacterial gene under control of theSV40 early regionpromoter.J. Mol.Appl. Genet. 1:327-341.
37. Villarreal, E., and D. M. Knipe. Unpublished results.
38. Wang, Y., and J. Hall. 1990. Characterization of a major DNA-bindingdomain in the herpes simplex virustype1 DNA-binding protein (ICP8). J. Virol. 64:2082-2089.
39. Weller, S. K., J.Lee, D. J. Sabourin, and P. A. Schaffer. 1983. Geneticanalysis of temperature-sensitivemutantswhich define thegenefor the major herpes simplex virus type-1 DNA-binding protein. J. Virol. 45:354-366.
40. Wu, C. A., N. J.Nelson, D. J. McGeoch, and M. D. Challberg. 1988. Identification of herpes simplex virus type 1 genes re-quired for origin-dependent DNA synthesis. J. Virol. 62:435-443.
VOL. 67,1993