Vol. 64, No. 10
Structure-Function Studies of the Herpes Simplex Virus Type
1DNA
Polymerase
MARYL. HAFFEY,l* JIRI NOVOTNY,2 ROBERT E. BRUCCOLERI,2 ROBERTD. CARROLL,' JOHN T. STEVENS,' ANDJAMES T. MATTHEWS'
Departments of Virology' and Macromolecular Modeling,2 The Squibb Institute for Medical Research,
Princeton,
NewJersey
085434000Received 4 April 1990/Accepted 10 July 1990
The analysisofthe deduced amino acidsequenceof the herpes simplex virustype1(HSV-1) DNA polymerase reported heresuggeststhat the polymerasestructureconsists of domains carryingseparatebiological functions. TheHSV-1enzymeis knowntopossess5'-3'-exonuclease(RNase H),3'-5'-exonuclease, andDNA polymerase
catalytic activities. Sequence analysis suggests an arrangement of these activities into distinct domains resembling the organizationofEscherichia colipolymeraseI. In ordertomorepreciselydefinethestructure andC-terminal limits ofa putative catalytic domain responsible for the DNApolymerization activity of the
HSV-1enzyme, wehave undertaken in vitromutagenesisandcomputermodelingstudies oftheHSV-1DNA
polymerasegene.Sequence analysis predictsthat themajorDNApolymerizationdomain of the HSV-1enzyme
willbecontained between residues 690and1100,andwepresentathree-dimensional model of this region,on
thebasis of theX-ray crystallographic structureofthe E. coli polymerase I. Consistent withthesestructural
andmodeling studies,deletionanalysis byinvitromutagenesisof the HSV-1 DNApolymerasegeneexpressed inSaccharomycescerevisiae hasconfirmedthatcertainamino acidsfromtheCterminus(residues1073to1144 and 1177 to 1235)can be deleted withoutdestroyingHSV-1 DNA polymerase catalytic activityand that the extremeN-terminal 227residues arealsonotrequiredfor thisactivity.
Several complementary lines of research have been fo-cused on defining the structural-functional organization of the herpes simplexvirus type 1 (HSV-1) DNA polymerase (poi). These studies include comparative sequence analysis
(1, 2, 4, 5, 18, 19, 21, 26-29, 32, 34, 41, 42, 45, 47, 48, 50), productionofantibodiestodefinedregions ofthepol protein (28, 36, 46, 49), and in vitro mutagenesis of the HSV-1pol gene (35). We have beenparticularly interested in defining and elucidating features ofthe HSV-1pol catalytic domain because it is themajortargetforavariety ofpotentantiviral drugs (9, 10, 13, 16, 22, 31, 40).
Ithas beenproposed forseveralyearsthat theCterminus
of HSV-1 polencodes the substrate-binding domain of the
enzyme (19). This proposal was based on comparative
sequence analysis ofHSV-1pol drug-resistant point
muta-tions and regions of protein sequence homology among
various DNApol enzymes. Consistent with this proposal,
studiesutilizing antibodies todefinedregions of HSV-1pol have revealed that antibodies raised against C-terminal HSV-1 pol sequences can neutralize enzyme activity,
whereasantibodies directedatN-terminalsequences cannot (28, 36, 46, 49).Inaddition,astudy evaluating truncationsor
deletions of the HSV-1polgenetranscribedand translated in
vitro revealed that C-terminal truncations could destroy enzyme activity, while N-terminal truncations removingup
to 67residues could retain enzymeactivity (14).
Structure-function studies of HSV-1 pol, such as those
described above, could benefit from amino acid sequence
analysis andathree-dimensional (3D)model ofthe enzyme or apartthereof. The HSV-1polenzyme belongstoa group
of eucaryotic and procaryotic polymerases having amino acid sequence homology to the human DNA pol a., in
contrast toafamilyofpolymerases bearingsequence
homol-*Corresponding author.
ogytothe EscherichiacoliDNApol I (1, 2, 4, 5, 26, 34, 35). Only minor amino acid sequence homologies have been reported between the two pol families, primarily in the region of the putative 3'-5'-exonuclease domain (4, 15). Although nothingisknown about the 3Dstructureof HSV-1 polor the relatedfamily ofpolymerases, the crystal struc-tureof the Klenowfragmentof the E. colipolIenzymehas been resolved (39). The lack ofreadily distinguishable
se-quence conservation between pol enzymes from the two families has madeextrapolationfrom the bacterialpol struc-ture difficult; however, computer modeling studies under-taken inourlaboratories foundasurprising degreeof
poten-tial 3D structural similarity between the bacterial and the viralenzymes.
In this report,we presentanalignmentof the amino acid
sequenceof HSV-1polinto three distinctcatalyticdomains (RNase H, 3'-5'-exonuclease,and DNApolymerase, respec-tively), evaluate theC-terminal limits of the DNA polymer-ization domainusingdeletion analysis byin vitro mutagen-esis of the HSV-1polgene in a Saccharomyces cerevisiae expression system, and present a 3D model ofthe DNA polymerase catalyticdomain. The deletion studies described in thisreportreveal that previouslyunidentifiedportions of both the N and C termini are not required for HSV-1 pol catalytic activity, consistent with predictions of the
se-quenceanalysisandmodel.
MATERIALS AND METHODS
Protein sequenceanalysis and computer modelingstudies. The computer program of Novotny and Auffray (38) was
used to calculate and display the amino acid sequence profiles of hydrophobicity, formal side chain electric charges, and secondary structure propensities (a-helix,
,-sheet, and
turn).
The amino acidsequences of the E. coli polIKlenowfragmentand the HSV-1(strain KOS)DNApol genewereobtained from theProteinIdentification Resource5008
JOURNALOFVIROLOGY, Oct. 1990,p. 5008-5018 0022-538X/90/105008-11$02.00/0
Copyright ©1990, American Society for Microbiology
on November 10, 2019 by guest
http://jvi.asm.org/
Database, National Biomedical Research Foundation, Georgetown University Medical Center, Washington, D.C., and werealigned on the basis of sequence homology. Special
attention was paid to amino acid side chain identities, side
chain chemical similarities, and the correspondence of
sec-ondary structural elements (a-helices and ,-strands) be-tween the two sequences.
Theatomic coordinates of the backbone a-carbons of the
Klenow fragmentwere obtained from the Brookhaven Pro-tein Data Bank, Brookhaven, N.Y. Building of the HSV-1
catalytic domain structure, structural manipulations, chain insertions, and deletions was done by using the computer programs CONGEN (6) and PEER (R. E. Bruccoleri,
un-published work). Color-coded screen representations of
backbone modelsweredisplayed with theuseofthe PEER
interactive graphics program and the Silicon Graphics Iris
4D/80GT workstation.
Materials. Molecularcloning proceduresutilized T4 DNA
ligase, T4 DNApolymerase, and restriction endonucleases
purchased from New England BioLabs or Bethesda
Re-search Laboratories. [3H]TTP (15 Ci/mmol)waspurchased
from ICN Biomedicals, Inc. Chemicalswereobtained from
Sigma
Chemical
Co. unless otherwise noted. The plasmid pNN3wasobtained from M. Challberg, National Institutes ofHealth, Bethesda, Md., and has been described previ-ously (8).Strains and media. TheE. coli strains usedfor this study
were XL-1 Blue (Stratagene Cloning Systems) and CJ236
(Bio-Rad Laboratories). The yeast strain wasY294, which hasbeenpreviously described (7, 20). Bacteriaweregrown inLuria broth(37)containing50
jig
ofampicillinperml or 30 ,ugofchloramphenicolperml whereappropriate; yeastcells were grown in minimal medium(0.7%
yeast nitrogen base[Difco Laboratories]) containing requiredamino acids (20to
30
,ug/ml)
and2% raffinoseor2%galactose. Transformationofyeast cells with the recombinant
plasmids
wasaccom-plishedaspreviously described (3, 20, 35)orbyan
alterna-tivetechnique (23) usinglithiumacetate.
Oligonucleotides.
Oligonucleotides
used for this studywere
synthesized
on an Applied Biosystems model 380BDNA synthesizer using
f-cyanoethyl
phosphoramiditechemistry. Oligonucleotides
werepurified by
chromotogra-phyon oligonucleotide
purification cartridges (Applied
Bio-systemsProduct Bulletin, January 1988). The sequences of
theoligonucleotides usedwere asfollows: pRC204, 5'-ccgg gggtcacacttcggg-3';pRC210, 5'-cttggcgttattccctcaggcctacagg gccttgaatgt-3'; pRC211, 5'-ccccaacaggtgggatcaggcctagtaata gtccgt-3';pRC212, 5'-gcggggcttggacgctcaggcctatcccggggggtc ggc-3'; pRC213, 5'-cgcgtacggcatgcgcgcggagct-3' and5'-ccgc gcgcatgccgta-3';pRC216, 5'-gatccccgaaggcctgca-3' and5'-gg ccttcggg-3'; and pRC218, 5'-gaggccgcggcacaggatgatgcgcatg gaata-3'.
Mutagenesis.
Oligonucleotide-directed mutagenesis
wasconducted
according
totheprocedure
of Kunkel(30),
using
bacteriophage M13mpl9 (51)and reagents
purchased
inakitsupplied by Bio-Rad;
additional enzymes were from U.S.BiochemicalorNewEnglandBioLabs. In ordertofacilitate
mutagenesis and reconstruction of the HSV pol gene, a
PstI-KpnIrestriction endonucleasefragmentfrompNN3(8),
containing
the C terminus of the HSV-1 pol gene anddownstream untranslated sequences, was subcloned into
M13mpl9.
Uracil-containing single-stranded DNAwaspre-pared
from thisbacteriophage
vectoraftergrowth
inE. coli CJ236and usedas atemplate
for themutagenesis
reaction. The double-stranded product of this reaction was used totransform E. coli XL-1 Blue.
Phage plaques
wereinitially
screenedfor correct mutations byrestrictionendonuclease digestion when possible, and mutants were confirmed by DNA sequenceanalysis usingthedideoxy-chaintermination method (44) when necessary. Unless otherwise noted,
recombinant DNA methodologies were performed utilizing
standard procedures as described by Maniatis et al. (33). When appropriate, a PstI-KpnI restriction endonuclease
fragment containingthecorrectly mutated HSV-1pol DNA from M13mpl9 and two fragments (MluI-PstI and
KpnI-MluI) constituting
the remaining portionof the appropriateparental vector DNA (lacking the mutated fragment) were
isolated from 1%agarosegelsandjoinedinathree-fragment ligation, usingT4 DNAligaseto createthemutatedvectors. Construction ofHSV-1 pol deletionexpressionvectors.The
parentalvectors(pMH101,
pMH202,
pRC203, andpRC205)were described previously (20, 35). An initial series of deletions were created in the
parental
HSV-1(strain
17)expression
vectorpMH202
by
restriction endonucleasedi-gestion
andreligation.
The first mutant, pJS1, was createdby isolation of the BglII-HindIII restriction endonuclease fragment containingthe HSV-1pol genefrompMH202and insertion of that
fragment
into theexpression
vectorpMH101 between the BamHI andHindlIl restriction endo-nucleasesites in the
multiple cloning
site(MCS).
ThepJS1
vector was
predicted
toinitiateHSVpolprotein synthesis
at residue 228, the first methionine downstream of the pro-moter,andwasdeleted forall HSV-1 sequences upstreamofresidue 223. The first two C-terminal
deletions, pJS2
andpJS3,
were frameshift mutationscreated,
respectively, by
deletion of a BamHI restriction endonuclease
fragment
between residues 1072 and 1145 and
by
deletion ofaPstIfragment spanning
theregion
downstream ofresidue 1008into the MCS of the vector. The
pJS2
andpJS3
deletions were predicted to frameshift after residues 1072 and 1008,respectively.
Subsequent
studiesutilizedtwomodifiedparental
vectors,pRC203and
pRC205,
eachcontaining
anHSV-1polgeneaspreviously
described(35).
These vectors encode thepre-dicted HSV-1
(strain KOS)
aminoacidsequence, exceptthatresidue 33 isa serine not
glycine.
ThepRC205
vector wasderived from the
pRC203
parentby
deletion of a PstIrestriction endonuclease site from theMCS ofthevector to
facilitate construction ofsomeofthe mutatedvectors. Because of
negative
results obtained with theoriginal
C-terminal deletion vectors
(detailed
in thefollowing
sec-tions),
we chose to introduce more conservativemutationsinto the
parental
vectorby
the insertion ofstop codons atresidues
111% (pRC204),
1177(pRC210),
1162(pRC211),
and1130
(pRC212).
Thesevectors wereprepared
frompRC205
by
using
oligonucleotide-directed
mutagenesis. Anaddi-tional C-terminal truncation
(pRC303)
was created frompRC203 by
deletionof sequences between the NaeIrestric-tion endonuclease site at residue 1216 and the HindIII restriction endonuclease site in the MCS of the vector.
pRC303
waspredicted
to result in a frameshift at residue1216anda
predicted
terminationofthepolprotein
ataTAAstop codon in the
synthetic
transcription
and translationtermination sequence.
The
pRC213
vector was madeby
deletion of sequencesbetween the MluI and SstI restriction sites in
pRC205
and introductionofasynthetic
oligonucleotide adaptor
designed
tomaintain the properreading
frame.Thisvectorremovedalarge
portion
(residues
165to596)
of the N terminus of the HSV-1 polprotein.
ThepRC214
vector was createdby
restriction endonuclease
digestion
ofthewild-type
vector,pRC205,
withBamHI,
filling
out the 5'overhanging
endson November 10, 2019 by guest
http://jvi.asm.org/
5010 HAFFEY ET AL.
with bacteriophage T4 DNA polymerase and religating the blunt ends to maintain the correct reading frame while
deletingresidues 1073to1144.ThepRC215vectorcontained thesamedeletion of residues 1073 to1144andwasprepared in a similarmanner as the pRC214 vector, except that the
starting
vector was pRC210, a C-terminal deletion mutantdescribed in the preceding paragraph which contains a
terminationcodon after residue 1177.
The pRC216 vector was prepared by restriction
endonu-clease digestion of pRC205 with PstI and BamHI and
reli-gation,
usingasyntheticoligonucleotideadaptordesignedtomaintainthe correct HSV-1 pol readingframe, while
intro-ducing
aninternal deletion ofresidues 1009 to 1144. Finally,thepRC218 vector wascreatedfrompRC205by
oligonucle-otide-directeddeletion of the sequence between residues 884 and892, while maintaining the correct reading frame.
Protein analysis and immunoblotting. Protein
determina-tions were performed by using a Coomassie blue protein
assaykit
(Bio-Rad).
Polyacrylamide gel electrophoresis andimmunoblots were performed as previously described (20,
35, 36). The filters were processed, usingthe Immuno-Blot assay kit
(Bio-Rad),
essentially as instructed by themanu-facturer except that bovine serum albumin was substituted for
gelatin.
Thedilution ofprimary antibodywas 1:200, and the secondary antibody was diluted 1:1,000 (alkalinephos-phatase-conjugated swine anti-rabbit immunoglobulin G
[IgG] [Dako Corporation]). Theantigenusedforpreparation of the anti-P5rabbit antiserumwas asynthetic oligopeptide, Tyr-Gly-Asp-Thr-Asp-Ser-Ile-Phe-Val-Tyr, passively ad-sorbed to polyvinylpyrrolidone; otherwise, preparation of theP5antiserum was asdescribed forpreparationof the P3
antiserum (35, 36).
Preparation of enzyme extracts. Yeast extracts were
pre-pared
asdescribedpreviously (20, 35)from 100-mlovernightcultures grown at 30°C. Yeast cells were pelleted at 4°C, frozenondry
ice,
andsuspendedin 50mMTrishydrochlo-ride (pH 8.0)-50 mM NaCl-1% dimethyl sulfoxide-10%
(vol/vol)
glycerol-5mM,-mercaptoethanol-10 mMEDTA-2 mM benzamidine-1 mMphenylmethylsulfonyl fluoride at
4°C. Lysis
was achieved by repeated vortexing of the cellsuspensions,
in the presenceof acid-washedglass beads, for 30-s intervals followedby cooling on ice until >70% lysis was achieved as judged by light microscopy. The lysed extracts were transferred to 1.5-ml microcentrifuge tubesand
centrifuged
at 12,000 x g for 10 min. Extracts werestored at -80°Cin 15% glyceroluntil use.Thefinal protein
concentrationswere approximately Smglml.
DNApol assays. The assays for endogenous yeast DNA
pol and HSV-1 DNA pol were performed as previously
described (20, 35). Endogenous yeast DNA pol reaction mixtures contained S ,ul of celllysate(25to30 ,ugofprotein) in 10mMTrishydrochloride(pH7.5)-10mMMgCl2-10 mM
ammonium sulfate-0.1 mM dithiothreitol-100 ,uM (each)
dATP,
dCTP, dGTP, and[3H]TTP
(27 cpm/pmol)-40 ,ugofnickedcalfthymusDNA perml-2 mM spermidine in a final volume of 50 ,l. HSV-1DNApolymerase assayscontained 5,u of enzymeextract(about 25
pug
ofprotein) in 50 mM Trishydrochloride
(pH 8.0)-5 mM MgCl2-100 mM ammoniumsulfate-1 mM dithiothreitol-5 ,uM (each) dATP, dCTP,
dGTP,
and [3H]TTP (540 cpm/pmol)-30 ,ug of nicked calfthymus
DNA perml-100 ,ugof bovine serum albumin per mlinafinal volume of 50
,ul.
All DNApolymerase assays were conducted induplicate andincubatedat37°C for 15 min.Neutralizationstudies. Enzyme extracts (5
RI)
were incu-bated with theappropriate IgG (4 ,ug per ml) for 1 h on icebefore theaddition of the other reaction components of the
DNApol
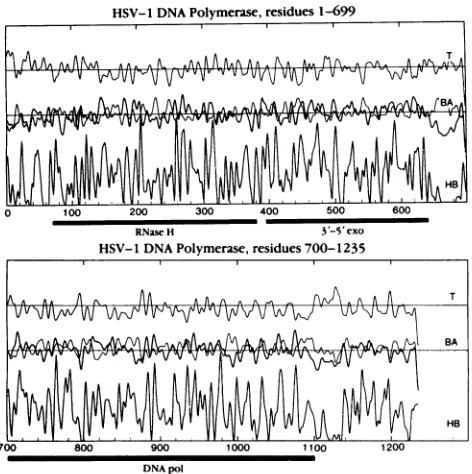
FIG. 1. Amino acid sequence profiles of theHSV-1 DNA poly-merase computed and smoothed as described previously (38). In each panel,the upperprofileshowsreverse-turn propensity(T);the middleprofilesare thoseof (x-helix(thinline, A) andp-sheet(heavy line, B) propensities; and the lower profile shows hydrophobicity (HB). The solid bars underthe panels denotethe putativelocation (within 10residuesateither end) of theRNaseH,3'-5'-exonuclease (3'-5' exo), and polymerase catalytic(DNA pol) domains.
HSV-1 DNA polymerase assay, as previously described
(36).
RESULTS
Structural analysis and computer modeling studies. Struc-turalanalysis of the HSV-1pol enzyme was performed on theassumptionthat structure-function studies under way in ourlaboratories using in vitro mutagenesis of the HSV-1pol
gene in a yeast expression system could benefit from such information. Figure 1 shows the resultant profiles of the HSV-1 pol amino acid sequence. The large size of the protein (1,235 residues) (19) suggested that its structure wouldconsist of several independent domains,and sequence profiles were used to identify polypeptide chain segments that separate the putative domains. Generally, interdomain segments of proteins are expected to consist of mostlypolar side chains and are conspicuous by a lack ofhydrophobicity. In contrast, the hydrophobic side chains are believed to be primarilyresponsible for protein folding and domain organi-zation (43). Prominent breaks in the hydrophobicity profiles of the HSV-1pol were found around residues 60, 660 to 690, and 1100 to 1140. Ofthese, the segment spanning residues 660 to 690 is the most remarkable, containing clusters of negatively charged residues (aspartates and glutamates) in-terspersed with positively charged lysines and arginines. This segment is likely to exist outside compact globular domainsbecause of its extreme charge.
Thedomain organization of the HSV-1pol which emerged from this analysis can be summarized as follows. (i) The N-terminal 65 residues are probably not integrated into the rest of the structure. (ii) The C-terminal 130 to 135 residues probablyrepresent a separate domain. (iii) Two segments, 65 J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.321.557.70.307.2]to 660 and 690 to 1100, are the best candidates for the principal structural domains. (iv) It is possible to subdivide the segment from residues 65 to 660 into two smaller domains and hypothetically assign the 5'-3'- (RNase H [11]) and 3'-5'-exonucleolytic activities (28, 40, 49) of HSV-1pol to residues 65 to 390 and 390 to 660, respectively, on the basis of size and structural similarities of the corresponding domains of the E. coli pol I Klenow fragment (25). (v) The 690 to 1100 domain corresponds in size and structural profile to the catalytic domain of the Klenow fragment, containing short runs of positively charged side chains (lysine and arginine) which are prerequisites for binding the strongly negatively charged polyphosphate backbone of the DNA. This domain (HSV-1 residues 690 to 1100) is the most likely candidate for the catalytic domain of HSV-1pol. Consistent with this assignment, many point mutations conferring resis-tance to antiviral drugs (nucleotide analogs) map to this region of the HSV-1 sequence (18, 28, 32).
Operating on the assumption that conservation of DNA
pol function might reflect conservation of structure, the HSV-1pol sequence was compared with that of the E. coli
pol
I enzyme. After secondary structural analysis, an amino acid alignment of the E. coli and HSV-1 putative catalytic domains was performed. Although the alignment showed only a low level of sequence conservation (about 10% identical residues and about 21% homologous residues) as previously reported (1, 2, 4, 5, 34), some of its features are supportive of a distant structural homology between the two domains. For example, (i) the majority of the conserved residues occur in a-helices and 1-strands, as opposed to unstructured loops; (ii) conservative amino acid replace-ments tend to cluster around invariant residues; (iii) virtually all insertions or deletions occur in surface loops; and (iv) the patterns of amino acid conservation and homologous re-placements conform to the character of the secondary ture found at that place in the X-ray crystallographic struc-ture of the Klenow fragment (i.e., n+1 pattern of residue conservation in the,B-strands,
conservation of prolines and glycines in loops) (Fig. 2).With use of the amino acid sequence alignment, a 3D tracing of the polypeptide chain was constructed in the computer, on the basis of the knowna-carboncoordinates of the Klenow fragment (Brookhaven Database). In the poly-merase catalytic domain of the Klenow fragment (residues 520 to 928 of the E.colipol I sequence), two segments of the backbone (residues 574 to 622 and 780 to 787) are not visible, probably because of local crystalline disorder. The X-ray structure of the Klenow fragment thus technically consists of three separate polypeptides, as does our HSV-1pol catalytic domain model (Fig. 2 and 3). Specifically, the Klenow fragment polypeptides containing residues 521 to 574, 620 to 774, and 790 to 928 approximately align with HSV-1 residues 698 to 749, 794 to 961, and 969 to 1106, respectively. Using the programs CONGEN and PEER, we first generated coordinates for the conserved parts of the catalytic domain backbone by copying the Klenow fragment coordinates into the HSV-1pol catalytic domain sequence. After completion of this step, the interactive graphics program PEER was used to model the rest of the backbone (insertions and deletions). The next steps involved (i) changes of chain topology involving the Klenow fragment region around a-helices J to K. In HSV-1 pol, the longer J helix connects to a13-strand homologous to strand 13 of the Klenow fragment and proceeds, via a turn, into strand 9. (ii) N-terminal extension of a-helix L by about two helical turns. (iii) Deletion of the irregular loop at residues 742 to 753 in the
Klenow fragment. (iv)Introduction of a few minor deletions and insertions in a-helix Q and the surrounding loops. The final approximate model of the HSV-1 DNA pol major catalytic domain is displayed in Fig. 3. The prominant upward groove is thepotentialDNA-binding site.
Expression of mutant polproteins. Predictions of the se-quence analysis described above were evaluated against
results obtained in our laboratories by using in vitro muta-genesis of the HSV-1pol gene. The S. cerevisiaeexpression
system used for themutagenesis studies has beendescribed
in detail elsewhere and has been shown to produce active HSV-1 pol (20). In vitro mutagenesis of the HSV-1 pol produced inyeast cells hasdemonstrated that apoint muta-tionintroduced into the gene canaffect thedrug resistanceor sensitivity of HSVpol enzyme activity and that the HSV-1 pol expression system in yeast cells can be used for struc-ture-function analysis of theenzyme (35).
Asummary of the mutantexpression vectors used for this study is shown in Fig. 4. Each of theexpressionvectors was evaluated for production of HSV-1 pol protein and for
HSV-1-specific DNA polymerase activity. The preparation
and analysis of yeast lysates was performed as previously
described (20, 35). Expression of the HSV-1 pol gene in yeast vectors is under the tight control of the yeast GAL] promoter (20, 24, 35). The GAL] promoter is active only
when cultures are grown in the presence of the inducing
sugar, galactose. When yeast cultures are grown in the presence of the noninducing sugar, raffinose, the GAL] promoter is not active and no HSV-1 pol protein is pro-duced.
Figure 4 summarizes the results of immunoblot assaysrun on lysates from galactose-induced cultures of the deletion mutants in order to detect the expression ofHSV-1pol. The immunoblot assays were performedas previously described
(35, 36), using an antipeptide antiserum to the HSV-1 pol
epitope, P3 (residues 548 to 557). An alternate
antipeptide
antiserum (P5) directed against residues 884 to892
(unpub-lished data) was used to evaluate expression of the HSV-1 pol gene in the pRC213 vectorwhichlacked the P3 sequence and to confirm the deletion of the P5 epitope in pRC218 as well as expression of HSV-1 pol in all of the other lysates
studied. Examples of the immunoblot assays
analyzing
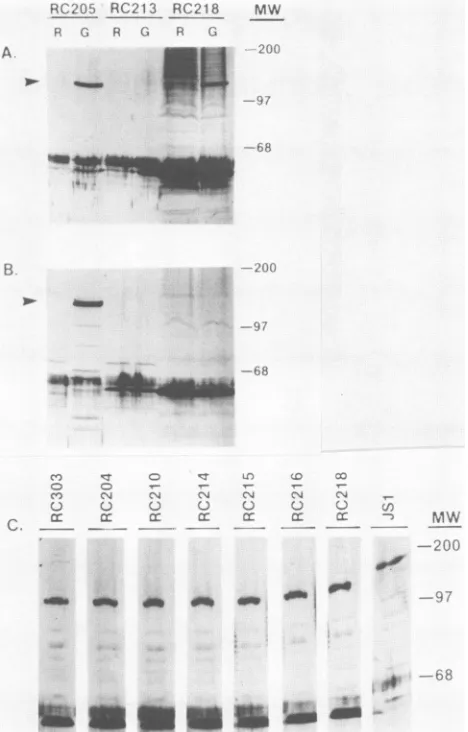
ly-sates from various recombinant yeast culturesare shown in Fig. 5. Panels A and B demonstrate that(i)the P5 antiserum appeared to recognize the sameprotein as the P3 antiserum
inlysates fromwild-typepRC205 cells grown under
inducing
conditions (in galactose), (ii) as expected, the P5 antiserum (but not the P3 antiserum) failed to detect the HSV-1 pol
protein expressed in the pRC218-derived lysate because of
thespecificdeletion of the P5 sequence, and
(iii)
neitherthe P3 nor theP5 antiserum could detect HSV-1polexpression
in control lysates of yeast cells grown under
noninducing
conditions (raffinose) or in the lysate ofthe
pRC213-trans-formed cells grown under inducing conditions. Panel C reveals that lysates from pJS1, pRC303,
pRC204,
pRC210,
pRC214, pRC215, pRC216, and pRC218 grown in the pres-ence of theinducing sugar,galactose, produced immunoblot-reactive HSV-1pol polypeptides.
Somewhat surprisingly, four of the C-terminal deletions (pJS2, pJS3, pRC211, andpRC212)failedto
produce
detect-able HSV-1 pol protein as measuredby
immunoblot assay using either P3 or P5 antisera(datanotshown).
Ofparticular
interest was the finding that termination of the HSV-1pol at residue 1177 (pRC210) did notgrossly affect
protein
expres-sion, whereas removal of 15 or 47 additional residues(ter-mination after residue 1162 [pRC211] or 1130
[pRC212],
on November 10, 2019 by guest
http://jvi.asm.org/
J.VIROL. 5012 HAFFEY ET AL.
521 530 940 550
Kisns 0LYPROLEOASN SALPE ASH ARTPROLO SVALPRO SAL ARE AO ASH SALLYE ASPPROLYE
698 710 720
Hsvp1 0LYALALYS vAL LEUASPPROTARSER 0L1 PE HISVALASHPROVAL VAL VALPM ASPPH ALASZRLO TSYRPROSER rIL
560 570
KiHAP VAL LEO HISAsn HISSER GL0 LEUTAIRLZUARE LEO ALAG01 LEO LYSLYE ALA HIIS G IL - - -
-730 740 750
ISVpI GLNALA HISASI LEOCYS PEzSERTER LEU SERLEUARE ALA ASP ALAVALALA HIS LU 0LUALA GLY LYSASPTYRLEO ILK
aaaaaa-aa-aa a-sz--- ^
a---ss---a-622 630 640 650
Riean PROLEU PRO LYS VALILZLO 00U TSYR ARE 0LYLEO ALALYSLEO LES SERTARTYR TARASPLYS LEO PROLW ARTILK ASH PROLYS
796 800 610 820
ElSVp1 SERPROGLU ALAVAL LEO LEO ASPLYSGLI ALAALA LYS VSALVALCYSASH SRVALTSYR0LYPAR TAR - - 0LYVAL
660 670
RlInH TER 0LY AREVALHISTA1R SERTYRHIS13W ALAVALTARALATAR0LY AREL SER - - -
-830 840 850
RSVpi GLNHISGLYLEO LEOPROCYSLEU HISVALALA ALATARVALTERTAR US CLYARE0LUHUTLEU LEUALATARAR 0WTSYR VALHIS
bbbbbbbbbbbbbbbbbbbbbbbb
.7. "8"
672 680 690
Kle U - TARASP PROASH LEO ALN ASHILEPROVAXLARG ASKCW 0W 1LYARG ARGILE ARG WLAALAPHI ILKALAPRtO
860 870 880
RSVpl ALAARCTRP ALA ALA PERGLO GLELEO LRE ALA ASPPMEPROGLU ALA ALA ASPHTAROALA PROCLYPROTSYRSARMET ARGrL1 rLE
697 700 710
KEIEn - - 0LUASP TYR VAL ILEVALSERALAASP - - - TYRSER ELNILreG0LEO ARG L MTALAHIS LEW
884 890 900 910
ASVpI TYR0LYASPTERASPSER ILEPEEVALLEUCYS ARG G YLEOTHR ALAALAGLYLEOTRRALAMUT0LYASPLYSNRTALASERHIS ILK
720 730 740
KEIEA SERARGASP LYS0LYLEOLEUTERALAPEEALAOLUGLY LYSASP ILEHISARGALATER ALA ALA0LUVALPEE0LY LEOPROLEU01
920 930 937
HSVpl SERARGALA LEUPEELEU PROPROILELYSLEU GLUCYSGL0LYSTER PRE TERLYSLEuLEULZUrLzALA - - -
-750 760 770
Flonr TER VALTER SER0L1 GLNAREARGSERALALYSALAILzASHPEA01 - - LEO IL TYRGLY - MSTSERALAPEEGLYLEOALA
-940 950 960
RSVpi - LYS LYS LYS TYRILzGLYVALILzTYRGLY0LYLYSMETLEO ILZ LYS0LYVALASPLEO7VAL ARGLYSASH -.0.
790 800 810
Kiss? LEUTYRPEAGLUARGTYR PRO0LYVALLEU0L1 TYRNRT 010 ARETERARGALAOLNALALYS010 080LYTSYR VAL01. TARLEUASP
970 980 990
IlSVpl TERSZRAROALALEuVALASPLEOLEU PEETYRASPASPTERvALSER 0LYALAALAALAALALEUALA010AREPROALA0W010TAP
a^a^-aaaaaaaaaaaaav-^---aaa^a^^aaaaaasa aaa _
.P.
10-620 830 840
Klns 0LYARGARELE TSYR LEU PROASPrLZLYI SERSERASH0LYALA ARG ARG ALAALA ALA01 AREALA ALA ILZASHALAPROHET OLN
1000 1010 1020
HSVpI LZU ALAARE - PROLEUPRO0LU 0LYLEUGOL ALAPEEGLY ALA - VALLEOVAL ASP ALA HISARE ARE rLZTARASPPRO0LU ARE bbbbbbbbbbbbbbbbbbbb
"11" "9.
850 860 870
KIC1? 0LY TAR ALA ALA ASP ILK LYS ARE - ALAMRTILzALAVALASPALA TAPLEU GLNALA 0LU GLE PRO ARE VALARE ARTILK ART
1030 1040 1050
8SVpi ASP ILz ASP PEE VALLEuTAR ALA LEUSERARE HISPRO ARE ALATYR THRASS LYSARE LEUALAHISLEUTER VALTYRTYR
a^-^aavaas--a-a -s s a kaa-a^a^as^a--a-aa-aaaaaaabaaaaaaaaaaaahbbbbbbbbbbbbbbbbbbb
.Q. "12"
880 890 900
Kliss GLNVAL HIS ASPG0. LEO VAL PEE VAL HISLYS ASP ASP VAL ASP - ALAVAL ALA LYS GLNILE HISGLN LEU ARTGLU
1060 1070 1080
lSVpI LYS LEO ARTALA ARE AREALA VAL PROSERILELYSASP ARE ILE PROTYRVAL ILEVALALA TAR ARE GLUVAL0LU GLU
910 920
ILUs? ASHCYSTARARELEOASPVALPROLEULEIJ VALGLUVALGLYSERGLYGL. ASETRP ASPGLNALAHIS
1090 1100 1110
lSVpi TARVAL ALAARELEUALAALA - - LEUAREGLULEU ASP ALA ALA ALA PROGLY ASP0LUPROALAPRO PROALAALA bbbbbbbbbbbbbbbbbbbbbbbbbbb"18"
Rw .6
on November 10, 2019 by guest
http://jvi.asm.org/
B
.o)e
1O6.
'{./8 -L
-:Q~
*e:c~~~~~~~~~ ,,,~
'<96 f__O6
*l:-~ ~ ~ ~ ~ 8
la L
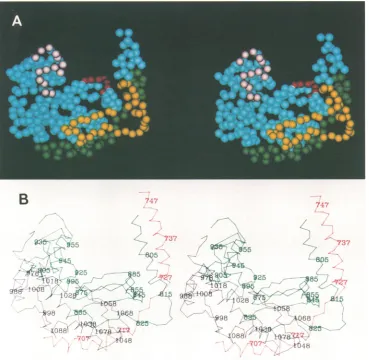
FIG. 3. (A)Stereoscopicrepresentation ofthecomputer-generatedmodelof the putative HSV-1DNApolymerasemajorcatalytic domain (refertoDNApol domain in Fig. 1). Amino acid residuesareschematicallyrepresentedasspheres with diameters comparabletothelength ofapeptideunit(0.38nm).Sequenceregions(I,II,IIl, andV)known tobeconservedin HSV-1 and other DNApolymerases describedby Gibbsetal.(18) andWang et al.(48)arecolorcodedasfollows:green,region II(residues694 to 736);yellow,regionIII(residues805 to845); red, regionI (residues882 to 889); and pink,region V (residues949 to963). Theprominant upward groove in themodel isthe putative DNA-bindingsite.ConservedregionVI(residues772 to791) (18, 48)isnotincludedin themodel becauseitalignswith aregion notresolved in the Klenow fragmentX-ray structure. (B) Alpha-carbon tracing ofthemodel of the putative HSV-1DNApolmajorcatalytic domain presented in panelA.The threeseparatepolypeptide chainsegments arecolor codedred,green, andblack. Theconsecutivenumbering of everytenthresidue is indicated and is consistent withthenumberingforHSV-1 polresiduesinFig. 2andreference 19.
respectively) resulted in lack of detectable HSV-1 pol pro-tein. Taken together, the results suggest that expression and/or detection of HSV-1 pol
protein
was affected by deletion of sequences between residues 1162 and 1177, atleast in the yeast expression system. Therefore, vectors
pRC214 and pRC215 were created, leaving intact residues
1162to 1177andadjacentupstream residues(1145 to1162),
which are conserved among the herpesvirus group (29), while deleting less-conserved sequences further upstream, residues 1073 to1144. Both thepRC214 and pRC215vectors
produced HSV-1 pol proteins recognized by immunoblot assay.
The pJS1, 227-residue N-terminal deletion produced
im-munoreactive protein but, disappointingly, an additional N-terminal deletion, pRC213, failed to produce detectable
HSV polprotein asmeasured in ourassay (Fig. 5). One unresolved result which emergedfrom the immuno-blot analysiswas alack ofapparent shift in electrophoretic mobility ofHSV-1pol immunoreactive
proteins
detected in the lysates derived from the HSV-1 pol deletion mutants (Fig. SC). We are attemptingto alterthegelconditionsand partiallypurify theexpressed polproteins inorderto inves-tigate this anamoly. Restriction endonuclease and/or DNA sequence analysis of the expressionplasmids,
NorthernFIG. 2. Amino acid sequence alignment of the putative catalytic domains of the E. coli pol I (KlenF) and HSV-1 (HSVpl) DNA polymerases.Eachof the sequences is numberedconsecutivelyfrom the NtotheCterminus.Thepositionsofsecondarystructureelements identifiedin theX-raycrystallographicstructureofthe Klenowfragment (39)aredenotedby"aaaa" (a-helices)and"bbbb"(P-sheets),and thelabeling convention (capitalletters andnumbers)is thatof Ollisetal. (39).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.123.492.80.440.2]5014 HAFFEY ET AL.
1 200 400 600 600 1000 1200 AA#
PEPTIDE# P3 P5 P6 P7
pJS1 *
2
3 .
pRC303 1
204 * *
210 1 *
211 * *
212 * A
213
1
*.-214 1*a
215 * 0
216 * * Ca
218
.cz.
AEXPRESSION
PROTEINIACTIVITY
+ I +
- I
-- I
-+ I +
+ I + + I +
_ I _
_ I
+ I + + I + + I
+ I
-RC205 RC213 RC218 MW
PR
; G R C'
A
t
^!
Vo _ aW
''.f (1
__EE
_0
i..4
FIG. 4. SummaryofHSV-1 DNApolymerase deletion studies. Schematic diagram ofthe deduced amino acid (AA) sequence of HSV-1pol(19), residues 1 to1235,is shown(horizontalbarattop). The regions of the enzyme determined in this study not to be requiredforenzyme activity are indicated in black. Thepositions (0) and designations (P3, P5, P6, or P7) of synthetic peptide sequences used togenerate antibodies used in this study arealso shown. Yeast expression vectors designated pJS- or pRC- and containingthe mutatedHSV-1DNApolgenearedescribedindetail in Materials and Methods, and the predictedopen reading frames are illustrated schematically (horizontal bars). Atthe right ofthe diagramis asummary ofthe resultsof expressionstudies,indicating whetheryeastcellscontainingeachvector expressed(+) orfailedto express(-)detectable HSV-1polproteinby immunoblot assayand showing HSV-1polenzymeactivity,measured asdescribedin the text.Resultsofassaysrunongalactose-inducedcultures are shown.
hybridization studies on RNA produced from the mutated vectors, and in vitro transcription and translation of the mutated pol genes does, however, reveal the expected size variations between wild-type and deleted constructs (data notshown). In addition, immunologicdatausingantiserato P5 inan immunoblotassay (Fig. 5) or antiserato P6 andP7 in the neutralization studies discussed below (Fig. 6) re-vealed thatdeletion of the cognate peptide sequences in the gene (pRC218, pRC214, orpRC204, respectively) results in expression of HSV-1 pol proteins which no longer are recognized by their respective antipeptide sera.
Detection of
HSV-l-specific
DNApolactivity. The HSV-1-specific DNA pol activity in yeastlysates was measured in the presence of 100 mM ammonium sulfate, as previously described; under thesehigh salt conditions the endogenous yeast DNA polymerases are not detected (20, 35). Table 1 andFig.4summarize the results of theDNApolassays run on the uninduced (raffinose) and induced (galactose) yeastcultures,
as well as control lysates from uninfected andHSV-1-infected HeLa cells. All of the
lysates
analyzed contained comparable amounts of total protein and produced comparable amounts of DNA pol activity when assayed under conditions in which the endogenous yeast enzymes were active (ot-pol assay). As expected, none of the four yeast lysates (pJS2, pJS3, pRC211, and pRC212) which failedto producedetectable HSV-1 pol protein by immuno-blot assay containedHSV-1-specific
DNA pol enzymeac-tivity. Two yeastlysateswhich produced HSV-1 pol protein
by immunoblot assay failed to produce detectable
HSV-1-specific DNA pol enzyme activity (pRC218 and pRC216). pRC218 contains an internal in-frame deletion of residues 884to892,aregion contained in the highly conserved region Iof DNApolymerasesdescribed by Wang et al. (48) refer to
C v o c
-.-
-_-CO CGq C'4 ^,A C'1 (N (N
() C) C) C C C
cc cr- ::a::. X o
C . _ _ ,MW
.C..0 Gi
.
. -,
[image:7.612.69.304.74.233.2]Emi
E~
el
"~
1fi
_
FIG. 5. Immunoblot assayofHSV-1 pol proteins in lysates of yeast cells containing thewild-type HSV-1polgene (pRC205) or
deletionmutants(pRC204, -210, -213, -214,-215, -216, -218, -303,or
-JS1[refertoFig. 4])growninraffinose(R)orgalactose (G) medium. Molecularsizesareindicated(MW)inkilodaltons.Arrowindicates mobilityof theHSV-1polprotein.(A)Immunoblotusing antiserum
againstthe P3peptide. (B)Immunoblotusingantiserumagainstthe P5peptide. (C) Composite ofimmunoblots on lysatesfrom
galac-tose-induced recombinantyeastculturesusingantiserumagainstthe P3 peptide. Assays were performed and the antisera used were preparedasdescribedinMaterialsandMethodsandby Matthewset
al. (36), respectively. Theamount ofyeastlysate loaded per lane
wasadjustedtobeequivalenttoapproximately 50to60,ugoftotal proteinonthebasis ofprotein assayresults.
Fig. 3),
and the P5antiserumepitope. pRC216containedaninternal in-frame deletion of residues 1009to 1144.
All of theremainingconstructswhichproduced detectable HSV-1
pol
proteinalsoproduced readilydetectable levels ofHSV-1-specific DNA
pol
activity. Taken together, these resultsdemonstrate thatlarge regionsofthe N terminus and certainregions
of the CterminusarenotrequiredforHSV-1 DNApol
enzyme activity. In contrast, sequences between residues 1009 and 1073(comparepRC215 with pRC216) and between 884 and 892 (pRC218) are apparently required for HSV-1pol-specific
activity, asmeasured inourassay.Neutralization studies. Previously, we had described the
productionoftwoantipeptide antisera capable of neutraliz-J. VIROL.
... (
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.325.559.75.441.2]%Control 100- m
80
60
40
0
4-pRC205 pRC204 IgG (4pg/ml)
IgG
* None
O Control
,P3
0 P6
P7
pRC214
FIG. 6. Resultsof neutralization studies on wild-type (pRC205) and mutated(pRC204 [P7 deletion] and pRC214 [P6 deletion]) HSV DNApolymerases expressed in yeast cells. IgG (4p.g/ml)was added to each reactionmixture and the assay was performed as described inMaterials and Methods. Control value (100%) was derived from assay runwithout IgG. Control IgG (derived from a rabbit immu-nizedwith bovineserumalbumin-thyroglobulincarrier protein) and the P3, P6, and P7 IgGs wereprepared as described previously (36).
ing the enzyme activity of purified HSV-1 DNA pol from infected HeLa cells (36). Peptide sequences P6 and P7 contain residues 1100 to 1108 and 1216 to 1224, respectively. These sequences were chosen in an effort to develop
[image:8.612.69.303.65.228.2]HSV-specific reagents and were not reported in other DNA polymerases. The structural analysis and modeling studies predicted, and the deletion studies clearly showed, that the P6 and P7 sequences could be deleted without destroying HSV-1-specific pol catalytic activity. Thus, these sequences were not required for substrate binding. We tested the
TABLE 1. Results of polassays
DNApolactivityasdeterminedbyb:
Extract' HSV polassay asPsaassay
Raffinose Galactose Galactose
Yeastcells
pRC205 0.28 9.88 117.7
pJSl 0.32 12.51 145.7
pJS2 0.36 0.31 138.7
pJS3 0.52 0.36 148.5
pRC204 0.24 10.00 137.5
pRC210 0.67 10.21 141.3
pRC211 0.29 0.37 156.1
pRC212 0.41 0.56 124.7
pRC213 0.37 0.41 143.0
pRC214 0.37 12.00 131.5
pRC215 0.33 12.14 135.9
pRC216 0.30 0.48 139.5
pRC218 0.44 0.44 151.0
HeLacells 0.33c 8.95d 109.5d
a Extractswerepreparedfromyeastcultures grown in either2%raffinose
or2%galactose mediumorfrom uninfectedorHSV-1-infectedHeLacultures,
asdescribed in Materials and Methods.
b DNApolassays wereperformedasdescribed in Materials and Methods
(20). All values are the averages ofduplicateassays andare expressedas
picomolesof[3H]TMPincorporatedper reaction.
cUninfected.
dInfected.
prediction that loss of these sequences would prevent the ability of the pRC214- andpRC204-encoded polymerases to be neutralized by the P6 and P7 IgGs, respectively. As previously reported for both crude and purified HSV-1 pol produced from HSV-1-infected HeLa cells (36), Fig. 6 re-veals that purified IgGs from both the P6 and P7 antisera were able to neutralize the HSV-1-specific pol enzyme activity in lysates from yeast cells producing the pRC205-encoded, wild-type HSV-1 pol. However, the HSV-1 pol activity in lysates from the yeast strain containing pRC214, which lacks P6 by virtue of a deletion of residues 1073 to 1144,and the strain containing pRC204, which lacks the P7 sequencebecauseof premature termination of the protein at residue 1196 (refer to Fig. 4), was not neutralized by anti-bodies directed at the deleted sequences. pRC214-encoded polcanbe neutralized by P7 but not by P6 IgG; conversely, pRC204-encoded pol can be neutralized by P6 IgG but not by P7 IgG. These studies confirm both the presence of the predicted deletions and the sequence specificity of neutrali-zation by these IgGs. Inaddition, these results demonstrate thatalthough the P6 and P7 sequences are specific targets for neutralizing antibodies, they are not strictly required for
catalytic activity asmeasured inourassay.
DISCUSSION
Structural analysis, computer modeling, and in vitro mu-tagenesis studies described in this report begin to define the C-terminal limit and structure of the HSV-1 DNA pol
catalytic domain. The model ofthis catalytic domain pre-sented in Fig. 3 is based on the crystal structure of the Klenowfragment ofE.colipolIand shares many features in common with the Klenow enzyme. Previous studies have proposed the involvement of this region of HSV-1 pol in substrate binding and catalysis. Gibbs et al. (18, 19) pro-posed that the region from residues 597 to 961 might foldto
formpartofadomaincontainingsubstrate anddrugbinding sites, onthebasis ofmapping ofHSV-1 poldrug resistance
mutations. A cluster of acyclovir-resistant mutations is
found in the lower right hand corner of the model (HSV
residues 719, 724, 813, 815, 821, 841, and 842 [28] Fig. 3).
Unfortunately, the model is not of sufficient resolution to evaluate the effects ofpoint mutations ontheoverall struc-ture of the enzyme. On the basis of amino acid sequence homology, Wangetal.(48)identified six segments that show a high degree of sequence conservation among viral and
eucaryotic DNA polymerases. Five of these segments map totheputativeHSV-1 polcatalytic domaindescribed in this report,andfour of them(regions I, II,III, andV)participate
in formation of the DNA-binding cleft (refer to Fig. 3).
Conserved region VI (HSV residues 772 to 791) was not included because italignswithadiscontinuityin the Klenow structure around residue 600. While ourmanuscript was in
preparation,Bernadetal. (4)predictedasimilarlocationfor theHSV-1polcatalyticdomain,aswellas anassignment of
the3'-5'-and5'-3'-exonuclease domainscomparabletothat described in further detail here. Conserved regions IV(48)
and A (18), which are not included in our model of the HSV-1polcatalyticdomain, arepredicted tobe part ofthe 3'-5'-exonuclease site (thisreport and Bernadetal.
[4]).
The modeling studies are supported by the view that conservation of function reflects 3D structure in lieu of extensive sequence conservation. The
validity
of thisview-pointwithrespecttopolenzymesisconfirmed
by
therecentreport of the 3D structure of the E. coliRNA
polymerase
holoenzyme determined by electron
crystallography (12),
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.62.301.454.654.2]5016 HAFFEY ET AL.
which exhibits remarkable similarity to the 3D structure of the E. coli Klenow fragment despite onlyasmall degreeof amino acid sequence conservation. Consistent with the structural analysis andthe model for HSV-1pol proposedin thisreport,wehave demonstrated thatregionsN terminalto residue 228 and certain regions C terminal to residue 1073
are not essential for HSV-1 pol catalytic activity as
mea-sured in ourassay. Because of difficulties in detection ofa
larger N-terminal deletion, wehavenotbeen abletofurther define the N-terminal limit of thecatalyticdomain. Infact,it
may notbe possibleto delete some of these sequences and maintainproteinconformation and stability, as was
experi-enced in deletions of the C-terminal residues between 1162 and 1177 described in this report. It is noteworthy that separation of the 3'-5'-exonucleaseandpolymerasedomains of the Klenow fragment adversely affected the stability of the polymerasedomain(15)and that the T4 DNA
polymer-asecannot yetbe physically separated into exonuclease and polymerase domains,andanoverlap of theT4domainshas been suggested (45). One could imaginethat the polymeriza-tion and exonuclease catalyticsitesareseparatebut that the sites requiredforbinding the DNA substrateorfoldingthe pol protein may overlap. Consistent with this concept,
Becker has reported that HSV-1polresidues between 431 and638maybeinvolvedinbindingtothe DNAtemplate (2). An N-terminal sequence proposed from the structural
analysistobeaseparatefoldingdomain(residues 1to65)is notessential forcatalytic activity. Thisis evidencedbythe predicted 227-residue, N-terminal (pJS1) deletion in this study and the predicted 27- and 67-residue deletions in an
earlier in vitro transcription-translation study (14).
Sequence structuralanalysisrevealed that the C-terminal 130to135residuesprobablyrepresentaseparatedomain. In contrast toresults fromaprevious study (14)and consistent
with this prediction, certain C-terminal sequences down-stream of residue 1100 (residues 1073 to 1144 and 1177 to 1235) can be deleted without grossly affecting HSV-1 pol catalytic activity despitethe fact thattheyarerecognized by
antibodiescapableofneutralizingpol activity. Antibodiesto these regions (P6 and P7 sequences) may accomplish
en-zyme inactivation by sterically blocking access of DNA
substratetothe active siteorby perturbingthe3Dstructure of the enzyme. If the former is true, it is possible that antibodies which prevent access of DNA substrate could affect all three catalytic activities of the enzyme. This hypothesis couldbe tested with the P6 and P7 antibodies and might explain results ofaprevious study (28, 49) in which antibodies directed at C-terminal sequences ofthe HSV-1
pol neutralized both pol and 3'-5'-exonuclease activities. Additional workwillbe requiredtodetermine whether P6or
P7orbothneutralizing antibodies sterically block binding of
DNA substrate or whether deletion of these sequences
affects exonucleaseactivity.
Conservation of certain sequences between HSV-1 and HSV-2 and not other viruses (including the P6 and P7 epitopes) suggests a requirement for these regions in the
specific context of HSV replication in vivo. The possible involvementof thesesequencesin interaction withthe other viral orcellularcomponents involvedin origin-specific
rep-lication (8)willbe investigated using the deletion mutants.
We have also shown that sequences between 1162 and
1177 may be required for stable expression of the pol protein, at least in the yeast expression system. This
se-quenceisnotaclassic nuclear localization orprotein
stabil-ity sequence, isgenerally hydrophobic, and could, alterna-tively, be involved in intranuclear localization or
intramolecularprotein folding.Arequirementfor thisregion
forcorrect intramolecularprotein foldingcould explainthe difference betweenourresult and that ofa
previous study by
Dorsky and Crumpacker (14). In that
study,
using
in vitrotranscription-translation,
itwasreported
thattermination of the HSVpol geneat aBamHI restriction endonuclease site coincident with residue 1073 of the HSVpol protein pro-ducedapolypeptide lackingHSVpolenzymeactivity. It ispossiblethat the HSVpolprotein expressedin theirsystem
was not properly folded because of the loss of residues between 1162 and 1177 which were
found,
in thestudy
reported here,tobe
required
fordetectionof stable HSVpolexpression
andactivity.
Theregion
between residues 1162and 1177 has
previously
beenreported
to be conservedamongDNA
polymerases
from such members of theherpes-virus group asEpstein-Barr
virus,
humancytomegalovirus,
and varicella-zoster virus
(2, 29; M.L.H.,
unpublished
ob-servations),
suggesting
aconservationoffunction,
albeitnotnecessarily
forcatalysis.
In
general,
regions
whicharepoorly
conserved among theherpesvirus
group (29) atboth the N and C termini canbedeleted without
destroying
HSV-1 polcatalytic
activity
or the ability to detectprotein expression,
consistent withprevious proposals that sequence conservationis relatedto conservation offunction. While deletion ofthe sequences upstream of residue 228 which are conserved among the
herpesvirusgroup
(residues
176to 221) does not affectpol catalyticactivity,
it ispossible
thatthis deletion may affect the 5'-3'-exonucleaseactivity
of the HSV-1 pol which ispredicted
to map to thisregion
(4; thisreport).
A modestsequence
homology
of thisregion
of the HSV-1polprotein
(residues
171 to 214)to the 5'-3'-exonuclease domainofE.coli
pol
I hasalso beenreported previously (2).
The loss of enzyme
activity,
as measured inoursystem,by
deletion of residues 1009 to 1144(pRC216)
but not residues 1073 to 1144(pRC215)
suggests that theregion
between 1009 and 1073 is
required
forcatalysis,
and the structuralanalysis
and the model would alsopredict
this.This
region
of the HSV-1 sequencealigns roughly
withhelixQand strands12and 13of the Klenow
fragment.
Thisregion
of the E. coli enzyme has been
proposed
as part of thepolymerase
active site(25).
Conservation of thisregion
inthe sequencesof the
herpesvirus polymerases
also supports afunctional role for thisregion (29).
Inordertodefinitively
address therequirement
for thisregion,
it will be necessaryto
purify
theexpressed pRC216protein
andevaluate itspolactivity
andability
tobind substrate underavariety
of assayconditions.
Finally,
deletion ofthehighly
conservedregion
I sequence(48)
(HSV-1
residues 884 to 892)destroyed
cata-lytic activity,
as measured in our assay. This result isconsistent with
predictions
of an essential role for this sequence (1,48, 50)
and results ofaprevious study using
invitro translation(14)inwhich deletion of residues 881to958
also
destroyed
HSV-1polactivity.
While this report focused on
defining
the HSV-1 polcatalytic domain,
the structuralanalysis
alsopredicted
anassignment
for the 5'-3'- and 3'-5'-exonuclease domainsby
analogy
withtheE. colipol
Iholoenzyme. Anumberof theconstructscreated for this
study
will be analyzedfor these additional activities. The structural analysis reported heremight predict
thatthepJS1
N-terminaldeletion would result in loss of 5'-3'-exonucleaseactivity
orthat thepRC216andpRC218C-terminal deletions which lackpolcatalytic
activ-ity might
still retainboth exonuclease activities.Mutagene-sis of the
putative
3'-5'-exonuclease domain is in progresstodirectly
addresspredictions
of this functionalassignment.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Severalof the mutations created for this study should also be testedfor the ability to be stimulated by another HSV-1 gene required for origin-specific replication, UL42 (17).
Because the model of the HSV-1polenzyme presented in Fig. 3 is 3D and closely resembles the Klenow fragment 3D structure, additional predictions andcomparisonsof the two enzymes can be made. Specific residues in the Klenow enzyme have been identified (25, 39) which bind nucleotide or DNA substrates (i.e., Arg-690, Lys-758, and Tyr-766); although these residues do not directly align with identical residues in the HSV-1 sequence, mutagenesis of the analo-gousregions of HSV-1polcould be performed to determine whether the HSV-1 sequences have similar properties. Other HSV-1 regions may be predicted from the model to be accessible to DNA or exposed on the exterior of the mole-cule and available for interaction with other components of the replication system. Both sorts of prediction are amenable to experimental testing. The 3D model of HSV-1 pol re-ported here is an approximate one and will certainly need refining; for example, we have shown that HSV-1 sequences between residues 1073 and 1100 are not essential for cataly-sis and thus need not be included. It is hoped that the model may serve as a starting point forfurther studies designed to elucidate the structure and function of this vital HSV-1 replicative enzyme. In addition, through a similar process, approximate 3D structures of other eucaryoticpolenzymes could be derived. Comparisons of the predicted 3D structure of the HSV-1 pol with other viral or cellular polymerases could also prove tobe enlightening.
ACKNOWLEDGMENTS
We gratefully acknowledge A. K. Field for support, encourage-ment, and critical reading of the manuscript. We thank M. Hagen and K. Mazina for preparation of the oligonucleotides and J. Alcantara and J. Danielson for creating some of the illustrations. We thank M. D. Challberg for the generous gift of the pNN3 vector and B. J. Terry for supplying HSV-1-infected and uninfected HeLa cell extracts.
LITERATURE CITED
1. Argos, P. 1988. A sequence motif in many polymerases. Nucleic Acids Res. 16:9909-9916.
2. Becker, Y. 1988. Computer-assisted primary and secondary structure analyses of DNA polymerases of herpes simplex, Epstein-Barr and varicella zoster viruses reveal conserved domains with some homology to the DNA binding domain in E. coli DNA polI. Virus Genes 1:351-367.
3. Beggs, J. D. 1978. Transformation of yeast by a replicating hybrid plasmid. Nature (London) 275:104-109.
4. Bernad, A., L. Blanco, J. M. Lazaro, G. Martin, and M. Salas. 1989. A conserved 3'-5'exonuclease active site in prokaryotic and eukaryotic polymerases. Cell 59:219-228.
5. Bernad, A., A. Zaballos, M. Salas, and L. Blanco. 1987. Struc-tural and functional relationships between procaryotic and eu-caryotic DNA polymerases. EMBO J. 6:4219-4225.
6. Bruccoleri, R. E., and M. Karplus. 1987. Prediction of the folding of short polypeptide segments by uniform conforma-tional sampling. Biopolymers 26:137-168.
7. Brugge, J. S., G. Jarosik, J. Andersen, A. Queral-Lustig, M. Fedor-Chaiken, and J. R. Broach. 1987. Expression of Rous sarcoma virus transforming proteinpp6v-src in Saccharomyces cerevisiae cells. Mol. Cell. Biol. 7:2180-2187.
8. Challberg, M. D. 1986. A method for identifying the viral genes required for herpesvirus DNA replication. Proc. Natl. Acad. Sci. USA83:9094-9098.
9. Chartrand, P., C. S. Crumpacker, P. A. Schaffer, and N. M.
Wilkie. 1980. Physical and genetic analysis of the herpes sim-plex DNA polymerase locus. J. Virol. 103:311-326.
10. Coen, D. M., H. E. Fleming, L. K. Leslie, and M. J. Retondo.
1985. Sensitivity of arabinosyladenine-resistantmutants of her-pes simplex virus to otherantiviraldrugs andmapping of drug hypersensitivity mutations to the DNA polymerase locus. J. Virol. 53:477-488.
11. Crute, J. J., and I.R. Lehman. 1989. Herpes simplex-1 DNA polymerase. J. Biol. Chem. 264:19266-19270.
12. Darst, S. A., E. W. Kubalek, and R. D. Kornberg. 1989. Three-dimensional structure ofEscherichiacoliRNA polymer-ase holoenzyme determined by electron crystallography. Na-ture (London) 340:730-732.
13. Derse, D., K. F. Bastow, and Y.-C. Cheng. 1982. Characteriza-tion of the DNA polymerases induced by a group of herpes simplex type 1 variants selected forgrowth in the presence of phosphonoformic acid. J. Biol. Chem.257:10251-10260. 14. Dorsky, D. I., and C. S. Crumpacker. 1988. Expression of
herpes simplex virus type 1 DNApolymerase gene by in vitro translation andeffects ofgene deletions on activity. J. Virol. 62:3224-3232.
15. Freemont, P.S., D. L. Ollis, T. A. Steitz, and C. M. Joyce.1986. A domain of the Klenow fragment of Escherichia coli DNA polymerase I has polymerase but no exonuclease activity. Proteins Struct. Funct. Genet. 1:66-73.
16. Furman, P. A., M.H. St.Clair, J. A. Fyfe, J. L. Rideout, P. M. Keller, and G. B. Elion. 1979. Inhibition of herpes simplex virus-inducedDNApolymerase activity and viral replication by 9-(2-hydroxymethyl)guanine and its triphosphate. J. Virol. 32: 72-77.
17. Gallo, M. L., D.I. Dorsky, C. S.Crumpacker,and D. S. Parris. 1989. The essential 65-kilodalton DNA-binding protein of her-pes simplex virus stimulates the virus-encoded DNA polymer-ase. J. Virol. 63:5023-5029.
18. Gibbs, J. S., H. C. Chiou,K.F.Bastow, Y.-C. Cheng, and D.M.
Coen. 1988. Identification ofamino acids inherpessimplexvirus DNA polymerase involved in substrate and drug recognition. Proc. Natl. Acad. Sci. USA 85:6672-6676.
19. Gibbs, J. S., H. C. Chiou, J. D. Hall, D. W. Mount, M. J. Retondo, S. K. Weller, and D. M. Coen. 1985. Sequence and mapping analysis of the herpes simplex virus DNA polymerase genepredict aC-terminal substrate binding domain. Proc. Natl. Acad. Sci. USA 82:7969-7973.
20. Haffey, M. L., J. S. Stevens, B. J. Terry, D. I. Dorsky, C. S. Crumpacker, S.M.Weitstock, W. T. Ruyechan, and A. K. Field. 1988. Expression of herpessimplexvirus type 1 DNA polymer-ase inSaccharomyces cerevisiaeand detection ofvirus-specific enzyme activity incell-free lysates. J. Virol. 62:4493-4498. 21. Hall, J. D. 1988.Modelingfunctional sites in DNApolymerases.
Trends Genet. 4:42-45.
22. Honess, R. W., and D. H. Watson. 1977. Herpes simplexvirus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584-600.
23. Ito,H., Y.Fukuda, K. Murata, and A. Kimura. 1983. Transfor-mation of intact yeast cells treated with alkali cations. J. Bacteriol. 153:163-168.
24. Johnson, M., and R. W. Davis. 1984.Sequencesthatregulatethe divergentGALI-GALIOpromoter inSaccharomycescerevisiae. Mol. Cell. Biol. 4:1440-1448.
25. Joyce, C. M., and T. A. Steitz. 1987. DNA polymeraseI: from crystal structure tofunction viagenetics. Trends Biochem. Sci. 12:288-292.
26. Jung, G., M. Leavitt, J.-C. Hsieh, and J. Ito. 1987. Bacterio-phage PRD1 DNApolymerase: evolutionof DNApolymerases. Proc. Natl. Acad. Sci. USA84:8287-8291.
27. Knopf, C. W. 1987. The herpes simplex virus type 1 DNA polymerase gene: site of phosphonoacetic acid resistance mu-tation in strain Angelotti is highly conserved. J. Gen. Virol. 68:1429-1433.
28. Knopf, C. W., and K. Weisshart. 1988. Theherpessimplexvirus DNApolymerase: analysis of the functional domains. Biochim. Biophys. Acta. 951:298-314.
29. Kouzarides, T., A. T. Bankier, S. C. Satchwell, K. Weston, P.
Tomlinson, and B. G. Barrell. 1987.Sequenceandtranscription
analysis of the humancytomegalovirus DNA polymerasegene. J. Virol. 61:125-133.
on November 10, 2019 by guest
http://jvi.asm.org/
5018 HAFFEY ET AL.
30. Kunkel, T. A. 1985.Rapid andefficient site-specific mutagenesis without phenotype selection. Proc. Natl. Acad. Sci. USA 82:488-492.
31. Larder, B. A., and G. Darby. 1985. Selection and characteriza-tion of acyclovir-resistant herpes simplex virus type 1 mutants inducingaltered DNA polymeraseactivities. Virology 146:262-271.
32. Larder, B. A., S. D. Kemp, and G. Darby. 1987. Related functionaldomains in virus DNA polymerases. EMBO J. 6:169-175.
33. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: alaboratorymanual. Cold Spring Harbor Laboratory, ColdSpring Harbor, N.Y.
34. Matsumoto, K., T. Hiroyoshi, C.I. Kim, and H. Hirokawa. 1989. Primary structure of bacteriophage M2 DNA polymerase: con-served segments withinprotein-priming DNA polymerases and DNApolymerase I of Escherichia coli. Gene 84:247-255. 35. Matthews, J. T., R. D. Carroll, J. T. Stevens, and M. L. Haffey.
1989. Invitro mutagenesis ofthe herpes simplexvirus type 1 DNApolymerase gene results in altered drug sensitivity of the enzyme. J. Virol.63:4913-4918.
36. Matthews, J. T., J. T. Stevens, B. J. Terry, C. W. Cianci, and M. L. Haffey. 1990. Neutralization of purified herpes simplex virus DNA polymerase by two antipeptide sera. Virus Genes 3:343-354.
37. Miller, J. H. 1972. Experiments in molecular genetics, p. 433. ColdSpringHarborLaboratory, Cold Spring Harbor, N.Y. 38. Novotny, J., and C.Auffray. 1984. A programfor prediction of
protein secondary structure from nucleotide sequence data: application tohistocompatibility antigens. Nucleic Acids Res. 12:243-255.
39. Ollis, D. L.,P.Brick, R. Hamlin, N. G. Xuong, and T. A. Steitz. 1985. Structure of large fragment of Escherichia coli DNA polymeraseIcomplexed with dTMP. Nature (London) 313:762-766.
40. Ostrander, M., and Y.-C. Cheng. 1980. Properties ofherpes simplex virus type 1 and type 2 DNApolymerase. Biochim. Biophys. Acta 609:232-245.
41. Pizzagalli, A., P. Valsasnini, P. Plevani, and G. Lucchini. 1988.
DNApolymerase I gene ofSaccharomycescerevisiae: nucleo-tide sequence, mapping of a temperature sensitive mutation, and protein homology with other DNA polymerases. Proc.Natl. Acad. Sci. USA 85:3772-3776.
42. Reha-Krantz, L. J. 1988. Amino acidchanges coded by bacte-riophage T4 DNA polymerase mutator mutants. Relating
struc-ture to function. J. Mol. Biol.202:711-724.
43. Rose, G. D., and S. Roy. 1980.Hydrophobic basis of packing in globular proteins. Proc. Natl. Acad. Sci. USA 77:4643-4647. 44. Sanger, F., S. Mikien, and A. R. Coulson. 1977. DNA
sequenc-ing with chain terminatsequenc-ing inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
45. Spicer, E. K., J. Rush, C. Fung, L. J. Reha-Krantz, J. D. Karam, and W. H. Konigsberg. 1988. Primary structure of T4 DNApolymerase. J. Biol. Chem. 263:7478-7486.
46. Thomas, M. S., L. M. Banks, D. J. M. Purifoy, and K. L. Powell. 1988. Production of antibodies of predetermined specificity against herpes simplex virus DNA polymerase and their use in characterization of the enzyme. J. Virol. 62:1550-1557. 47. Tsurumi, T., K. Maeno, and Y. Nishiyama. 1987. Asingle-base
change within the DNA polymerase locus of herpes simplex virus type 2 can confer resistance to aphidicolin. J. Virol. 61:388-394.
48. Wang, T.S.-F., S. W. Wong, and D. Korn. 1989. Human DNA polymerase oa: predicted functional domains and relationships with viral DNA polymerases. FASEB J. 3:14-21.
49. Weisshart, K., and C. W. Knopf. 1988. The herpes simplex virus type 1 DNA polymerase. Polypeptide structure and antigenic domains. Eur. J.Biochem. 174:707-716.
50. Wong, S. W., A. F. Wahl, P.-M. Yuan, N. Arai, B. E. Pearson, K.-I. Arai, D. Korn, M. W. Hunkapiller, and T. S.-F. Wang. 1988. Human DNApolymerase ao gene expression is cell prolif-eration dependent and itsprimary structure is similar to both procaryotic and eucaryotic replicative polymerases. EMBO J. 7:37-47.
51. Yanisch-Perron, C., J. Vieira, and J. Messing. 1985. Improved M13 phage cloning vectors and host strains: nucleotide se-quencesof theM13mpl8 and pUC19 vectors. Gene 33:103-119. J. VIROL.
![FIG. 6.assayandtoinnizedtheDNA Materials each Results of neutralization studies on wild-type (pRC205) mutated (pRC204 [P7 deletion] and pRC214 [P6 deletion]) HSV polymerases expressed in yeast cells](https://thumb-us.123doks.com/thumbv2/123dok_us/1319064.85498/8.612.62.301.454.654/assayandtoinnizedthedna-materials-results-neutralization-deletion-deletion-polymerases-expressed.webp)
