Vol. 64, No. 10
Analysis of Mutations in
the
Integration Function of
Moloney
Murine Leukemia
Virus: Effects
on
DNA
Binding and Cutting
MONICA J. ROTH,1 PAMELA SCHWARTZBERG,2 NAOKO TANESE,2t ANDSTEPHEN P. GOFF2*
DepartmentofBiochemistry, University of Medicine andDentistryofNewJerseylRobert Wood Johnson Medical School,
Piscataway, New Jersey 08854,1 andDepartments of Biochemistry and MolecularBiophysics and Microbiology,
Columbia University College of Physicians andSurgeons, 630 West168th Street, New York, New York100322
Received5April 1990/Accepted 20June 1990
The3' terminus of thepolgene of Moloney murine leukemia virus encodes the integration (IN) protein,
required for the establishment of the integrated provirus. A series of six linker insertion mutationsand two
single-base substitutions were generated within the region encoding the IN protein. Mutationswereinitially
generatedwithinanEscherichia coliplasmid expressing the IN protein, and the resulting variantswereassayed
for DNA-binding activity. Mutations which altered conserved cysteine residues within a potential DNA
finger-binding motif resulted in lowerorvariable DNA binding, which appearedto be the resultof variable
protein folding. Upon renaturation, these proteinswereabletononspecifically bind DNA ina mannersimilar
tothat of the othermutantINproteins and theparent.Whenreconstructed back into full-length virus,seven of theeight mutations werelethal.All mutantsproducedastable IN protein in virions and mediated normal
conversion ofthe retroviral RNAtoits three DNA forms. Fine-structure analysis of the linear double-stranded
viral DNA indicated that all seven lethal alterations within the IN protein blocked the formation of the3'
recessedterminithat normally precedes integration.
The key step in the retroviral life cycle that ensures the
persistence of the viral genome in the infected cell and its
transmission todaughter cells is the integration ofa
double-stranded DNAcopyofthegenomeintothe DNAof the host
cell (for recent reviews, see references 21, 40, and 53).
Integration of the viral DNA is an efficient and orderly
process: specific sites neartheends of the viralgenome are
joined to the host DNA, so that every integrated provirus
has a similarstructure and organization (9, 25). These sites
consist ofshortinvertedrepeats,oftenimperfect,presentat
the terminiof thelongterminalrepeats(LTRs)that flank the
viral genes (55). During the integration reaction, two
char-acteristic events occur: the two terminal base pairs ofthe inverted repeatsare lost, and a small numberofbase pairs
from thetargetsequence areduplicated andultimatelyflank
the integrated provirus (24, 51, 52). These features are
sharedby retroviruses, otherretroposons, and many other
transposable elements (50).
A single viral gene product, the integration protein (IN),
has been identified as essentialforthe establishment of the
integrated provirus (11, 41, 44, 49). The IN function is
encoded nearthe 3' end of the viralpolgene; mutations in
this region have no effect on reverse transcription butcan
block integration of the viral DNA and subsequent
expres-sion of viralgenes. The IN region is initially expressed as
part of the large gag-pol precursor protein (28, 29). In the mammalian retroviruses this precursor is cleaved during
virion assemblyto generateaseparateINprotein of about 40
to45kilodaltons(kDa) (58). Thisproteinhas been shownto
exhibitatleastnonspecific DNA-binding activity (34, 46). In the avianretroviruses,thegag-polprecursorisonly partially
cleaved, and the IN function exists both as a free protein
termed pp32 and as part of the beta subunit of reverse
*Correspondingauthor.
tPresent address: Department of Biochemistry, University of
California, Berkeley,CA 94720.
transcriptase (14, 48).Thepurified avian proteins have been
shownto exhibit DNA binding (22, 39) and DNA
endonu-clease activity (18-20, 23, 32, 59), with specificity for the termini ofthe viral LTR (7, 12, 13, 27). Thepresence ofa
zincfinger motif-a cysteine-containingsequencecapableof
folding around a chelated zinc-has been noted and
pro-posedasimportantfor DNAbinding (26). Mutagenesisof the
Moloney murine leukemia virus (M-MuLV)IN protein has
been previously described, but detailed analysis of the
biochemical effects of thesemutationshas notbeen carried
out(10).
Recent work in the M-MuLVsystemhasprovideda more
detailed definition of the structures and sequences of the DNA intermediates in the integration reaction. Direct
anal-ysisof theunintegratedlinearviral DNA has shown that the
two ends are a mixture oftwo structures: full-length blunt
ends, and termini with the 3' ends recessed bytwo
nucleo-tides (5, 45). Kinetic analysis suggested that the terminal nucleotides on the 3' ends are cleaved in vivo to form recessed 3' ends and that the IN function isrequiredfor this
step. More information has followed from in vitro reactions
which recapitulate the integration of viral DNA, either
endogenousorexogenous,into added target DNA(3, 4, 15,
16). These studies have shown that the linear form of the viralDNA,notthe circularforms,is the directprecursorto
the provirus; that linear DNAs with recessed 3' ends are
activeprecursors; and that these 3' endsaredirectly joined
totargetDNA in the strand transfer step, leaving protruding
5' endsonthe viral DNA.
Tohelpdefine the role of the INproteininintegration,we
generateda series of linker insertion andsingle-base
substi-tutionsthroughout the domain of thepolgene encodingthe
M-MuLVINfunction. The effects of these mutationsonthe
in vitro DNA-binding activity of the protein and on the
formation of the recessed 3' termini of newly synthesized
linear viral DNA were examined. Mutations located in the
cysteine region, affecting the proposed zinc finger domain,
apparently altered the folding of the protein but did not
4709 JOURNALOFVIROLOGY, Oct. 1990,p.4709-4717
0022-538X/90/104709-09$02.00/0
Copyright© 1990, AmericanSociety for Microbiology
on November 10, 2019 by guest
http://jvi.asm.org/
completely abolish the nonspecificDNAbinding activityin
vitro. Most of these mutations were lethal to virus
replica-tion, blocking early events in infection. All mutations that
blocked viral integration werefoundtoblockthe formation
of the recessedtermini on the linear DNA formed in vivo.
MATERIALS AND METHODS
Cells, viruses,andinfections.NIH 3T3and Rat2cellswere
grown in Dulbecco modified Eagle medium (GIBCO)
sup-plemented with 10% (vol/vol) calf serum (Hyclone).
Wild-type virus was derived from clone pTll (33) orpNCA (8).
MutantsofM-MuLVweretested forreplicationcompetence
by the XC assay (47) andby reverse transcriptaseassay of
the supernatant medium (17) after transformation ofNIH
3T3 cells by the DEAE-dextran method (37) with cloned
DNA (0.25 ,ug per 105cells). dl5401viruswasobtained from
an existing NIH 3T3 producer line (49). Cloned Rat2 cell
linesexpressing IN- mutant genomeswereestablished after
either Polybrene- or calcium phosphate-mediated
cotrans-formation(61) withpSV2neo DNA(54). Individualcolonies
were selected for growth in thepresence ofthedrug G418
(GIBCO; 400 ,ug/ml) and assayed for release of reverse
transcriptase activity (17). Virus used for infection was
collected from 10-cm dishes containing 5 ml of medium
every 12 h. Infections ofRat2 cells were carried out in the
presence of Polybrene (8 ,ug/ml).
Bacteria, plasmids, and extracts. Escherichia coli HB101
was used as the host for propagation of all plasmids. The
bacterial IN protein expression plasmid pSCB150-18 was
described previously (46). Preparation of lysates and
isola-tion of insoluble protein fractions ofE. coli were done as
describedpreviously (57).
Electroblot transfer and binding assay. Southwestern
(DNA-protein) blot analysis was performed as described
previously (46).Insomeexperiments,afterelectroblot
trans-fer,thenitrocellulose filtercarryingtheproteinswassoaked
in buffercontaining 50 mMTris hydrochloride(pH 8.3), 50
mMdithiothreitol, 1 mMEDTA, and 8Mguanidine
hydro-chloridefor1htodenature theproteins.The filterwasthen
washed for 1 h at room temperature in dilution buffer (50 mM
Tris-HCl[pH 7.5], 2 mMEDTA, 2 mMdithiothreitol,0.5 M
NaCl, 0.1% Nonidet P-40, and 10% [wt/vol] glycerol),
fol-lowedby anadditional wash at 4°C for at least 24 h in the
samebuffer, to permitrenaturation.
Mutagenesis of pSCB150-18 plasmid DNA. (i) Bisulfite
mutagenesis. Plasmid pSCB150-18 DNA was partially
di-gested with BspMI in the presence of 1 ,ug of ethidium
bromideper ml (42), and the full-length linear DNAs were
isolated after electrophoresis (60). The DNA was treated
with 2 M sodium bisulfite for 3 h at 37°C and dialyzed as
described before (43). The plasmidDNAwas recircularized
by ligation and used to transform E. coli BD1528 (ung).
Colonies were screened for loss of either the ApaLI or
Hindlll sites which overlapped the BspMI cleavage site in
the pol gene.
(ii) Insertional mutagenesis. After limited digestion of
plasmidpSCB150-18 withAluI,thelinear DNA was isolated
by agarose gel electrophoresis. Then, 12-base-pair (bp)
EcoRIlinkers(sequence CCGGAATTCCGG; New England
BioLabs)wereadded to the termini with T4 DNA ligase. The
DNAwas digested with EcoRI, treated with T4 DNA ligase
to form circles, and used to transform bacteria. Mutants
werenamedby the nucleotide position of the mutation, using
thenumbering system in which the left edge of the left LTR
is nucleotide 1.
0 2 4 6 8 kb
Wild type
b15346-1
3-12 1 f * j557-12
247-12 /
ThrCy*LysAb Cys Al ---CCTCAAA GCT T,GT GCA C
-BspMI Hindlil ApaLI Val
---- ACCTGCAAAt TGT GCA C
-T r
b15349-1 ----ACCTGCAAAGCT T
GCAC---Ala GlyIh Pro Ala
in5345-12 AccGGATaC AAA-ATTCOcrG q TGT GCA C -EcoRI
FIG. 1. Positions of mutations affecting the IN protein. M-MuLV proviralDNAisshown at the top; thetwoboxedregions
atboth ends of theDNArepresentLTR sequences.Enlargement of
theINregion is shown below.Open triangles above the line indicate positions of 12-bp EcoRI (RI) linker insertion mutations. Aminoacid
and DNA sequences in thecysteineregion areshown inthelower half of thefigure. RT,Reverse transcriptase; PR,protease.
Antisera and immunoprecipitations. Anti-Pol serum was
prepared in rabbitsby immunizationwith thebacterialfusion
protein encoded by plasmid pSC1(58). Goatanti-M-MuLV
(serum 81S-107) was obtained from the National Cancer
Institute. To detect virion proteins, cultures were labeled
with [35S]methionine (Amersham), and the virions were
collected, pelleted through a 25% sucrose cushion, and
immunoprecipitatedasdescribedpreviously (58).Prestained
molecular weight markers were purchased from Sigma
Chemical Co.
DNA electroblotting and hybridization. Electroblotting of
DNA fromsequencing gels and hybridizationwith
oligonu-cleotide probes were performed as described before (45).
The U5 probe D contains the sequence 5'-GAGGAGACT
CACTAAC-3'.
RESULTS
Construction of linker insertionmutations in the IN domain
andanalysis of theirDNA-binding activity. Inpreviouswork,
we haveexpressedtheIN protein ofM-MuLVinanE. coli
expression system and demonstrated that both the
bacteri-ally derived protein and the natural gene product exhibit
DNA-binding activity (46). To identify regions of the IN
protein required forbindingtoDNA,weconstructeda setof
derivatives of bacterialplasmid pSCB150-18 expressingthe
INprotein byinsertinga12-bpEcoRI linker into AluI sites.
Six clones with different inserts in the IN region were
identified andmapped (Fig. 1). The presence ofonlyasingle
linker in each mutantwasconfirmed by detailed restriction
mapping, ensuring that eachmutationresulted in the
inser-tion ofonly fouramino acids(Table 1).
To testthe the ability of the mutant plasmids toencode
stableproteins,extracts werepreparedafterinduction of the
trp promoter, and proteins in the insoluble fraction were
analyzed by sodium dodecyl sulfate (SDS)-polyacrylamide
gel electrophoresis (Fig. 2). Ineach case, the IN protein of
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.315.555.65.262.2]M-MuLV IN PROTEIN MUTATIONS 4711 TABLE 1. Summary of mutations in IN domain
Amino acids Reverse
Virus Wild Viability' transcriptase
type Mutant activityb
Uninfected Rat 2 -
-cells (control)
in5233-12 QL QPEFRL - +
in5247-12 LSF LSRNSGF - + +
in5345-12 KAC KAGIPAC - + +
in5557-12 KL KPEFRL - +
in5939-12 QAH QAGIPAH - + +
in6161-12 AAW AAGIPAW - ++
bi5346-1 A V + +++
bi5349-1 C Y - +
Wild type + +++
a Viabilitywasdeterminedby detection ofXCplaques and reverse tran-scriptaseactivity aftertransient transformationof cells with theindicatedviral
DNAs (seeMaterialsandMethods).
b Reverse transcriptase activity present in culture medium from stable
producer celllinesexpressing theindicated viral DNAs. +++, Wild-type levels;++, 10 to25% of wild-type levels;+, 4 to10%oofwild-type levels; -,
nodetectableactivity.
Mr 46,000 could be readily detected by Coomassie blue
staining (Fig. 2A); in some casesthemobilityof themutant
protein was slightly altered from that of the wild type,
suggesting that significant structural changes had been
caused by themutation (e.g., Fig. 2A, lane6).
Each of the mutant proteins was tested for its ability
to bind a mixture ofsingle- and double-stranded DNA by
the Southwestern blotting procedure (2, 38). Briefly,
pro-teins were separated by electrophoresis, blotted to
nitro-cellulose, and challenged with labeled DNA. Most of the
mutant proteins retained the parent protein's ability to
bind DNA, either with added
Mg2+
as the divalent cation(Fig.2B1) or in the presenceof EDTA(Fig.2B2). Ingeneral,
there was stronger binding with EDTA than with
Mg2+.
There were significant variations in the amount of DNA
bound by differentmutant proteins, notcorrespondingwith
the amount of protein produced by different clones. The
protein from one mutant, in5557-12, consistently bound
more DNA than those from the other mutants and the
wild-type parent (lanes 6). Only one mutant, in5345-12,
showed a significant defect in binding when normalized to
the amountofprotein(lanes 5). Similar resultswereobtained
when the binding was performed in higher saltconditions,
although here weak binding by mutant in5345-12 could be
detected (Fig. 2C1 and C2). The mutation in this construct
resulted in the insertion offour new amino acids nearthe
N-terminus, increasing the spacing between two cysteine
residues from two six amino acids. This region is highly
conserved among homologous retroviral pol proteins (26)
and has the potential to form a zinc finger DNA-binding
structure, although not a canonical finger structure (1).
These results suggest that this region maybe important for
binding activityinthisassay,affectingeither therefolding of
theIN protein orits intrinsicbinding activity.
Constructionof twopointmutations near the5'end of the
IN region and further analysis of DNA-binding activity.
Be-causeanalysis ofthe insertion mutations suggestedthat the
amino-terminal region of the IN protein was potentially
involved in formation ofa DNA-binding site, point
muta-tions in this area were also tested. Two mutants were
constructed: bi5346-1, bearingahighlyconservative
Ala-to-Val substitution between the two cysteine residues
men-tionedabove, andbi5349-1, carryingachange of the second
Cysresidueto aTyr
(Fig.
1).Theproteins
encodedby
thesenew mutantplasmids, by thelinker insertion
plasmids,
andbythe parentwereanalyzedfor
DNA-binding
activity
afterelectroblotting
(Fig.
2Dand E). The INproteins
of thetwopoint mutants bound DNA
well,
both with(panel D1)
andwithout (panel D2) divalent cation; the Cys residue was
therefore not essential for this activity.
Experiments
withmutant in5345-12 gave variable results,
depending
on theparticular
preparation
ofelectroblotted filters(Fig. 2).
Thevariation in the binding
activity
ofthis mutantcould reflectvariable refolding ofthis
protein; depending
ontheexperi-ment, the mutant demonstrated no
binding
(panels
B1andB2), weak
binding
(panels Cl andC2),
orgood
binding
(panels Dl andD2).
To test whether variations in the renaturation of the
proteinswere responsiblefor these results,
duplicate gels
ofthe variousmutant proteins were
prepared
andelectroblot-ted as usual. One copy was incubated
overnight
and thenused directlyin a
binding
assay, while anotherwas treatedwith guanidine
hydrochloride
and thenexposed torenatur-ation conditions before the assay. Controlled
experiments
done either
directly
(Fig.
2E1) or after denaturation andrenaturation
(Fig.
2E2) showed that the INproteins
withalterations in the
cysteines
did not refoldcorrectly
tore-cover
activity.
We conclude that under someconditions,
thesemutant
proteins
arecapable
ofbinding DNA; however,
the ability of the in5345-12
protein
to renature,compared
with that of the proteins from the wild type and the other
mutants, is sometimes
impaired.
Whenfoldedcorrectly,
themutant proteins still exhibitbinding
activity.
Viability ofviralgenomeswith mutations in the INdomain.
Totest the
infectivity
of viral genomescarrying
site-specific
mutations in the IN
region, fragments carrying
eachmuta-tionwereexcisedfromthe
expression
constructsandusedtoreplacethe
equivalent fragment
ofafull-length
clone of theviral genome. Theresulting cloned DNAs were introduced
into NIH 3T3 cells by the DEAE-dextran method
(37).
Production of
replication-competent
virus was detectedby
the XC
plaque
assay (47) andby
measurement ofvirion-associated reverse
transcriptase
activity (17).
None of the12-bp insertion mutants gave rise to viable viruses after
transfectionat
37°C,
asjudged by
theXCplaque
assayorthereverse transcriptase assayof the medium (Table 1).
Trans-fection
experiments
with thetwopoint
mutantsshowed thatthe
bi5349-1
mutation, which resulted in the change ofthecysteine
residue totyrosine,
wassimilarly
lethal.However,
mutantbi5346-1,
with theconservativechange ofanalanineresidue to valine, was
viable;
the number of XCsyncytia
induced after DNA transfection was similar to that of the
wild-type virus. The level ofreverse
transcriptase
activity
detected in the medium was
indistinguishable
from that inthe medium collected from the
wild-type producer
cells.These results
suggested
thatalthoughseveral12-bp
insertionmutations did not affect the
DNA-binding
activity
of thefusion
protein
invitro,
these mutations were neverthelesslethal to the virus in vivo. In
addition,
the mutation thatchangedthe
cysteine
residuetotyrosine
was lethal invivo;
in thenitrocellulose-binding
assay of the INprotein
ex-pressed
in E.coli,
however, thecysteine
residue did notappeartobecriticalfor DNA
binding.
Therefore,
alterationsin the IN
protein
that havelittle effecton theDNA-binding
activity
asassayed
in vitro canblock its function in vivo.The functions of the IN
protein
inreplication
areclearly
more
demanding
than thebinding
ofDNA as measured in vitro.Construction of cell lines
producing
IN- virions. Tochar-VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
4712 ROTH ET AL.
A
!
M 1 2 3 4 5 6 7 8 M205~ POOl"
66mm- "se-n
45u.--
~~~
-
29w-BI
1 2 3 4 5 6 7 8
.
- ,w
B2
1 2 3 4 5 6 7 8 k@
-.c205
_w116
-m974
40mm"1_X '. 66
_ _h* s_ -w45
_r0
e.-mc29
C2
3 4 5 6 7 8 1 2 3 4 5 6 7 8
Vp
a-.1-.-...*
D2
El
1 2 3 4
!R
1 2 3 4 5c205~~~~~~~~~~~~~~~~~~~~~~~~~.
.mc116
I.
--a97A.-~ -ac66
_k
""C45_-> 044am -- 29
E2
1 2 3 4 5 kQ
-w18O
*c 116
m 84
"mc58
mA48.5
14
-- 36.5 _126.6
Cl
1 2
-c205 ._wl6
-am974
ef
DI
1 2 3 4
* ,,
-_92K
lonson-
.."l-,q
AMEMERL.-J. VIROL.
.4
INIMPIP"
AdkL-"ROW
on November 10, 2019 by guest
http://jvi.asm.org/
M-MuLV IN PROTEIN MUTATIONS 4713
FIG. 2. DNA-binding activity ofIN proteins with insertion and point mutations. (AtoC) Insoluble proteinswereprepared fromvarious
bacteriallysates; proteinsweresolubilized inSDS and analyzed by electrophoresisonSDS-polyacrylamide gels. (A)Coomassie stain of the
protein gel. Inlanes 2 to 8,the 46-kDa(kD)INprotein is indicated by the arrowhead. (B andC) Nitrocellulose-binding assayof the IN
proteins. Following electrophoresis,proteins wereblottedtonitrocellulose, denatured,renatured, andassayed forDNA-binding activityin
thepresenceof50 mMNaCl and5mMMgCl2(Bi),50 mMNaCl and10 mMEDTA(B2), 150mMNaCl and 5mMMgCl2(Cl),or150 mM
NaCl and10 mM EDTA(C2). Samples inpanels A toC: lanes1,pATH3; lanes2,wild-typeplasmidpSCB150-18; lanes 3, in5233-12; lanes
4,in5247-12;lanes 5,in5345-12;lanes 6,in5557-12; lanes7,in5939-12;lanes8,in6161-12;lanesM,sizemarkers. Positions of the size standards
are indicated. (D) Analysis of proteinsfrom added mutants. Proteins on blots were denatured, renatured, andassayed forDNA-binding activity inthe presence of 50 mMNaCl and5mM MgC12 (Dl)or 50 mMNaCl and10 mM EDTA(D2).Lanes1,pATH3controlcells; lanes 2, pSCB150-18; lanes3,in5345-12; lanes 4,bi5349-1. (E)Proteinswereelectroblottedasbeforeandincubated indilution buffer aloneprior
totheDNA-binding assay(El)or denatured andrenatured (E2). Each lane containsinsolubleprotein pelletsfrom thefollowing plasmids:lane
1, pATH3;lane 2, pSCB150-18;lane 3, in5345-12;lane 4,bi5349-1;lane 5, bi5346-1.
acterizein more detail the block to replication caused by the
new mutations in the IN domain, we established cell lines
expressingthemutantgenomes. The complete proviral DNA
bearing each mutation was introduced into Rat2 cells by
cotransformation with pSV2neo plasmid DNA (54).
Individ-ual G418-resistantclones were tested forproduction of virus
byassays ofreverse transcriptase activity released into the
medium.Allthe mutant genomes were able to induce release
of virion-associated reverse transcriptase activity,
suggest-ingthat late stages of the life cycle were normal (Table 1).
Virus was collected from these lines and used without
dilutiontoinfectfresh NIH 3T3 cells; the number of virions
in the preparation, judged from the level of reverse
tran-scriptase activity detected, was approximately 5- to 10-fold
less than in a similarpreparation of wild-type virus.
Medium washarvested from the infected cells and tested
for the appearance of progeny virus by reverse transcriptase
assays. Whereas cells infected with wild-type virus became
producers ofprogeny within 1 to 2days, cells infected with
the mutants did notrelease detectable progeny virus for up
to a month postinfection. These results suggested that the
mutant virions were unable toestablish aproductive
infec-tion andwereblockedinthe early phase of the life cycle.
Analysis of the virion proteins from IN- virus. Toexamine
the viral proteins present in the IN- mutant virions,
pro-ducer cellsweremetabolically labeled with [35S]methionine overnight, thevirionswereharvested,and the viral proteins
wereanalyzed by SDS gelelectrophoresisafter
immunopre-cipitationwith an antiserum raisedagainst whole M-MuLV
virions (Fig. 3). The abundant gag proteins of the virus are
the major proteins recognized by this antiserum. In
wild-type virions, the initial gag gene product,
Pr659'9,
isnor-mally processed to four smaller cleavage products by the
viral protease; in these experiments, high levels of the
mature CA protein were detected, showing that, as
ex-pected, cleavage oftheprecursor was essentially complete
(Fig. 3, lane 10). In most ofthe IN- mutant virions, the
predominantgagproteindetected was also the CA product.
The quantity of gag protein detected was highly variable
from producer linetoline; thisvariation may reflect both the
inherent efficiencyof assembly of the mutant virion and the
levelof expression from the particular integrated provirus.
Inseveralof the IN- mutantvirions,the Pr659'9precursor
proteincouldstillbedetected along with variousprocessing
intermediates(Fig.3, lanes 2 to 6 and9). A reduced rate and
final extent of cleavage of the gag proteins have been
reported previously for several mutants with alterations in
thepolgene (49). For mutantin5557-12, the cleavage was so
limited that the levels of the precursor were higher than
those of the mature proteins. These results indicate that
changes in many portions ofthepol gene can have some
effect on the activity or action of the viral protease. The
inability of these mutant viruses to process every gag
protein in the viral particle, however, is probably not the
causeof the lethality of these mutations. These virus were
capable of successfully infecting and carrying out reverse
transcriptionafter infection of fresh cells (seebelow).
Itis alsoimportant to note that all the mutants produced
functional levels of env gene products. Because of overlap
between the IN coding region and 5' portions of the env
mRNA, the two 3'-proximal mutations constructed here
could have affected env mRNA expression. In particular,
mutation in5939-12 is an insertion of 12 bp precisely at the
acceptor splice site for the formation of the singly spliced
env mRNA(31) and might well have disrupted formation of
the mRNA and protein. The fact that these two mutants
expressed normal amounts of envelope protein and could
entercells and carry out reversetranscriptionshows that the
mutant mRNAs containing the insertions are fully
func-tional.
Analysis of pol gene products of the virus. One trivial
explanationfor the lethality ofthe IN mutationsis thatthe
mutant IN protein might be unstable and not persist in the
CS N N N CY C4 in Co, CzNU)a
_ 4>N OC N c.
CM-LO LO Lo into n I'D
E 5 57E .E 1:5
1 2 3 4 5 6 7 8 9 1 0
kD
4-116
- 84
Pr659ag_
a_9*
= 58
4-CA (P30)_p 36
2 27
Anti-Moloney MuLV Sera
FIG. 3. Analysis of virion proteins. Cells were metabolically labeled with [35S]methionine, and virus was pelleted from the
medium. Virion proteins were immunoprecipitated with anti-M-MuLVantiserumandanalyzedbySDS-polyacrylamidegel
electro-phoresisandfluorography. Lane1, Rat2 cells, uninfectedcontrol;
lane2,d15401inNIH 3T3cells;lanes 3 to9, mutantsasindicated;
lane10, wild-typeM-MuLV. Molecularmassofproteinsize mark-ers are shownatright (in kilodaltons).Arrowsatleft indicate the
position ofthe majorcapsid protein CA and the gag polyprotein precursor
PM65gag.
VOL.64,1990
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.333.549.420.628.2]CM N C4 N N4
-- - #-
-~
-'-7
u) C' Cm -st t '> (D >
° ,) N 0) c4 C.) C)
-t
u) WU U) U In e)
OX.- .: .s . . :k ,
o 7
L?C1
IOU,c
oT0
f
N N N cN
0) ) - - c0 _
, teCto o
) N N U) 0)
e o e U) U, ID
C)
CQoScE i:s
-1 2 3 4 5 6 7 8 9 10
kD
80-v
_
_0
16_- X
84- u _ - * RT
(P80)
58 _
48 (P46)
_m _o _ *-4 N (P46)
3 6
_t
_ftw
jAnti-Moloney PQI Sera
FIG. 4. Analysis of virionpol gene products. Cells were
meta-bolicallylabeled with [35S]methionine, virus was pelletedfromthe
medium, and virion proteins were immunoprecipitated with rabbit
anti-pSC1antiserum and analyzed by SDS-polyacrylamide gel elec-trophoresis. Lane 1, Rat2 cells, uninfected control; lane 2,d15401in NIH 3T3cells;lanes 3 to 9, mutants asindicated;lane 10,wild-type
M-MuLV. Positions of protein size markers are shown at left (in
kilodaltons). Arrows atright indicate the positions ofthe reverse
transcriptase(RT)and IN proteins.
mature virion. To examine this possibility, the pol gene
products within the virions were examineddirectly. Proteins
weremetabolically labeled with[35S]methionine, viral
parti-cleswereisolated, and the virionproteinswere
immunopre-cipitated with sera which recognize both the reverse
tran-scriptase and the IN pol gene products (Fig. 4). Wild-type virions exhibited the expected pattern of an equimolar
amountofthefully-processedreversetranscriptase (ofabout
80 kDa) and mature IN proteins (46 kDa); none of the
gag-pol precursor, Pr200Bg-Pl, was detected (Fig. 4, lane
10). In sixoftheseven mutantvirions,thematureINprotein
was readily detected and was present at approximately
equimolaramountswith themature reversetranscriptase. In
some experiments the IN proteins of both mutants and
wild-type virions migrated as a doublet. The basis for this
behavioris unknown, but itisprobably not due to
phosphor-ylation (58). Forinsertionmutant in5557-12, theprocessing
of the pol precursor was incomplete; in some experiments
theprecursorpredominated andthecleaved INprotein was
barelyvisible (Fig. 4, lane 3), while in others thematureIN
protein was readily detected. These results show that the
new mutant IN proteins are synthesized, are generally
processed correctly, and persist stably inthemutantvirions.
As previously described, virions of the deletion mutant
d15401contained no detectable IN protein (58).
Analysis of the viral DNA synthesized after infection by
IN-virus. Todetermine whether the mutant virions could initiate
an infection and mediated the reverse transcription of the viral RNA into DNA, we examined the viral DNA in recently infected cells. Virus preparations of the various
IN- mutants were collected from the stable producer cells
and used to infect fresh Rat2 cells. The
low-molecular-1 2 3 4 5 6 7 8 9 1 0 1 1
%t=8:1§ FORM 11
_
- 4---Linear3w "3 4-=8.8 FORM
8.2
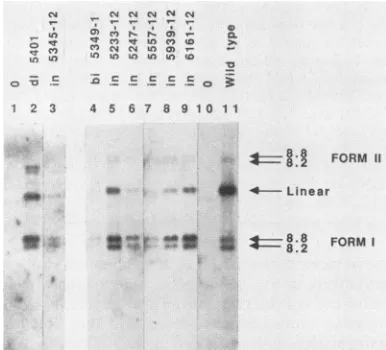
FIG. 5. Southern blot analysis of unintegratedviral DNA.Virus
washarvested from producer celllinesand used toinfect freshRat2
cells. Low-molecular-weightDNAwasisolated, separated by elec-trophoresis on agarose, transferred to nitrocellulose, and probed
with M-MuLV sequences. Cells were infected with supernatants from thefollowing lines: lane 1, Rat2 uninfectedcontrol; lane 2, d15401in NIH 3T3cells;lanes 3 to 9, mutants asindicated; lane 10, mock-infectedcontrol;lane11,wild-typeM-MuLVcontrol.Lanes 1 to 3 were a7-dayexposure. Lanes 4 to 11 were a1-dayexposure.
Sizesareindicatedattheright (in kilobases).
weight DNA was isolated 30 h postinfection, separated by
electrophoresis, and analyzed by Southern blot
hybridiza-tion(Fig. 5).
Inawild-type M-MuLV infection, three viral DNA
prod-ucts weredetected:an8.8-kb linearDNA,an8.8-kbcircular
DNA, and an8.2-kb circularspecies (Fig. 5, lane 11). The
linearspecieswasthepredominant formatthisearlytime in
infection; the amount of circular forms increased at later
time points (data not shown). Infection with the deletion
mutant dl5401 generated the same three proviral DNA
forms, although the ratio of circular to linear forms was
higherthanin the wild type(lane2), asdescribedpreviously
(45, 49). All of the IN- linker insertion and point mutants
were capable of infecting cells and synthesizing the three
proviralDNAproducts (lanes3to9). Theratio of circularto
linear DNAspecies againwas generallyincreased, favoring
the circular forms, as for mutant dl5401. Similar
observa-tions have been made after infectionbymutantsdefectivein
integration due to changes in the LTR termini (8). For
mutantin5557-12, theincomplete processing of the gag and
pol geneproducts apparentlyhad littleeffectontheabilityof
the virusto reversetranscribe its RNA genome(Fig. 5, lane
7).Interestingly, althoughthevirions of the insertionmutant
in5345-12 contained high levels of reverse transcriptase
activityaswellas reversetranscriptaseprotein (Fig. 4, lane
7), thismutantconsistently synthesizedadisproportionately
lowlevel ofproviral DNA.
Analysis ofthe viral DNA formed after infectionby the
various mutants showed that all the mutations were still
present. All the viral DNAscontaining the linker insertions
were sensitive todigestion withEcoRI,and the DNAs from
thebisulfitemutants were resistanttodigestionwith ApaLI
(datanotshown).The results show that all the IN- mutants
were genetically stable and at least largely competent to
carry out reversetranscription of all the viral DNA forms.
Analysisof the termini of linear viral DNAsynthesized after
infectionbyIN- virus.Recentanalyses ofunintegratedlinear
1
on November 10, 2019 by guest
http://jvi.asm.org/
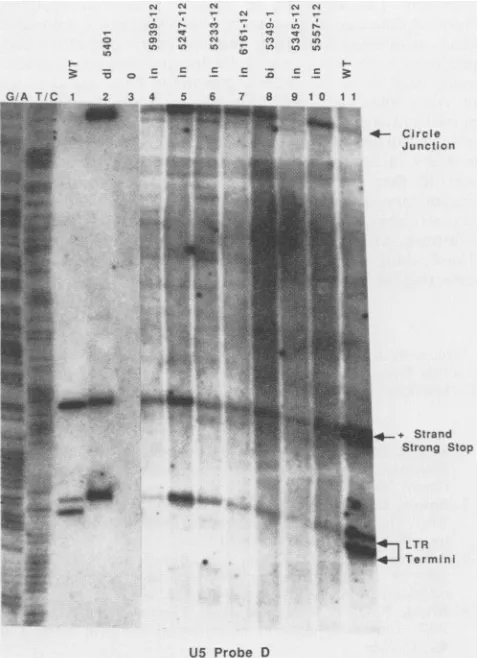
[image:6.612.340.533.72.247.2] [image:6.612.70.289.73.295.2]M-MuLV IN PROTEIN MUTATIONS 4715
retroviral DNAs have demonstrated that a mixture of two
terminal structures are present: some termini are
blunt-endedandothers have 3' ends recessed by two nucleotides
(5, 45). The IN- deletion mutant d15401 was found to be
unable to form the recessed ends, suggesting that the IN
protein was required for and might directly mediate the
cleavage ofthese strands. To test whether the new mutant
IN proteins could mediate this early step in the integration process, we examined the terminal structures of the linear
DNAsformed after infection by the IN- viruses.
The low-molecular-weight DNA from freshly infected
cellswas first isolated and digested with restriction enzymes
to generate small terminal fragments. The resulting
frag-ments werethen separated by electrophoresison a
sequenc-ing gel, electroblotted onto nylon filters, and probed with
oligonucleotides specific for each of the strands at the two
termini of the linear DNA (Fig. 6). DNA from wild-type
virus showed the expectedmixture ofblunt and recessed 3'
ends (lanes 1, 11); deletion mutantd15401 showed only the
blunt ends, as seen previously (lane2). DNA fromeach of
the six linker insertion mutants and the defective point
mutant bi5349-1 showed only the blunt termini, with no
detectable formationofthe 3' recessed ends(lanes 4 to10).
Thus, every IN- mutant was defective in the cleavage
process, the first detectable step leading to the integrated
provirus. No region of the protein was singled out as
dispensable for this early eventbut neededfor later events;
therewere nopartially functionalmutants, as tested at37°C.
In confirmation ofthe Southern blot
analysis above,
theIN- viruses generally directed the formation of a higher
proportion of theLTR-LTRjunction fragmentthanterminal
fragments (lanes 4 to 10). The
higher
abundance of thatfragment reflects the higher abundance of circular DNAs
overlinear seenin the absence of
integration.
DISCUSSION
Inthese studies,we haveexamined the effects of various
mutations inthe M-MuLV INprotein, bothin vitro, with a
DNA-bindingassay, andinvivo, by measuringthe
ability
ofthe mutant viruses toproceedthroughthelife
cycle.
Muta-tions altering the INprotein had
complicated
effectsonthebinding activity. Most ofthe mutations showed very little
effectonthe potent,
nonspecific
DNA-binding activity
oftheprotein. Two mutations, both
mapping
in the zincfinger
region near the N-terminal third ofthe
protein,
did reduce thebindingin some assays, althoughtwoother mutations in this regionhad no effect. Alterationschanging
the distancebetween two cysteines (in5345-12) or
eliminating
one ofthese two cysteines (bi5349-1) often
profoundly
decreasedbinding, suggestingthat the
cysteines
may function inform-ingaDNA-bindingstructure, buttheir effectswere variable andcritically dependentontheway the
protein
washandledbefore assay, as is often seen for renatured enzymes
(30).
Thus, themutations may act
largely by
affecting
theability
of the
protein
torenaturerather
thanby
directly
affecting
theinherent binding of the protein. Furthermore, the
activity
assayed after
blotting
mayrequire only
partial refolding
andmay not reflect the true sequence
specificity
of the nativeprotein. Otherassays may be required to define functional
domains ofthe
protein.
The effects of these mutations as
judged by
our otherassays in vivowere more severe. All of thelinker insertion
mutations were lethal to the
virus,
suggesting
that the INprotein carries out far more
specific
functions than thesimple DNA binding measured in vitro. Of the bisulfite
CD t- N
aN
uV
3: 1-0 c .- ic
) In
-w 4n to
'- zEe E 3
._ n _ N_
1 1
~
4**"-, -- Circle
Junction
-+ Strand
Strong Stop
] LTR
Termini
U5 Probe D
FIG. 6.
Analysis
of termini from a collection of mutant IN-viruses.Wild-typeM-MuLV andmutantIN-viruseswerecollected fromproducer
cells and usedtoinfectRat2cells.Cellswerelysed
30h postinfection,and terminal DNA structures were
analyzed
afterblotting as described in Materials and Methods. The filter was
probedtodetect the 3' terminus ofUS.For IN-virus,
proviral
DNAfrom
approximately
6 x 107 to 9 x 107 cells wasanalyzed;
forwild-type virus,DNAfrom1 x 107to2 x 107cellswasused.Lanes 1and11,
Wild-type
(WT)virus. Lane2,d15401virus(IN-).Lane3, DNA isolated after a mock infection. Lanes 4 to 10, Mutants as indicated. The identities ofthe variousDNAspecies,
basedonthesize ofthe
fragments,
areindicatedattheside of thepanel.
MarkerDNAs derived from Maxam and Gilbert
sequencing
reactions(36) (G+Aand T+Clanes)are atthe left.mutations,
only
theextremely
conservative Ala-to-Valmu-tation had no effecton virus
replication.
In all the mutants,the
ability
of the virus to enter the cell andsynthesize
thelinear viral double-stranded DNA was not
altered;
integra-tionitselfwas
apparently
blocked. Fine-structureanalysis
ofthe termini ofthe viral DNAformed invivo indicatedthat
thelinear molecules from
IN-
mutant viruswereallblunt-ended. These results
support
the model(5,
45)
that the INprotein
isrequired
for the formation of the 3' recessedtermini and that thisstructureis inturn
required
forintegra-tionof the DNA.
The process of
integration
seemstobedivisibleintotwosteps:
cleavage
of the termini of the viralDNA,
and thenstrand transfer ofthe 3' ends into the
target
DNA,
leaving
nicks in the target
(5,
16).
Allof themutantsanalyzed
herewereblocked in the firststep,the
cleavage
of the viralDNA,
and we have not been able to
separate
these twosteps
by
VOL.64, 1990
W,Oql,
,z,
if
*.
1 2 3 4
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.320.559.74.403.2]mutation. The
approach
ofcreating
mutations scatteredalong
anentirecoding regionhasbeensuccessfulwith othergenes in
defining
functional domains of therespective
geneproducts (6, 35, 56); although
INdomainsmayexist, neitherassay used
here-nonspecific binding
in vitro and cleavagein vivo-allowed a definition of any such domains with
partial
activities. It ispossible
that the INprotein
is foldedasa
single
functionalprotein.
Wearecurrently
analyzingthesemutants in an in vitro
integration
reaction to determinewhether
they
areblockedat separate stages ofthe process.Preliminary
data suggest that most of these mutants areseverely impaired
in theirability
tointegrate
anexogenoussubstrate,
even when the recessed 3' ends are preformed.Theseresultssuggest that themutants wehave
generated
aredefectivefor both steps of the
integration
process.ACKNOWLEDGMENTS
Thisworkwassupported byPublic Health ServicegrantCA30488
from the National Cancer Institute.M.J.Risaspecialfellow of the LeukemiaSocietyofAmerica, Inc.
LITERATURE CITED
1. Berg,J.M.1990.Zincfingersand othermetal-bindingdomains.
Elements for interactions between macromolecules. J. Biol. Chem. 265:6513-6516.
2. Bowen, B., J. Steinberg, U. K. Laemmli, and H. Weintraub.
1980. The detection ofDNA-binding proteins by protein
blot-ting. NucleicAcids Res. 8:1-20.
3. Bowerman,B.,P. 0.Brown, J. M.Bishop, and H. E. Varmus.
1989. A nucleoprotein complex mediates the integration of retroviralDNA. Genes Dev. 3:469-478.
4. Brown,P.O., B.Bowerman,H. E. Varmus,andJ.M.Bishop.
1987. Correct integration of retroviral DNA in vitro. Cell
49:347-356.
5. Brown, P. O.,B.Bowerman,H. E. Varmus,andJ.M.Bishop.
1989. Retroviral integration: structure of the initial covalent
productandits precursor, andarolefor the viral IN protein.
Proc. Natl. Acad. Sci. USA 86:2525-2529.
6. Chen, M., and M. S. Horwitz. 1989. Dissection of functional
domains of adenovirus DNA polymerase by linker-insertion
mutagenesis.Proc. Natl.Acad. Sci. USA86:6116-6120.
7. Cobrinik, D.,R.Katz,R.Terry,A. M.Skalka,andJ.Leis.1987.
Aviansarcomaand leukosisviruspol-endonuclease recognition
of the tandem long terminal repeatjunction: minimum site
requiredforcleavageisalsorequiredfor viralgrowth.J.Virol.
61:1999-2008.
8. Colicelli,J.,andS. P. Goff.1985. Mutantsandpseudorevertants
of Moloney murine leukemia virus with alterations at the
integrationsite. Cell 42:573-580.
9. Dhar, R., W. McClements, L. Enquist, andG. Vande Woude.
1980. Nucleotide sequences of integrated Moloney sarcoma
provirus longterminalrepeatsandtheirhost and viraljunctions.
Proc. Natl. Acad. Sci. USA77:3937-3941.
10. Donehower, L. A. 1988. Analysis ofmutant Moloney murine
leukemiavirusescontaininglinker insertion mutationsin the 3'
regionofpol. J. Virol. 62:3958-3964.
11. Donehower,L. A., and H. E. Varmus. 1984. A mutantmurine
leukemiavirus withasinglemissense codon inpol is defective
inafunctionaffecting integration. Proc. Natl. Acad. Sci. USA
81:6461-6465.
12. Duyk, G., J. Leis, M. Longiaru, and A. M. Skalka. 1983.
Selective cleavage inthe avian retrovirallong terminal repeat sequence by the endonuclease associated with the alpha-beta
form of avianreversetranscriptase.Proc. Natl. Acad.Sci.USA
80:6745-6749.
13. Duyk, G., M. Longiaru, D. Cobrinik, R.Kowal, P. D.Haseth,
A.M.Skalka, andJ.Leis. 1985. Circleswithtwotandemlong
terminalrepeatsarespecifically cleavedby pol gene-associated endonuclease fromavian sarcomaandleukosis viruses:
nucle-otide sequences required for site-specific cleavage. J. Virol.
56:586-599.
14. Eisenman, R.N.,W. S.Mason,and M. Linial. 1980. Synthesis andprocessing of polymerase proteinsofwild-typeand mutant avianretroviruses. J. Virol. 36:62-78.
15. Fujiwara,T., and R. Craigie.1989.Integration ofmini-retroviral DNA: acell-free reaction for biochemicalanalysis of retroviral integration.Proc. Natl. Acad. Sci. USA 86:3065-3069.
16. Fujiwara, T., and K. Mizuuchi. 1988. RetroviralDNA integra-tion:structureofanintegration intermediate. Cell54:497-504. 17. Goff,S. P., P.Traktman, and D. Baltimore.1981. Isolation and
properties of Moloney murine leukemia virusmutants:useofa
rapidassayfor release of virionreversetranscriptase. J. Virol. 38:239-248.
18. Golomb, M., and D. P. Grandgenett. 1979. Endonuclease
activ-ity of purified RNA-directed DNA polymerase from avian myeloblastosisvirus. J. Biol. Chem. 254:1606-1613.
19. Golomb, M., D. P. Grandgenett, and W. Mason. 1981.
Virus-coded DNA endonuclease from avian retrovirus. J. Virol.
38:548-555.
20. Grandgenett, D. P., M. Golomb, and A. C. Vora. 1980.
Activa-tionofan
Mg2"-dependent
DNAendonuclease of avian myelo-blastosis virusalpha-betaDNApolymerase by in vitroproteo-lytic cleavage. J.Virol. 33:264-271.
21. Grandgenett, D. P., and S. R. Mumm. 1990. Unraveling
retro-virusintegration. Cell60:3-4.
22. Grandgenett, D. P., A. C. Vora, and R. D. Schiff. 1978. A
32,000-dalton nucleic acid-binding protein from avianretrovirus corespossesses DNAendonucleaseactivity. Virology
89:119-132.
23. Grandgenett,D.P.,A.C.Vora,R.Swanstrom,andJ.C. Olsen.
1986. Nuclease mechanism of the avian retrovirus pp32 endo-nuclease. J.Virol. 58:970-974.
24. Hughes, S. H., A.Mutschler,J. M. Bishop, and H. E. Varmus.
1981.A Rous sarcomavirusprovirus is flanked by short direct
repeats ofacellularDNAsequence presentin onlyonecopy
priortointegration. Proc. Natl.Acad. Sci. USA 78:4299-4303.
25. Hughes, S. H., P. R. Shank,D. H. Spector,H.-J. Kung,J. M. Bishop, H. E. Varmus, P. K. Vogt, and M. L. Breitman. 1978.
Proviruses of avian sarcoma virus are terminally redundant,
co-extensive with unintegrated linear DNA, and integratedat many sites. Cell15:1397-1410.
26. Johnson, M.S.,M.A.McClure,D.-F.Feng, J. Gray, and R. F. Doolittle. 1986. Computer analysis of retroviral pol genes:
assignment ofenzymatic functions to specific sequences and
homologies with nonviral sequences. Proc. Natl. Acad. Sci.
USA 83:7648-7652.
27. Katzman,M., R. A. Katz, A. M.Skalka,andJ. Leis. 1989. The
avian retroviral integration protein cleaves the terminal se-quencesof linear viralDNA at theinvivo sites ofintegration.J.
Virol.63:5319-5327.
28. Kopchick, J. J., G. A. Jamjoom, K. F. Watson, and R. B. Arlinghaus. 1978. Biosynthesis ofreverse transcriptase from
Rauschermurine leukemia virusbysynthesis and cleavageof a
gag-pol readthrough viral precursor polyprotein. Proc. Natl. Acad. Sci. USA75:2016-2020.
29. Kopchick, J. J.,W. L. Karshin, and R. B. Arlinghaus. 1979. Tryptic peptide analysis ofgagand gag-polgene products of
Rauscher murine leukemiavirus. J. Virol. 30:610-623. 30. Lacks, S. A., and S. S. Springhorn. 1980. Renaturation of
enzymes afterpolyacrylamide gel electrophoresis in the pres-enceof sodiumdodecylsulphate.J.Biol. Chem. 255:7467-7471.
31. Lazo,P.A.,V.Prasad,and P. N. Tsichlis.1987.Spliceacceptor
sitefor theenvmessageofMoloneymurine leukemia virus.J. Virol. 61:2038-2041.
32. Leis,J., G. Duyk, S. Johnson, M. Longiaru, and A. M. Skalka.
1983. Mechanism of actionoftheendonucleaseassociatedwith thealpha-beta and beta-beta forms of avianRNA tumorvirus
reversetranscriptase.J. Virol.45:727-739.
33. Lobel, L. I., andS.P. Goff. 1984. Construction of mutants of
Moloneymurineleukemia virusby suppressor-linker insertional mutagenesis: positionsof viableinsertion mutations.Proc.Natl. Acad. Sci. USA 81:4149-4153.
34. Luc, K.-C., T. D. Gilmore, and A. T. Panganiban. 1987. The
on November 10, 2019 by guest
http://jvi.asm.org/
M-MuLV IN PROTEIN MUTATIONS 4717
spleen necrosis virusint geneproduct expressed in E.colihas DNA binding activity and mediates att and U5-specific DNA multimerformation in vitro. Virology 157:127-136.
35. Lyman, S. D., and L. R. Rohrschneider. 1987. Analysis of
functional domains of the v-fms-encoded protein of Susan McDonough strain feline sarcoma virus by linker insertion mutagenesis. Mol. Cell. Biol. 7:3287-3296.
36. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNA withbase-specificchemicalcleavages. Methods Enzymol.
65:499-560.
37. McCutchan, J. H., and J. S. Pagano.1968.Enhancement of the
infectivityof simian virus 40 deoxyribonucleic acid with
dieth-ylaminoethyl-dextran. J. Natl.CancerInst. 41:351-357.
38. Miskimins, W. K., M. P. Roberts, A. McCleUland, and F. H. Ruddle.1985.Useofaprotein-blottingprocedure and a specific
DNA probe to identify nuclear proteins that recognize the promoter region of the transferrin receptor gene. Proc. Natl.
Acad. Sci. USA 82:6741-6744.
39. Misra, T. K., D. P. Grandgenett, and J. T. Parsons. 1982. Avian
retroviruspp32 DNA-bindingprotein.I. Recognitionof specific sequences onretrovirusDNAterminalrepeats. J.Virol. 44:330-343.
40. Panganiban, A. T. 1985. Retroviral DNA integration. Cell 42:5-6.
41. Panganiban, A. T., and H. M. Temin. 1984. The retroviruspol geneencodes aproduct required forDNAintegration: identifi-cation ofaretroviral intlocus. Proc. Natl. Acad. Sci. USA 81:7885-7889.
42. Parker, R. C., R. M. Watson, and J. Vinograd. 1977.Mapping of
cloned circular DNAs by cleavage with restriction endonu-cleases and calibration by agarose gel electrophoresis. Proc. Natl.Acad.Sci. USA74:851-855.
43. Peden, K. W. C., and D. Nathans. 1982. Local mutagenesis
within deletion loops of DNA heteroduplexes. Proc. Natl. Acad. Sci. USA 79:7214-7217.
44. Quinn, T. P., and D. P. Grandgenett. 1988. Genetic evidence
that the avian retrovirus DNA endonuclease domain ofpolis
necessaryfor viralintegration.J. Virol.62:2307-2312. 45. Roth, M. J., P. Schwartzberg, and S. P. Goff. 1989.Structure of
the termini ofDNAintermediates intheintegrationof retroviral DNA:dependenceonINfunction and terminalDNA sequence.
Cell58:47-54.
46. Roth, M.J., N. Tanese, and S. P. Goff. 1988.Gene product of Moloney murine leukemia virus required forproviral integration isaDNA-binding protein. J. Mol.Biol.203:131-139.
47. Rowe, W. P., W. E. Pugh, and J. W.Hartley.1970.Plaqueassay
techniques for murine leukemia viruses.Virology42:1136-1139. 48. Schiff,R.D., and D. P.Grandgenett.1978.Virus-encodedorigin ofa32,000-dalton protein from avian retroviruscores:structural relatedness ofp32 and thePpolypeptideof the avian retrovirus DNApolymerase.J. Virol. 28:279-291.
49. Schwartzberg, P., J. Colicelli, and S. P. Goff. 1984.Construction
and analysisof deletion mutations in the pol gene of Moloney murine leukemia virus: a new viral function required for pro-ductive infection. Cell 37:1043-1052.
50. Shimotohno, K., S. Mizutani, and H. M. Temin. 1980. Sequence of retrovirus provirusresembles thatof bacterial transposable
elements. Nature (London)285:550-554.
51. Shoemaker, C., S. Goff, E.Gilboa, M. Paskind, S. W. Mitra, and D. Baltimore. 1980. Structure of a cloned circular Moloney
murine leukemia virus DNA molecule containing aninverted segment: implications for retrovirus integration. Proc. Natl. Acad. Sci. USA 77:3932-3936.
52. Shoemaker, C., J. Hoffman, S. P. Goff, and D. Baltimore. 1981. Intramolecular integration within Moloney murine leukemia virus DNA. J. Virol. 40:164-172.
53. Skalka, A. M. 1989. Integrative recombination of retroviral DNA, p. 701-724. In R. S. Kucherlapati and G. R. Smith (ed.), Genetic recombination, American Society for Microbiology,
Washington, D.C.
54. Southern,P.J., and P. Berg. 1982.Transformationof mamma-lian cells to antibiotic resistance with a bacterial gene under control of the SV40early regionpromoter. J.Mol.Appl. Genet. 1:327-341.
55. Swanstrom,R., W. J.DeLorbe, J.M.Bishop,and H. E. Varmus. 1981. Nucleotide sequence of cloned unintegrated avian sar-coma virus DNA: viral DNA contains direct and inverted repeats similar to those in transposable elements. Proc. Natl. Acad. Sci. USA 78:124-128.
56. Tanese, N., and S. P. Goff. 1988. Domain structure of the
Moloney murine leukemia virus reverse transcriptase: muta-tionalanalysisand separateexpressionof the DNApolymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 85:1777-1781.
57. Tanese, N., M. J. Roth, and S. P. Goff. 1985. Expression of
enzymatically active reversetranscriptase inEscherichia coli. Proc. Natl. Acad. Sci. USA 82:4944 4948.
58. Tanese, N., M. J. Roth, and S. P. Goff. 1986. Analysis of retroviralpolgeneproducts with antisera raisedagainstfusion
proteins producedin Escherichiacoli. J.Virol. 59:328-340. 59. Terry, R., D. A.Soltis,M.Katzman,D.Cobrinik, J. Leis,and
A.M.Skalka. 1988. Propertiesof avian sarcoma-leukosis virus
pp32-related pol-endonucleasesproducedin Escherichiacoli.J.
Virol.62:2358-2365.
60. Vogelstein, B.,and D.Gillespie.1979.Preparativeandanalytical purificationof DNAfromagarose.Proc. Natl.Acad.Sci.USA
76:615-619.
61. Wigler, M.,R.Sweet,G. K.Sim,B.Wold,A.Pellicer,E.Lacy, T.Maniatis, S.Silverstein,and R. Axel.1979. Transformationof mammalian cellswithgenesfromprocaryotes andeucaryotes.
Cell16:777-785. VOL. 64, 1990