JOURNAL OFVIROLOGY, Dec. 2004, p. 13139–13152 Vol. 78, No. 23 0022-538X/04/$08.00⫹0 DOI: 10.1128/JVI.78.23.13139–13152.2004
Copyright © 2004, American Society for Microbiology. All Rights Reserved.
Spread and Replication of and Immune Response to
␥
1
34.5-Negative
Herpes Simplex Virus Type 1 Vectors in BALB/c Mice
Eeva K. Broberg,
1,2,3Jutta Peltoniemi,
1,3Michaela Nyga
˚rdas,
1Tero Vahlberg,
4Matias Ro
¨ytta
¨,
5and Veijo Hukkanen
1,2*
Department of Virology,1Department of Biostatistics,4Department of Pathology,5and MediCity Research Laboratory,2
University of Turku, and Turku Graduate School of Biomedical Sciences,3Turku, Finland
Received 13 May 2004/Accepted 8 July 2004
We have previously shown that intracranial infection of herpes simplex virus type 1 (HSV-1) vector R8306 expressing interleukin-4 (IL-4) can abolish symptoms of experimental autoimmune encephalomyelitis, which is used as a model for human multiple sclerosis (Broberg et al., Gene Ther. 8:769–777, 2001). The aim of the current study was to search for means other than intracranial injection to deliver HSV-derived vectors to the central nervous system of mice. We also aimed to study the replication efficiency of these vectors in nervous system tissues and to elucidate the effects of the viruses on the immune response. We studied the spread and replication of the following viruses with deletions in neurovirulence gene␥134.5: R3616, R849 (lacZtransgene),
R3659 (alpha-tk), R8306 (murine IL-4 transgene), and R8308 (murine IL-10 transgene). The samples were taken from trigeminal ganglia and brains of BALB/c mice after corneal, intralabial, and intranasal infection, and the viral load was examined by viral culture, HSV DNA PCR, and VP16 reverse transcription (RT)-PCR. The results show that (i) intranasal infection was the most efficient means of spread to the central nervous system (CNS) besides intracranial injection; (ii) the viruses did not grow in the culture from the brain samples, but the viral DNA persisted even until day 21 postinfection; (iii) viral replication, as observed by VP16 mRNA RT-PCR, occurred mainly on days 4 and 7 postinfection in trigeminal ganglia and to a low extent in brain; (iv) R3659, R8306, and R8308 showed reactivation from the trigeminal ganglia in explant cultures; (v) in the brain, the vectors spread to the midbrain more efficiently than to other brain areas; and (vi) the deletions in the R3659 genome significantly limited the ability of this virus to replicate in the nervous system. The immunological studies show that (i) the only recombinant to induce IL-4 mRNA expression in the brain was R8306, the gamma interferon response was very low in the brain for R3659 and R8306, and the IL-23p19 response to R8306 decreased by day 21 postinfection, unlike for the other viruses; (ii)⌬␥134.5 HSV vectors modulated the subsets
of the splenocytes differently depending on the transgene; (iii) R3659 infection of the nervous system induces expression and production of cytokines from the stimulated splenocytes; and (iv) HSV vectors expressing IL-4 or IL-10 induce expression and production of both of the Th2-type cytokines from splenocytes. We conclude that the intranasal route of infection is a possible means of delivery of⌬␥134.5 HSV vectors to the CNS in
addition to intracranial infection, although replication in the CNS remains minimal. The DNA of the HSV vectors is able to reside in the brain for at least 3 weeks. The features of the immune response to the vectors must be considered and may be exploited in gene therapy experiments with these vectors.
Herpes simplex virus type 1 (HSV-1)-derived vectors are attractive for gene delivery to the central nervous system (CNS) because of their ability to infect neurons and other cell types (i.e., glial cells) residing in the CNS tissue. In addition, the large DNA genome of HSV enables extensive modifica-tions of the genome for gene delivery purposes. HSV vectors have already been widely tested for treatment of malignant tumors (21, 26, 27, 34). Mutant G207, which has deletions of both copies of the␥134.5 gene and alacZinsertion that
inac-tivates the ICP6 gene (UL39) (30), has shown potential in
tumor treatment studies and has proved to be a safe vector. HSV vectors with a deletion in the ␥134.5 gene have been
considered neuroattenuated or even nonneurovirulent, where-as the wild-type virus strains are neurovirulent.
The wild-type virus replicates and spreads efficiently in mu-rine CNS cells and can cause encephalitis. The growth of
HSV-1 in the CNS depends, among other gene products, on the ␥134.5 gene products (11, 39). The ␥134.5 gene deletion
viruses have a lethal dose of⬎106PFU, whereas the wild-type
virus HSV-1(F) requires only 2⫻ 102to 4 ⫻102 PFU upon
intracranial administration (11, 28). Replication, establishment of latency, spread from mucosal sites to the CNS, and induced reactivation have been reported to be impaired in infections with ␥134.5 deletion viruses R3616 and R4009 (39). Other
studies have shown effective gene delivery even from footpad infection to dorsal root ganglia by ␥134.5-deleted vectors of
17⫹strain origin (12). In addition to␥134.5, other genes affect
the severity of productive viral infection in the rodent CNS. These include the nonessential genes US1 through US5 (35),
HSV DNA polymerase (31), and UL45 (39). Vectors lacking
additional genes besides␥134.5, such as ICP27, elicit minimal
damage in the CNS (17). Multiple deletions of the immediate-early genes reduce the cytotoxicity further in comparison to single deletions (22).
Immune response to wild-type HSV has been examined in animal models and also in patients with recurrent herpesvirus * Corresponding author. Mailing address: Department of Virology,
University of Turku, Kiinamyllynkatu 13, FIN-20520 Turku, Finland. Phone: 358-2-3337417. Fax: 358-2-2513303. E-mail: veijo.hukkanen @utu.fi.
13139
on November 8, 2019 by guest
http://jvi.asm.org/
infections. The immunological consequences of vector use have to be considered when planning HSV-based gene ther-apy. HSV-1 vectors can induce an immunological response which can decrease vector survival and decrease the replication or establishment of quiescent infection. Previous reports show that HSV-based gene therapy leads to the development of anti-HSV antibodies (19) and to a cellular immune response including infiltrating lymphocytes (29). Long-lasting transgene expression from HSV vectors has, however, been observed even up to 180 days when the transgene is located under the latency-associated promoter (36). Stereotactic injection of HSV vectors can induce long-lasting upregulation in T-cell and macrophage functions (40). The HSV vector encoding gamma interferon (IFN-␥) as well as the wild-type virus can induce enhanced replication in trigeminal ganglia and induce a Th1-type pattern of cytokine responses (14). Existing immunity to HSV does not influence gene transfer by an HSV vector in animal models (7, 8, 13, 25, 29). Still, existing immunity can induce a rapid increase in HSV-specific cytotoxic lymphocytes or seroconversion to HSV-1 after therapy. ICP34.5 has been shown to have a role in inhibiting the host interferon response (9, 10, 16) and in regulating major histocompatibility complex class II expression in vitro (38). However, the local and sys-temic cytokine response against␥134.5-negative mutants has
not been studied.
We have previously shown the abolishment of an experimen-tal autoimmune disease in BALB/c mice by a␥134.5-deleted
HSV vector encoding Th2-type cytokine IL-4 but not by one encoding IL-10 (4). We aim now at elucidating alternative routes of infection instead of the intracranial one used in the previous gene therapy experiments in order to find the most efficient route for the viruses to spread to the CNS of mice. We compared corneal, intralabial, and intranasal infection. The replication of these vectors is impaired in the CNS tissue be-cause of the␥134.5 deletion, but they replicate well in vitro.
Here we report the spread and replication of the vectors R3616, R849, and R3659 and the cytokine-expressing viruses R8306 and R8308 in BALB/c mouse peripheral nervous system (trigeminal ganglia) and CNS (brains). We also examined the changes in cytokine production in the CNS and by the immu-nological cells of the spleen.
MATERIALS AND METHODS
Viruses.The genetic arrangement of the recombinant viruses has been pub-lished earlier. The mutants are based on HSV-1(F). R3616 has a deletion in the
␥134.5 gene, but the rest of the genome is intact (11). R849 has an insertion of
theEscherichia coli lacZgene at the locus of the␥134.5 deletion of R3616 (37).
R8306 has an insertion of the gene for murine IL-4 and R8308 has an insertion of the gene for murine IL-10 at the deletion site (2). R3659 has a replacement of
StuI-BstEII fragment from the domains of ORF P and␥134.5 by the chimeric
p␣27-tk gene (23, 24). All the transgenes and mutations exist in the genome as
duplicates because of the inverted repeats of the HSV-1 original genome. All the viruses used were thymidine kinase positive and able to replicate in Vero cells,
where they were propagated to 109
PFU/ml. Growth curves in monolayer cul-tures for most of the viruses have been presented earlier (2). All the vectors were from Bernard Roizman, University of Chicago.
Mice, infections, and sample collection.Female BALB/c mice were used for all studies. The specific-pathogen-free mice were obtained from the Central Animal Laboratory, University of Turku, Turku, Finland. The mice were main-tained at the animal facility of the Microbiological Institute, University of Turku, under permit LSLH-2002-5757/Ym-23 of the Ethical Committee for Animal Experiments of the University of Turku and notification number 4/P/99 of the Board of Gene Technology, Finland.
Twenty 6- to 8-week-old female BALB/c mice were infected with 105PFU of
wild-type HSV-1 (strain F) corneally after corneal scarification, intranasally or intralabially under anesthesia. Mice were killed on days 5 and 14 after the infection. Viral DNA analysis and virus culture were performed as described below. In order to examine the sufficient infectious load of recombinant HSVs, we infected 6- to 8-week-old female mice intranasally under anesthesia with
either 106
or 107
PFU of R849 virus in the right nostril. Samples were collected
on days 3, 5, 7, 10, and 14 postinfection (n⫽4 per group). Thereafter, we
infected 155 6-week-old female BALB/c mice under anesthesia in groups of five mice per virus and per sampling day in the right nostril by Hamilton syringe and
26-gauge needle with 105
PFU of HSV-1(F) or 107
PFU in 10l of
phosphate-buffered saline of the viruses R3616, R849, R3659, R8306, and R8308.
The mice were killed under CO2anesthesia on days 4, 7, 10, 14, and 21 after
the infection. Day 21 was chosen as the latest time point for the recombinant viruses because the vectors are to be used in gene therapy of CNS autoimmune disease and this time point is relevant in such studies (4). Samples were also collected from nine uninfected control mice. For HSV-1(F), the latest time point was day 31 postinfection. Spleens were collected into RPMI 1640 medium (Gibco-BRL, Gaithersburg, Md.). Blood was collected by cardiac puncture, and serum was separated by centrifugation. The mice were perfused by sterile phos-phate-buffered saline after the cardiac puncture to remove traces of blood. The trigeminal ganglia were removed and cut into halves, which were used for the viral culture and for the viral DNA PCR. The olfactory bulbs and the anterior parts of the frontal lobes (mean weight, 20 mg) and parts of the lateral hip-pocampus (mean weight, 20 mg), midbrain (mean weight, 45 mg), and cerebel-lum (mean weight, 15 mg) were collected and snap frozen in dry ice and stored
at⫺70°C until preparation for the viral DNA PCR and the virus culture.
Virus culture.Virus culture of the homogenized trigeminal ganglia and brain samples was performed with a rapid culture described earlier by Ziegler et al. (41). The homogenized trigeminal ganglia samples were diluted 1:2 and the brain samples 1:10 in culture medium before overlaying on Vero cells in a 12-well culture plate. The staining method is based on immunoperoxidase staining of cultures by a monoclonal anti-gC-1 antibody. For verification of latency, explant
cultures of trigeminal ganglia were incubated for 5 days at 37°C in a 5% CO2
atmosphere.
DNA extraction, PCR, and time-resolved fluorescence assay detection.DNA extraction was performed for the trigeminal ganglia and brain samples with the Boehringer Mannheim Viral Nucleic Acid Purification kit as described by the
manufacturer. The elution volume was 50l. The PCR was done as described
earlier, as was the semiquantitative time-resolved fluorescence assay (6, 18). The HSV-1 DNA copy numbers were calculated on the basis of the equation derived
from the standard curve, which was generated from samples containing 10, 102
,
103and 104copies of HSV-1 DNA. Samples containing more than five copies of
HSV-1 DNA were considered positive. In order to standardize for the differences
in sample size, the copy numbers of viral DNA were standardized to the-actin
copy numbers on the basis of a-actin LightCycler real-time PCR (5, 6).
Splenocyte stimulation.Splenocyte stimulation was performed as described earlier (6). In brief, spleen cells were collected by centrifugation with Lympholyte (Cedarlane Laboratories, Hornby, Ontario, Canada). Collected spleen cells were cultured at a density of 250,000 cells/well in RPMI 1640 medium containing 10% fetal bovine serum, 20 mM HEPES, 0.03% glutamine, and 0.01 mg of gentamicin per ml in U-bottomed 96-well plates (Costar, Cambridge, Mass.). Four parallel
wells were prepared in each case. Inactivated HSV antigen (1g/ml) (20) or 2g
of concanavalin A (Sigma-Aldrich Corp., St. Louis, Mo.) per ml was used as the stimulating antigen and as the specific and positive internal control, respectively. As the negative control antigen, we used culture medium.
RNA extraction, cDNA reaction, and real-time PCR.RNA extraction, cDNA reaction, and LightCycler real-time RT-PCR of stimulated splenocytes were performed as described earlier (5, 6). In short, the mRNA of the splenocytes was extracted by the semiautomated RNA extractor KingFisher (Labsystems, Hel-sinki, Finland). The total RNA of the brain samples was extracted by the Trizol reagent as described by the manufacturer (Invitrogen Life Technologies, Carls-bad, Calif.). The cDNA reaction was performed as described earlier (6, 15). The other primers and probes have been published earlier (3, 5, 6, 32) except for the IL-4 primers, which were CATATCCACGGATGCGACA (sense) and GCTCA
CTCTCTGTGGTGTTCTT (antisense). VP16 was chosen as a marker of late (␥)
gene expression, and thus of viral replication, because it is expressed after initiation of viral DNA replication.
Enzyme immunoassay.Enzyme immunoassay analyses of the splenocyte su-pernatants were performed with the OptEIA Sets for mouse IL-4, IL-10, and
IFN-␥(Pharmingen, San Diego, Calif.) as instructed by the manufacturer. All
cytokines were detected as duplicates of the samples. The serum samples were diluted 1:6 in assay diluent, and the splenocyte supernatants were diluted 1:2
13140 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
before testing. The results are shown as calculated concentrations of the undi-luted samples.
Fluorescence-activated cell sorting.Fluorescence-activated cell sorting analy-sis was performed as described earlier (6, 33). All the antibodies were purchased from Pharmingen (San Diego, Calif.).
Statistical analysis.Comparisons of infection route and dose were statistically
evaluated by the Mann-WhitneyUtest. Intranasal infection was compared to
intralabial and corneal infections, and the dose of 107PFU was compared to that
of 106
PFU by the amount of HSV DNA as copy numbers in the brain or trigeminal ganglia. Calculations of the statistical relevance of all VP16 and
cytokine analyses were also performed by Mann-WhitneyUtest. Expression of
cytokines is shown in copy numbers, and the HSV vector-infected groups were compared to uninfected and HSV-1 (F)- and R3616-infected groups. In the case of brain cytokine analyses, selected vector-infected groups were compared to
R8306-infected (days 4 and 7 for IL-4 and day 21 for IFN-␥and IL-23p19). The
fluorescence-activated cell sorting analysis was done by factorial analysis of variance. The comparisons were made as percentages of specific cell populations within the total number of cells by comparing the different HSV vector-infected groups with the uninfected and wild-type HSV-1-infected groups.
Statistical analysis of the ganglia HSV DNA was performed by two-way anal-ysis of variance, where virus group and day were explanatory variables. Further analyses were done by one-way analysis of variance because the interaction between virus group and day was statistically significant. To show the statistical differences between the virus groups on different days, multiple comparisons with
Dunnett⬘s correction were used. Due to the skewed distribution, HSV DNA
values were log transformed for statistical analysis. The analysis of brain HSV DNA was performed with the number of copies of viral DNA and the ratio of
viral DNA and-actin values, which were used as dependent variables in logistic
regression analysis. Dependent variables were divided into two categories (for
copies of viral DNA,⬎5 was considered positive; for ratio of viral DNA and
-actin values,⬎0 was considered positive). Virus group, day, and brain area
were explanatory variables in multivariate logistic regression analysis.
RESULTS
Spread and replication of⌬␥134.5 HSV vectors in the
ner-vous system.We first compared corneal, intralabial, and intra-nasal infection with a small amount of HSV-1 strain F. The spread of the virus to the CNS was tested by virus culture and viral DNA PCR. Virus culture resulted in positive cultures in Vero cell monolayers only after intranasal infection (n⫽4) on days 5 and 9 postinfection (except for one positive culture from intralabially infected mouse trigeminal ganglia on day 5 postin-fection). All trigeminal ganglia samples from mice infected intranasally with HSV-1(F) were culture positive (Table 1). Cultures showed HSV-1 loads from 3 to 28 PFU/mg in the cerebellum, 2 to 144 PFU/mg in the frontal lobe, up to 189 PFU/mg in the hippocampus, and up to 60 PFU/mg in the midbrain homogenates. The trigeminal ganglia of these mice showed HSV-1 loads of up to 800 PFU. Semiquantitative viral DNA PCR revealed that intranasal infection was the most
efficient means of delivery besides intracranial infection to the peripheral nervous system (trigeminal ganglia) and CNS (brain) of mice by HSV-1 (Fig. 1A). In brain, the wild-type virus spread more easily to the frontal lobe and cerebellum than to the hippocampus and midbrain. The virus was cleared in part from the trigeminal ganglia, and it spread first to the frontal lobe and then to other brain areas by day 14 postinfec-tion (Fig. 1).
In order to find out the suitable dose of ⌬␥134.5 viruses
for intranasal infection, corneal and intralabial infection of BALB/c mice with 106and 107PFU of recombinant R849 HSV
was performed. The R849 virus was chosen as a control be-cause it has the␥134.5 deletion in the genome but carrieslacZ
instead of a murine cytokine as a transgene. With the lower dose of R849, we could show viral DNA in the trigeminal ganglia or brain tissue only in some samples and in very small amounts (Table 2). Increase of the infectious load to 107PFU
caused a dramatic difference in the spread to both the trigem-inal ganglia and brain (P ⫽ 0.0023 for brain, Table 2). The virus R849 spread to both the trigeminal ganglia and brain of the mice. The distribution of the virus in the brain was not even; there were very low levels of viral DNA in the hippocam-pus (Table 2). On the basis of these pilot results, we continued, in a larger experiment, testing the spread and replication of five different ␥134.5-deleted HSV mutants in comparison to
HSV-1(F). Infections were made intranasally with 107PFU per
virus (for the wild-type virus, only 105PFU was administered).
Three of the HSV-1(F)- and R849- and one of the R8308-infected mice died before the sampling.
Virus culture and VP16 mRNA expression showed dimin-ished replication of all recombinant viruses in comparison to wild-type HSV-1 (Table 1 and Fig. 2 and 3). None of the mutants showed such abundant replication in the brain tissue that it would have been detected as replicating virus in the virus culture. However, the viruses showed some VP16 mRNA expression in the brain (Fig. 4G) as shown by the sensitive quantitative RT-PCR. R3616 and R3659 showed no or only minimal viral replication in the trigeminal ganglia (Table 1 and Fig. 3F). On day 4, R849- and R8308-infected samples resulted in 50% positive cultures. R8306 was the only mutant to repli-cate in all samples of trigeminal ganglia on day 4. Explant cultures after 21 days (or 31 days for the wild type) of infection showed reactivation from latency in HSV-1(F)-, R3659-, R8306-, and R8308-infected mice (Table 2).
We examined the existence of viral DNA in the nervous system by viral DNA PCR (Fig. 2 to 4). Samples with more than five copies of HSV DNA were considered positive. In trigeminal ganglia (Fig. 3), the difference in viral DNA load during the experiment between the different viruses was shown to be statistically significant (all days,P⫽0.0029; day 7,P⫽ 0.0034; day 10,P⫽0.0003; day 14,P⫽0.0122; and day 21,P⫽ 0.0078). R3616 was used as the basis for comparisons (no recombinant genes and no additional mutations in the ge-nome) in statistical analysis. On days 7 (P⫽0.0055), 10 (P⫽ 0.0002), and 21 (P⫽0.0484), R3659 differed significantly from R3616 in viral DNA load. The viral load of R849 differed from that of R3616 on day 14 (P⫽0.0101).
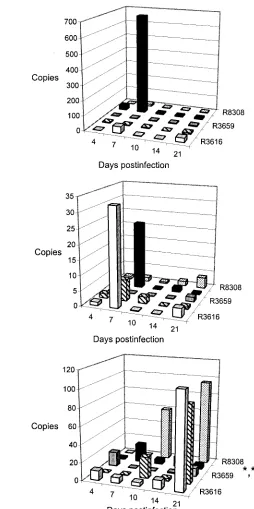
[image:3.585.43.284.97.205.2]The recombinant viral load in the brain is summarized in Fig. 4A and shown for different viruses in Fig. 4B to F. The viral DNA load for HSV-1 is shown in Fig. 2B. The viral DNA TABLE 1. Virus culture of trigeminal ganglia of BALB/c mice
infected intranasally with 105PFU of the HSV-1(F) or 107PFU of the⌬␥34.5 HSV vector
Virus
No. of positive samples/no. tested (% positive) Acute culture
(day 4–5) Explant culture
HSV-1 (F) 5/5 (100) 3/3 (100)
R3616 0/4 (0) 0/5 (0)
R849 (lacZ) 2/4 (50) 0/5 (0)
R3659 (alpha-tk) 0/4 (0) 2/5 (40)
R8306 (murine IL-4) 4/4 (100) 2/5 (40) R8308 (murine IL-10) 2/4 (50) 5/5 (100)
VOL. 78, 2004 SPREAD AND REPLICATION OF HSV VECTORS 13141
on November 8, 2019 by guest
http://jvi.asm.org/
copy numbers of the brain samples were corrected for the size of the brain sample by dividing by the-actin copy number of the same DNA sample. The logistic regression analysis showed differences in HSV DNA load with different recombinant vi-ruses (P ⫽ 0.0022) and in distribution of the HSV DNA to different brain areas (P ⬍ 0.0001). The viral DNA load of R3659 differed from that of R3616 (P ⫽0.0006; 95% confi-dence interval, 0.014 to 0.312). The viral DNA load of the midbrain was increased in comparison to all other brain areas examined (cerebellum,P⫽0.0004, confidence interval, 0.113 to 0.534; frontal lobe,P⬍0.0001, confidence interval, 0.064 to 0.365; hippocampus,P⬍0.0001, confidence interval, 0.088 to
0.437). The mutant viruses favored the midbrain localization, and virus R3659 showed only a minimal DNA load in the brain.
The expression of the HSV genes by the viruses in the brain was examined by VP16 real-time RT-PCR of the HSV DNA-positive samples. VP16 mRNA expression represented viral gene expression during the DNA replication cycle. The gamma gene expression of the Th2 cytokine-expressing vectors R8306 and R8308 was increased rather than reduced in comparison to the control vector R3616 on day 4 (Fig. 3F). The -galactosi-dase-expressing vector R849 showed a short peak of expression on day 7 which differed from all other viruses (P ⬍ 0.05). Neither of the two R3659 HSV DNA-positive samples showed VP16 mRNA expression, suggesting impaired replication of the virus in the CNS. The wild-type virus HSV-1 (F) showed dramatically higher expression of VP16 mRNA in comparison to the mutant viruses (Fig. 2C and 3F), although it had lower HSV DNA levels (Fig. 2B and 3).
[image:4.585.120.470.73.397.2]Expression of Th1 and Th2 cytokines in the CNS.The local expression of type 1 and 2 cytokines was studied in brain samples by LightCycler quantitative RT-PCR. IL-4 was studied as a marker of type 2 cytokine expression. The only recombi-nant vector that induced mRNA expression of IL-4 in relevant amounts was the IL-4 carrier vector R8306, and the expression FIG. 1. Viral DNA in the trigeminal ganglia and in different brain areas after corneal (n⫽2; black bars), intralabial (n⫽4; hatched bars), and intranasal (n⫽4; white bars) infections with 105PFU of HSV-1(F) on day 5 (A) and day 14 (B) postinfection. Trigeminal ganglia and brain were collected, the viral DNA was extracted, and PCR was performed as described in Materials and Methods. The copy numbers were calculated on the basis of standard curves. Each bar represents the mean⫾standard deviation. The statistical analysis was performed by the Mann-WhitneyU test by comparing the intralabial and intranasal infection routes.ⴱ,P⬍0.05.
TABLE 2. Mean HSV DNA copy numbers after intranasal infection of BALB/c mice with either 106or
107PFU of R849 on day 7 postinfection
Dose (PFU)
Mean copy no. Trigeminal
ganglia
(n⫽2)
Frontal lobe
(n⫽2)
Hippo-campus
(n⫽2)
Midbrain
(n⫽2)
Cere-bellum
(n⫽2)
Brain total
(n⫽8)
106 1.0 0.4 0.6 1.2 0.3 0.6
107
1,200 820 1.8 260 630 430a
aP⫽0.0023 by Mann-WhitneyUtest.
13142 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.585.43.284.646.717.2]of IL-4 in the R8306-infected group in the midbrain area was significantly different from that of the other groups (Fig. 5A, P⬍0.05 on day 7). IL-10 mRNA expression was induced by various vectors but mainly by R3616, R849, and R8306 on day 7 and in increasing copy numbers toward the later time points also by R8308 (Fig. 5B). The level of IL-10 mRNA expression was very low throughout.
IFN-␥was studied as a marker of type 1 cytokine produc-tion. IFN-␥expression was increasing at the latest time point after infections with R3616 and R849 and from earlier time points (days 10 to 21) after infections with R8308 (Fig. 5C). The vectors R3659 and R8306 induced minimal expression of IFN-␥in comparison to other recombinant vectors, especially on day 21 (P⬍0.05).
The family of IL-12 components are involved in the initia-tion and maintenance of the type 1 cytokine response. The components p35 and p40 showed various expression patterns at intermediate and low levels, respectively (Fig. 5D to E). R8308 seemed to decrease the expression of IL-12p35 and increase the expression of IL-12 p40 and p19 toward the later time points. On the contrary, R8306 infection resulted in di-minishing expression of IL-23 p19 during the later time points, unlike the other recombinant viruses (P ⬍ 0.05), which in-duced increasing expression of IL-23 p19 during the experi-ment in the infected mice (Fig. 5F).
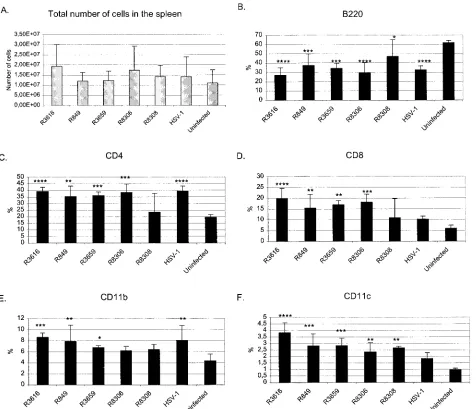
Systemic immune response to HSV vectors.In order to ex-amine the systemic immunological changes induced by the HSV vectors, we studied the lymphocyte and monocyte sub-types of the spleen and cytokine expression by these cells. Fluorescence-activated cell sorting was performed on day 10
postinfection of splenocytes of each mouse, which we have shown to be the peak time point of the response to an anti-genic stimulus in our BALB/c HSV-1 infection models (6). The groups were compared to each other, to uninfected BALB/c mice, and to HSV-1 wild-type-infected BALB/c mice. Of the splenocyte subtypes, we examined CD4⫹and CD8⫹T lympho-cytes, B220⫹B cells, CD11b⫹macrophages, and CD11c⫹ an-tigen-presenting cells. The results are shown as percentages of each cell population from the total splenocyte count of each spleen (Fig. 6). The results shown in Fig. 6 show an increase in all other cell types except B cells in infected mice.
The relative amounts of B220-positive B cells were down-regulated in the spleen by HSV vector infections. Wild-type HSV-1 caused a downregulation (P⫽0.0001), as did the vec-tors R3616 (P ⬍ 0.0001), R849 (P ⫽ 0.0008), R3659 (P ⫽ 0.0002), R8306 (P ⬍ 0.0001), and R8308 (P ⫽ 0.0394). Al-though R8308 also showed a statistically significant relative reduction of B cells in comparison to uninfected mice, R8308 induced more B cells than the virus R3616 (P⫽0.0071) or the IL-4-producing vector R8306 (P⫽0.0169).
CD4⫹lymphocytes were increased in HSV vector infections except for R8308 infection in comparison to uninfected mice for R3616 (P ⫽ 0.0001), R849 (P ⫽ 0.0013), R3659 (P ⫽ 0.0008), and R8306 (P⫽0.0002). R8308-infected mice did not differ from uninfected mice in CD4 T-cell count. The wild-type HSV-1 infection caused an induction of CD4⫹T cells (P ⫽ 0.0001).
The HSV vectors except for R8308 caused an upregulation of CD8⫹ T cells: R3616 (P ⬍ 0.0001), R849 (P ⫽ 0.0043), R3659 (P⫽0.0013), and R8306 (P⫽0.0005), although wild-FIG. 2. HSV DNA load and VP16 mRNA expression in the trigeminal ganglia (TG) (A, C) and brain (B, D) of BALB/c mice infected intranasally with 105PFU of of HSV-1(F). The HSV DNA and quantitative RT-PCR were performed as described in Materials and Methods. The copy numbers were calculated on the basis of the standard curve. Each bar represents the mean of three to five samples⫾standard deviation.
VOL. 78, 2004 SPREAD AND REPLICATION OF HSV VECTORS 13143
on November 8, 2019 by guest
http://jvi.asm.org/
type infection with HSV-1 strain F did not show any difference from uninfected mice on day 10. Intranasal HSV-1 infection induced increase of CD11b⫹macrophages (P⫽0.0025). Viral vectors R3616, R849, and R3659 also caused an upregulation of macrophages (P ⫽ 0.0006, P ⫽ 0.0035, and P ⫽ 0.0367, respectively). However, the macrophage count of the mice infected with Th2-type cytokine-producing vectors did not dif-fer from that of uninfected mice. IL-4 of R8306 might even
downregulate the macrophage induction caused by the vector (R8306 versus R3616,P⫽0.0354).
[image:6.585.77.507.69.453.2]CD11c⫹ antigen-presenting cells were upregulated in the spleen by intranasal HSV vector infection, R3616 (P⬍0.0001), R849 (P⫽0.0004), R3659 (P⫽0.0003), R8306 (P⫽0.0048), and R8308 (P⫽0.0018). Vectors without cytokine transgenes upregulated antigen-presenting cells more than wild-type HSV-1, R3616 (P⫽0.0002), R849 (P⫽0.0317), and R3659 (P FIG. 3. HSV DNA copy numbers in the trigeminal ganglia of BALB/c mice infected intranasally with 107PFU of viruses R3616 (A), R849 (B), R3659 (C), R8306 (D), and R8308 (E). The PCR was performed as described in Materials and Methods. The copy numbers were calculated on the basis of the standard curve. Each bar represents the mean of five samples⫾standard deviation. The statistical analysis was performed by variance analysis, and the viral DNA loads of different vectors were compared to that of virus R3616 (ⴱ,P⬍0.05;ⴱⴱ,P⬍0.01; andⴱⴱⴱ,P⬍0.001). Panel F shows VP16 mRNA expression in trigeminal ganglia during the different recombinant vector infections.
FIG. 4. (A) Total HSV DNA load (mean of DNA copy numbers at any brain locus) in the brains of⌬␥134.5 HSV vector-infected BALB/c mice on different days postinfection. R3616, white bars; R849, hatched bars; R3659, stippled bars; R8306, gray bars; and R8308, black bars. (B to F) Illustration of the relative HSV copy numbers in the different brain areas (frontal lobe, white bars; hippocampus, hatched bars; midbrain, gray bars; cerebellum, black bars) of BALB/c mice infected intranasally with 107PFU of viruses R3616 (B), R849 (C), R3659 (D), R8306 (E), and R8308 (F) on days 4 to 21 postinfection. The different anatomical loci of the brain were separated, viral DNA was prepared, and PCR was performed as described in Materials and Methods. The differences in the sizes of the original brain samples were standardized by dividing the viral DNA copy numbers by the-actin copies. Each bar represents the mean of five samples. Panel G shows VP16 mRNA expression in the brains of mice infected with the different recombinant vectors.
13144 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
13145
on November 8, 2019 by guest
http://jvi.asm.org/
⫽0.0292). R8306 and R8308 did induce production of antigen-presenting cells, but the level of production did not differ from wild-type HSV-1.
The conclusion from the fluorescence-activated cell sorting
[image:8.585.187.441.65.574.2]data was that HSV-1(F) and the ␥34.5-negative viruses de-creased the relative amount of B cells but inde-creased the num-ber of T cells and CD11b⫹macrophages and CD11c⫹ antigen-presenting cells. The IL-10 transgene seems to be able to FIG. 5. Cytokine mRNA expression in brains of mice infected with␥134.5-negative vectors R3616 (white), R849 (hatched), R3659 (gray), R8306 (black), and R8308 (stippled) at different time points of the infection. The copy numbers were summarized from the copy numbers of specific cytokines in the different anatomical loci. (A) IL-4, (B) IL-10, (C) IFN-␥, (D) IL-12 p35, (E) IL-12 p40, and (F) IL-23 p19. The expression profiles are shown on different scales due to the difference in expression levels of the individual cytokines. The statistical analysis was performed by Mann-WhitneyUtest. Statistical significance for expression of IL-4 (ⴱ, midbrain area of mice infected with R8306 versus other viruses on day 7;P⬍0.05), IFN-␥on day 21 (ⴱ, R8306 versus R3616 or R849,P⬍0.05;ⴱⴱ, R8306 versus R8308,P⫽0.001), and IL-23 p19 on day 21 (ⴱ, R8306 versus R3659,P⬍0.05;ⴱⴱ, R8306 versus R849,P⫽0.001).
13146 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
counteract these changes and the IL-4 transgene to downregu-late the macrophage induction.
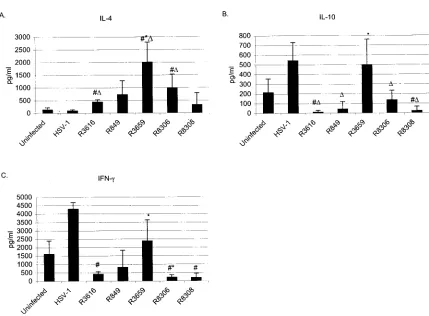
Spleen cell-derived cytokine production.We examined both cytokine mRNA expression (Fig. 7) and cytokine secretion (Fig. 8) from the stimulated splenocytes. The statistically sig-nificant increase of IL-4 mRNA expression in comparison to the uninfected was detected in R3616, R3659 and R8306 in-fected groups (P⬍0.05) stimulated with concanavalin A. The
[image:9.585.218.446.79.610.2]IL-4 expression was reduced in splenocytes of R849 and R3659 infected mice (P ⫽0.0339) but was increased in the spleno-cytes of R8306- and R8308 -infected mice in comparison to splenocytes of R3616-infected mice (Fig. 7). Surprisingly, the production of IL-4 was at its highest level in R3659-infected mice (P ⫽ 0.0209 compared to uninfected and P ⫽ 0.0339 compared to R3616, Fig. 8A). The IL-4 production was also significantly higher in R3616 (P⫽0.0339) and in R8306 (P⫽ FIG. 5—Continued.
VOL. 78, 2004 SPREAD AND REPLICATION OF HSV VECTORS 13147
on November 8, 2019 by guest
http://jvi.asm.org/
0.0209) infected in comparison to uninfected mice. The wild-type HSV-1(F) did not induce IL-4 production.
IL-10 mRNA expression was not detected in the splenocytes of R3616-, R849-, and R3659-infected mice, but was induced by the Th2-type cytokine-expressing vectors R8306 and R8308 in HSV-stimulated splenocytes (Fig. 7). In the concanavalin A-stimulated splenocytes, all groups expressed IL-10 mRNA at 10-fold higher levels than in the HSV-stimulated splenocytes. The secretion of IL-10 was at a low level in all groups but was highest in R3659-infected mice and in wild-type virus-infected mice (Fig. 8B). Surprisingly, the production of IL-10 was sig-nificantly decreased in R3616- and in R8308-infected mice compared to the uninfected and in mice infected with R849, R8306, and R8308 compared to wild-type infection (P⬍0.05, Fig. 8B).
The mRNA expression of IFN-␥was induced by the HSV vectors in the HSV stimulated splenocytes in comparison to the uninfected (P⬍0.05 in R3616 and R8306). No statistically significant difference was seen between the vector infected groups in the HSV-stimulated splenocytes. On the contrary, in the concanavalin A-stimulated splenocytes, the splenocytes of the uninfected expressed the highest amounts of IFN-␥mRNA, whereas in the R365g-, R8306-, and R8308-infected mice, the expression was reduced (P⬍0.05 in comparison to uninfected and R3616 infected). Wild-type HSV-1(F) infection induced production of IFN-␥(Fig. 8B). Although the IFN-␥production was increased in R3659 infected mice (P⫽0.0339, Fig. 8C) its mRNA expression was reduced at the same time in the con-canavalin A-stimulated splenocytes (P⫽0.0339). The produc-tion of IFN-␥was suppressed in R3616-, R8306-, and R8308-FIG. 6. (A) Total number of spleen cells on day 10 postinfection with⌬␥134.5 HSV vector or wild-type HSV-1 (strain F) in BALB/c mice. The standard deviations are shown. (B to F) Distribution of the cell subsets in the spleens on day 10 post-⌬␥134.5 HSV vector or wild-type HSV-1 (strain F) infection in BALB/c mice. The analysis was performed by flow cytometric analysis with antibodies defined in Materials and Methods. B cells (B220⫹), CD4⫹and CD8⫹T cells, macrophages (CD11b⫹), and antigen-presenting cells (CD11c⫹) were studied. Each bar represents the mean of quadruplicates⫾standard deviation. The statistical analysis was performed by analysis of variance factorial analysis by comparing virus-infected to uninfected samples.Pvalues:ⴱ,P⬍0.05;ⴱⴱ,P⬍0.01;ⴱⴱⴱ,P⬍0.001; andⴱⴱⴱⴱ,Pⱕ0.0001.
13148 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:10.585.57.533.66.475.2]infected mice when compared to the uninfected (P⬍0.05, Fig. 8).
IL-23 mRNA expression was increased in both HSV- and concanavalin A-stimulated splenocytes of cytokine-vector-in-fected mice (Fig. 7). In all other HSV-vector-incytokine-vector-in-fected groups, statistical significance (P⬍0.05) was reached except for con-canavalin A-stimulated R3659 and R8308 groups. We were not able to test the amounts of secreted IL-23, because no test was commercially available.
The cytokine content was tested also from the sera of the mice in order to investigate the peripheral immune response. Uninfected mice did not have any of these cytokines in their sera at any time point (days 4, 7, 10, and 21). The HSV-vector-infected mice showed variable cytokine secretion into serum during the infection, but the most consistent data were from day 10 (data not shown). IL-4 was secreted into serum very sparsely. IL-10 secretion was observed in R3659-, R8306-, and R8308-infected mice, but not in R3616- or R849-infected mice.
DISCUSSION
Wild-type HSV-1 spreads effectively in the murine CNS and causes encephalitis. It can infect a variety of other cell types in addition to the neurons, it replicates efficiently at the site of infection and spreads to the whole brain. The␥134.5 gene of
HSV-1 has been shown to enable the replication of the virus in CNS tissue (11, 39). Deletions in the gene of␥134.5 increase
the lethal dose of HSV in mouse CNS by over a thousand times and ␥134.5 negative mutants are considered to be the least
virulent mutants of HSV known. However, the mutants are able to replicate in vitro. The host range and the identity of
nervous system cells infected by ␥134.5 mutant R3616 have
been reported previously (28). Markovitz et al. (35) showed that R3616 infects a variety of cells in the CNS as well as wild-type HSV-1. R3616 uses retrograde transport to spread in neurons and destroys the infected cell, but it has an impaired replication capacity and inefficient spread from cell to cell. ␥134.5 negative virus replicates efficiently in malignant glioma
cells (1), which would suggest that factors associated with dys-regulated cell division would enhance the replication of ICP34.5-negative mutants.
We have previously shown that the intracranial delivery of ICP34.5-negative, IL-4-producing HSV vector R8306 can in-hibit the development of CNS autoimmune disorder in mice (4). The intracranial infection was effective in disease abolish-ment, but is itself damaging for the brain tissue. We therefore aimed at elucidating noninvasive means of vector delivery to murine CNS. We also wanted to investigate whether the vi-ruses are capable of replicating in the CNS tissue and of spreading from olfactory bulbs to other parts of the brain. We compared replication efficiency and spread of wild-type HSV-1(F) and R3616 to the vectors expressing transgenes in the place of␥134.5. The data generated in this study demonstrate
that intranasal infection is a possible means for delivering ␥134.5-deleted HSV vectors to CNS of mice. The replication of
the vectors R3616, R849, R3659, R8306, and R8308 is im-paired, and the increase in the infectious dose does not in-crease the replication efficiency.
The spread of the viral DNA was demonstrated by viral DNA PCR. The data show that all the different mutants tested spread to the peripheral nervous system (here, trigeminal gan-FIG. 7. Cytokine mRNA expression of herpes simplex virus antigen-stimulated (HSV, left) and concanavalin A-stimulated (ConA, right) splenocytes harvested on day 10 postinfection, shown as copy numbers of cytokine mRNAs on a logarithmic scale. The RT-PCR was performed with LightCycler real-time PCR. Originally, 250,000 cells were cultivated for each RNA sample. IL-4 is shown in black, IL-10 in hatched, IFN-␥ in white, and IL-23 in gray bars. Each bar represents the mean of four samples⫾standard deviation. The # symbols (compared to uninfected) and asterisks (ⴱ, compared to R3616) designate a statistical significance ofP⬍0.05 by Mann-WhitneyUtest.
VOL. 78, 2004 SPREAD AND REPLICATION OF HSV VECTORS 13149
on November 8, 2019 by guest
http://jvi.asm.org/
[image:11.585.49.534.85.314.2]glia) and to the CNS. The spread to the midbrain area of the CNS was emphasized for all other vectors except for R3659, which was present at very low viral DNA copy numbers. The viral DNA load of the R3659 was reduced already in the trigeminal ganglia and the spread from peripheral to central nervous system was almost inhibited. The differences in the efficiency of spread seem not to be due to the presence of the transgene in general, since the R849 lacZ-coding vector reached the highest individual viral DNA load in the brain, and as the R8306 infection yielded a similar level of viral DNA in the brain as the backbone virus R3616.
No viable R3659 was recovered from the brain during the acute infection, but the explant culture yielded some latent virus. Probably, the deletion in the naturaltkand UL24 genes
ameliorates the extent of the infection. The two brain samples containing viral DNA in the R3659 group were negative for mRNA expression of VP16. The sensitivity of the assay (⬍10 copies) (5) would support the interpretation that R3659 did not replicate in the brain tissue. The lower copy numbers of viral DNA in the HSV-1(F)-infected mice were possibly due to the two logs lower initial infectious loads given to the mice. However, the viral DNA load of R3659 was even lower than in wild-type HSV-1-infected mice.
To our knowledge, the local and systemic cytokine response of the␥134.5 vectors has not been investigated previously. We
have previously shown that wild-type HSV-1(F) induces strong IL-23 p19 expression in trigeminal ganglia and brains of
BALB/c mice after corneal infection (6). This induction was true also in our current intranasal infection model for the recombinant viruses, although R8306 seems to somewhat downregulate the expression during the later time points. The high level expression of IL-23 p19 mRNA was seen also in brain samples of our gene therapy of EAE with these vectors (3). Consistent with our previous data are also the IL-4 expres-sion induced only by the vector R8306. Our current data on the very low expression of IFN-␥in brain by vectors R3659 and R8306 would support the Th2-type cytokine response induced by these vectors, as suggested in our previous studies (3, 4).
The immune response mounted during the ␥134.5 vector
[image:12.585.77.506.60.376.2]infection resulted in alterations in the splenocyte subsets and the cytokines produced by the splenocytes. In comparison to our previous results from corneal infection (6), the intranasal infection route can have a different impact on the cell subsets in the spleen. For example, the intranasal infection induces a stronger CD4⫹response than the ocular infection. The intra-nasal R8308 infection did not result in an increase of CD8⫹ and CD4⫹T cells although all other vectors induced strong T-cell response. Nor did R8308 cause as strong a B-cell deple-tion as the other vectors and wild-type HSV-1 in intranasal infection. However, wild-type HSV-1 infection in the cornea has a similar effect on the B-cell count of the spleen as R8308 given intranasally. Overall, this would indicate an immunolog-ical downregulatory role for IL-10 produced by the vector R8308.
FIG. 8. Cytokine (A, IL-4; B, IL-10; and C, IFN-␥) production of concanavalin A-stimulated splenocytes harvested 10 days postinfection. Results are shown as concentrations. Each bar represents the mean of four samples⫾standard deviation. The statistical significance (P⬍0.05) was evaluated by Mann-WhitneyUtest (#, compared to uninfected;⌬, compared to HSV-1; andⴱ, compared to R3616).
13150 BROBERG ET AL. J. VIROL.
on November 8, 2019 by guest
http://jvi.asm.org/
Both of the Th2-type cytokines, IL-4 and IL-10, expressed by vectors R8306 and R8308 reduced the induction of CD11b⫹ macrophage and CD11c⫹antigen-presenting cells in compar-ison to R3616. The cytokine transgenes in R8306 and R8308 or the immunological activity of the produced cytokines would suppress the immune response elicited towards the viral vec-tor, which would also allow enhanced replication efficiency in comparison to the other vectors. The suppressed immune re-sponse by infections with R8308 and especially with R8306 suggests that these vectors might be applicable for treatment of diseases requiring anti-inflammatory therapy.
Interestingly, the vector R3616 and the wild-type virus in-fection led to different cell population profiles in the spleen, although the total number of the splenocytes was not altered in the R3616-infected mice. The CD4⫹ and CD8⫹ T-cell and CD11c⫹ antigen presenting cell counts were increased in R3616 infected mice in comparison to wild-type virus infected mice. This would indicate an immunomodulatory role of the ␥134.5 gene in addition to its roles in infectivity of CNS and in
the immune evasion mechanisms of HSV-1 in inhibiting pro-tein synthesis shutoff. Several studies have shown the role of ␥134.5 protein in inhibiting host interferon response (9, 10, 16).
Moreover, upregulation of major histocompatibility complex class II protein expression has been observed in cultured gli-oblastoma cells infected with R3616 (38).
The cytokines produced by the splenocytes showed a marked increase in the cytokine production of the R3659 vector, al-though no such increase in mRNA expression was seen. Rather, the cytokine-expressing viruses R8306 and R8308 in-creased the overall IL-4 and IL-23 mRNA expression in splenocytes. In our previous study, where we used R3659 as a control virus in the gene therapy of experimental allergic en-cephalomyelitis (4), we showed an induction of IL-4 or other Th2-type cytokines by R3659 (3). The present study confirms the earlier observations that R3659 alters the immunological response and that Th2-type cytokines can be produced as a response to this virus in the infected mice (3). The mechanisms of the infectivity of the CNS and the significance of induced cytokines to the vector survival require further investigation.
In conclusion, although the ␥134.5 negative HSV vectors
show impaired replication in the nervous system, the vectors have an impact on the immune response both locally in the CNS as well as systemically in the spleen. The immune re-sponse is dependent on the transgene and Th2-type cytokine transgenes can downregulate the immune response otherwise evoked against HSV-1 based␥134.5-negative vectors. ␥134.5
gene seems to have also a role in the type of immune response induced and elucidation of the mechanism would be the goal of our future studies.
Understanding the mechanisms of spread and replication of the viral vectors in the CNS may result in the development of more efficient viral vectors for usage in the CNS disorders. The immune response involved in the clearance of the viral vectors challenges the development of vectors for long-term expres-sion of transgenes.
ACKNOWLEDGMENTS
We thank Bernard Roizman and Bin He for R3616, R849, R3659, R8306, and R8308. We thank Leena Ruohonen and Inka Tulonen for technical assistance with the viral DNA PCRs and Noora Liuhanen,
Terhi Ma¨kela¨, and Johanna Va¨nni for assistance with the LightCycler PCRs.
We express our gratitude to the following financial supporters: the Turku Graduate School of Biomedical Sciences, the Academy of Fin-land (project 54050), the Paulo Foundation, the Finnish Cultural Foundation, and the Finnish MS Foundation.
REFERENCES
1.Advani, S., S. Chung, S. Yan, G. Gillespie, J. Markert, R. Whitley, B. Roizman, and R. Weichselbaum.1999. Replication-competent, nonneuroin-vasive genetically engineered herpes virus is highly effective in the treatment
of therapy-resistant experimental human tumors. Cancer Res.59:2055–2058.
2.Andreansky, S., B. He, J. van Cott, J. McGhee, J. Markert, G. Y. Gillespie, B. Roizman, and R. J. Whitley.1998. Treatment of intracranial gliomas in immunocompetent mice using herpes simplex viruses that express murine
interleukins. Gene Ther.5:121–130.
3.Broberg, E., A. Salmi, and V. Hukkanen.2004. IL-4 is the key regulator in
HSV-based gene therapy of BALB/c EAE. Neurosci. Lett.364:173–178.
4.Broberg, E., N. Seta¨la¨, M. Ro¨ytta¨, A. Salmi, J.-P. Era¨linna, B. He, B. Roiz-man, and V. Hukkanen.2001. Expression of interleukin-4 but not of inter-leukin-10 from a replicative herpes simplex virus type 1 viral vector precludes
experimental allergic encephalomyelitis. Gene Ther.8:769–777.
5.Broberg, E. K., M. Nyga˚rdas, A. A. Salmi, and V. Hukkanen.2003. Low copy number detection of herpes simplex virus type 1 mRNA and mouse Th1 type cytokine mRNAs by Light Cycler quantitative real-time PCR. J. Virol.
Meth-ods112:53–65.
6.Broberg, E. K., N. Seta¨la¨, J.-P. Era¨linna, A. Salmi, M. Ro¨ytta¨, and V. Hukkanen.2002. Herpes simplex virus type 1 infection induces upregulation of interleukin-23 (p19) mRNA expression in trigeminal ganglia of BALB/c
mice. J. Interferon Cytokine Res.22:641–651.
7.Brockman, M., and D. M. Knipe.2002. Herpes simplex virus vectors elicit durable immune responses in the presence of preexisting host immunity.
J. Virol.76:3678–3687.
8.Chahlavi, A., S. Rabkin, T. Todo, P. Sundaresan, and R. Martuza.1999. Effect of prior exposure to herpes simplex virus 1 on viral vector-mediated
tumor therapy in immunocompetent mice. Gene Ther.6:1751–1758.
9.Cheng, G., M. E. Brett, and B. He.2001. Val193and Phe195of the␥ 134.5
protein of herpes simplex virus 1 are required for viral resistance to
inter-feron-␣/. Virology290:115–120.
10.Cheng, G., K. Yang, and B. He.2003. Dephosphorylation of eIF-2␣mediated
by the␥134.5 protein of herpes simplex virus type 1 is required for viral
response to interferon but is not sufficient for efficient viral replication.
J. Virol.77:10154–10161.
11.Chou, J., E. Kern, R. Whitley, and B. Roizman.1990. Mapping of herpes
simplex virus-1 neurovirulence to␥134.5, a gene nonessential for growth in
culture. Science250:1262–1266.
12.Coffin, R. S., A. R. MacLean, D. S. Latchman, and S. M. Brown.1996. Gene delivery to the central and peripheral nervous systems of mice using HSV1
ICP34.5 deletion mutant vectors. Gene Ther.3:886–891.
13.Delman, K., J. Bennett, J. Zager, B. Burt, P. McAuliffe, H. Petrowsky, D. Kooby, W. Hawkins, B. Horsburgh, P. Johnson, and Y. Fong.2000. Effects of preexisting immunity on the response to herpes simplex-based oncolytic viral
therapy. Hum. Gen. Ther.11:2465–2472.
14.Ghiasi, H., Y. Osorio, Y. Hedvat, G. C. Perng, A. B. Nesburn, and S. L. Wechsler.2002. Infection of BALB/c mice with a herpes simplex virus type 1 recombinant virus expressing IFN-gamma driven by the LAT promoter.
Virology302:144–154.
15.Halminen, M., P. Klemetti, O. Vaarala, M. Hurme, and J. Ilonen.1997.
Interferon-␥production in antigen specific T cell response: Quantitation of
specific mRNA and secreted protein. Scand. J. Immunol.46:388–392.
16.He, B., M. Gross, and B. Roizman.1997. The␥134.5 protein of herpes
simplex virus 1 complexes with protein phosphatase 1␣to dephosphorylate
the␣subunit of the eukaryotic translation initiation factor 2 and preclude
the shutoff of protein synthesis by double-stranded RNA-activated protein
kinase. Proc. Natl. Acad. Sci. USA94:843–848.
17.Howard, M. K., T. Kershaw, B. Gibb, N. Storey, A. R. MacLean, B. Y. Zeng, B. C. Tel, P. Jenner, S. M. Brown, C. J. Woolf, P. N. Anderson, R. S. Coffin, and D. S. Latchman.1998. High efficiency gene transfer to the central nervous system of rodents and primates using herpes virus vectors lacking
functional ICP27 and ICP34.5. Gene Ther.5:1137–1147.
18.Hukkanen, V., T. Rehn, R. Kajander, M. Sjo¨roos, and M. Waris.2000. Time-resolved fluorescence PCR assay for detection of herpes simplex virus
in cerebrospinal fluid. J. Clin. Microbiol.38:3214–3218.
19.Hunter, W. D., R. L. Martuza, F. Feigenbaum, T. Todo, T. Mineta, T. Yazaki, M. Toda, J. T. Newsome, R. C. Platenberg, H. J. Manz, and S. D. Rabkin.
1999. Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation of intracerebral injection in nonhuman primates.
J. Virol.73:6319–6326.
20.Ilonen, J.1979. Lymphocyte blast transformation response of seropositive and seronegative subjects to herpes simplex, rubella, mumps and measles
virus antigens. Acta Pathol. Microbiol. Scand.87:151–157.
VOL. 78, 2004 SPREAD AND REPLICATION OF HSV VECTORS 13151
on November 8, 2019 by guest
http://jvi.asm.org/
21.Kramm, C. M., M. Chase, U. Herrlinger, A. Jacobs, P. Pechan, N. G. Rainov, M. Sena-Esteves, M. Aghi, F. H. Barnett, E. A. Chiocca, and X. O. Breake-field.1997. Therapeutic efficiency and safety of a second-generation repli-cation-conditional HSV1 vector for brain tumor gene therapy. Hum. Gen.
Ther.8:2057–2068.
22.Krisky, D. M., D. Wolfe, W. F. Goins, P. C. Marconi, R. Ramakrishnan, M. Mata, R. J. Rouse, D. J. Fink, and J. C. Glorioso.1998. Deletion of multiple immediate-early genes from herpes simplex virus reduces cytotoxicity and
permits long-term gene expression in neurons. Gene Ther.5:1593–1603.
23.Lagunoff, M., G. Randall, and B. Roizman.1996. Phenotypic properties of herpes simplex virus 1 containing a derepressed open reading frame P gene.
J. Virol.70:1810–1817.
24.Lagunoff, M., and B. Roizman.1995. The regulation of synthesis and prop-erties of the protein product of open reading frame P of the herpes simplex
virus 1 genome. J. Virol.69:3615–3623.
25.Lambright, E., E. Kang, S. Force, M. Lanuti, D. Caparrelli, L. Kaiser, S. Albelda, and K. Molnar-Kimber.2000. Effect of preexisting anti-herpes immunity on the efficacy of herpes simplex viral therapy in a murine
intra-peritoneal tumor model. Mol. Ther.2:387–393.
26.Lasner, T. M., S. Kesari, S. M. Brown, V. M. Lee, N. W. Fraser, and J. Q. Trojanowski.1996. Therapy of a murine model of pediatric brain tumors using a herpes simplex virus type-1 ICP34.5 mutant and demonstration of
viral replication within the CNS. J. Neuropathol. Exp. Neurol.55:1259–1269.
27.Markert, J., M. Medlock, S. Rabkin, G. Gillespie, T. Todo, W. Hunter, C. Palmer, F. Feigenbaum, C. Tornatore, F. Tufaro, and R. Martuza.2000. Conditionally replicating herpes simplex virus mutant, G207 for the
treat-ment of malignant glioma: results of a phase I trial. Gene Ther.7:867–874.
28.Markovitz, N., D. Baunoch, and B. Roizman.1997. The range and
distribu-tion of murine central nervous system cell infected with the␥134.5⫺mutant
of herpes simplex virus 1. J. Virol.71:5560–5569.
29.Miller, C., and N. Fraser.2000. Role of immune response during neuro-attenuated herpes simplex virus-mediated tumor destruction in a murine
intracranial melanoma model. Cancer Res.60:5714–5722.
30.Mineta, T., S. Rabkin, T. Yazaki, W. Hunter, and R. Martuza.1995. Atten-uated multi-mutated herpes simplex virus-1 for the treatment of malignant
gliomas. Nat. Med.1:938–943.
31.Pelosi, E., F. Rozenberg, D. M. Coen, and K. L. Tyler.1998. A herpes simplex virus DNA polymerase mutation that specifically attenuates neurovirulence
in mice. Virology20:364–372.
32.Peltoniemi, J., E. Broberg, A. Halenius, N. Seta¨la¨, J.-P. Era¨linna, A. Salmi,
M. Ro¨ytta¨, and V. Hukkanen.2004. Immunomodulation by roquinimex de-creases the expression of IL-23(p19) mRNA in the brains of herpes simplex
virus type 1 infected BALB/c mice. Clin. Exp. Immunol.137:305–312.
33.Peltoniemi, J., N. Seta¨la¨, E. Broberg, M. Ro¨ytta¨, V. Hukkanen, A. Salmi, and J.-P. Era¨linna.2002. Semliki Forest virus infection is enhanced in Th1-prone SJL mice but not in Th2-prone BALB/c mice during Linomide-induced
immunomodulation. J. Neuroimmunol.132:83–92.
34.Rampling, R., G. Cruickshank, V. Papanastassiou, J. Nicoll, D. Hadley, D. Brennan, R. Petty, A. MacLean, J. Harland, E. McKie, R. Mabbs, and M. Brown.2000. Toxicity evaluation of replication-competent herpes simplex virus (ICP 34.5 null mutant 1716) in patients with recurrent malignant
glioma. Gene Ther.7:859–866.
35.Rasty, S., P. Poliani, D. Fink, and J. Glorioso.1997. Deletion of the S
component inverted repeat sequence c⬘and the nonessential genes U(S)1
through U(S)5 from the herpes simplex virus type 1 genome substantially impairs productive viral infection in cell culture and pathogenesis in the rat
central nervous system. J. Neurovirol.3:247–264.
36.Scarpini, C., J. May, R. Lachmann, C. Preston, S. Dunnett, E. Torres, and S. Efstathiou.2001. Latency associated promoter transgene expression in the central nervous system after stereotaxic delivery of replication-defective
HSV-1-based vectors. Gene Ther.8:1057–1071.
37.Skelly, C. L., M. A. Curi, S. L. Meyerson, D. H. Woo, D. Hari, J. E. Vosicky, S. J. Advani, H. J. Mauceri, S. Glagov, B. Roizman, R. R. Weichselbaum, and L. B. Schwartz.2001. Prevention of restenosis by a herpes simplex virus mutant capable of controlled long-term expression in vascular tissue in vivo.
Gene Ther.8:1840–1846.
38.Trgovcich, J., D. Johnson, and B. Roizman.2002. Cell surface major histo-compatibility complex class II proteins are regulated by the products of the
␥134.5 and UL41 genes of herpes simplex virus 1. J. Virol.76:6974–6986.
39.Whitley, R., E. Kern, S. Chatterjee, J. Chou, and B. Roizman.1993. Repli-cation, establishment of latency, and induced reactivation of herpes simplex
virus␥134.5 deletion mutants in rodent models. J. Clin. Investig.91:2837–
2843.
40.Wood, M., A. Byrnes, D. Pfaff, S. Rabkin, and H. Charlton.1994. Inflam-matory effects of gene transfer into the CNS with defective HSV-1 vectors.
Gene Ther.1:283–291.
41.Ziegler, T., M. Waris, M. Rautiainen, and P. Arstila.1988. Herpes simplex virus detection by macroscopic reading after overnight incubation and
im-munoperoxidase staining. J. Clin. Microbiol.26:2013–2017.
13152 BROBERG ET AL. J. VIROL.