Vol.64, No. 1 JOURNALOFVIROLOGY,Jan. 1990, p.347-353
0022-538X/90/010347-07$02.00/0
Copyright C1990,American Society for Microbiology
Programmed Factor Binding to Simian Virus 40 GC-Box Replication
and Transcription Control Sequences
ROBERTL.BUCHANANt AND JAY D. GRALLA*
Departmentof Chemistry and Biochemistry and Molecular Biology Institute, University of California, Los
Angeles,
405Hilgard
Avenue,
LosAngeles,
California
90024
Received6 July 1989/Accepted 22 September 1989
Nuclearfootprinting revealed a temporal programinvolving factor binding to the repetitive GC-box DNA elements present in the simian virus 40 regulatory region. This program specified ordered and directional binding to these tandem regulatory
sequences
in vivoduring the late phase of infection. The program was interrupted by the DNA replication inhibitor aphidicolin or by inactivation of the viral replication factor simian virus 40 T antigen, suggesting a link between viral DNA replication and new factor binding. Measurements of DNAaccumulation in viruses lacking either the distal or proximal halves of the GC-box region suggested that theregion has a dual role in replication control. Overall, the datapoint toimportantrelationships between DNAreplication and factor binding to the GC-box DNA, a multifunctional regulatory region.Factor binding to mammalian regulatory DNA elements is
of central importance ingenecontrol. The cellular
distribu-tion and activity ofsuchfactors likely contributes to cell-specific patterns ofgeneexpression. Although many
DNA-binding factorshave beenidentified (see reference 26 for an example), the mechanisms that control their distribution and
activity are notunderstood. Many models relevant to such
mechanisms center onprogrammed factorbinding to DNA
and especiallyon theinfluence of DNAreplication on such programs (see, for example,reference 5).
Study of mammalian DNA tumor viruses has revealed well-defined genetic programs that depend directly or
indi-rectly on DNAreplication (45). In simian virus 40 (SV40) and adenovirus,componentsofthereplicationand
transcrip-tion machineryinvolved insuchprograms have beenpurified (14, 26). Most of the protein components are host cell proteins, which suggests that regulation of viral programs has important features incommon with cellularregulation. Among these proteins are cell factors which control SV40 replication and transcription through common DNA ele-ments(10).AcriticalDNA targetof these cellular
proteins
is the repeated motif(GGGCGG)6 orthe GC box which is aregulatory element adjacent to the viral core origin of replication that influences DNA replication (24, 31, 32), as
wellasearly (1,11)andlatetranscription(4,13).Therole of
thesefactors in uninfected cells is alsolikelytobe involved with gene regulation, since GC boxes are often associated
with regulatory DNA (27, 38).
In situ DNase I footprinting of nuclear SV40 templates isolated from infected CV-1monkey cellsshowed abundant
factorbindingtothe SV40GCboxesat48 h
postinfection,
butneithertheidentity ofthe factorsnortheirfunctionalroleisknown (6). Inthis report, we
identify
agenetic
programinvolvingthis factor andpresentexperiments
suggesting
that the programmed binding requires templates competent to replicate. Since GC boxesandrepetitive
DNAarerelatively
common in cells, these observations may haveinteresting
implicationsfor
programming
interactions with cellularreg-ulatoryDNA.
*
Corresponding
author.tPresentaddress:Division ofBiology
156-29,
CaliforniaInstitute ofTechnology, Pasadena, CA 91125.MATERIALS ANDMETHODS
Viral infections, preparation of nuclei, and DNA purifica-tion. SV40 strain tsA58 and parental strain VA45-54 were
obtained fromPeterTegtmeyer. Ingeneral, CV-1 cellswere
infected with1 to5 PFUofSV40 per cell and maintained in Dulbecco modified Eagle medium plus 2% calf serum plus antibiotics. Enough cells wereinfected to provide approxi-mately0.1 to 0.5 ,ugof SV40DNAperfootprinting reaction. Isotonic nuclei lysed with Nonidet P-40 (Sigma Chemical Co.) wereprepared andnicked with DNase I aspreviously described (6).DNA waspurifiedaspreviously described(6), except that the order ofRNase Aand proteinase K diges-tions was reversed to use endogenous RNA as acarrier in DNAprecipitations.
In some experiments, DNA replication was inhibited
before isolationof infected nuclei. At32 hpostinfection,the
cell culture medium was brought to 5 ,ug of
aphidicolin
(Sigma) per ml andinfections werecontinued forup to 16 h
(29).
Primer extension and gel electrophoresis. The sites of
DNase Inickingweredeterminedby hybridizing
5'-end32P-labeled
oligonucleotide
primers to denatured samples and extending them to breaks with Klenow DNA polymerase essentially aspreviously
described (6, 20), except that allextension reactions weredone at50to
52°C.
Base denatur-ation ofviral DNAtemplates
gavealowerbackground
than heat denaturation for samples isolated before 40 h. Basedenaturationandsubsequent
primer
extension havealready
been
reported
(3).Quantitationofreplicatedviral DNA. ConfluentCV-1 cells were infected with equal titers ofwild-type SV40 mutant virus strain 776, SV-P7, or PXS7.After 2h, infected
plates
were washed three or four times with warm Dulbecco modified Eagle mediumtoreduce theamountof unattached virus. Viral DNA wasprepared
as described above andelectrophoresed on 1% agarose
gels
after linearization withEcoRI. Full-length nick-translated
([32P]dATP)
FIII SV40DNA wasusedto
probe
DNA from 12-to24-hpostinfection
samplesbySouthern
hybridization.
DNAthathybridized
to thelabeled SV40probe
wasvisualizedby
autoradiography
with Cronex film at room temperature. Several
autoradio-gramsatdifferentexposureswerescanned
by densitometry.
Viral DNAsamples
prepared
fromexperiments
doneat24 347on November 10, 2019 by guest
http://jvi.asm.org/
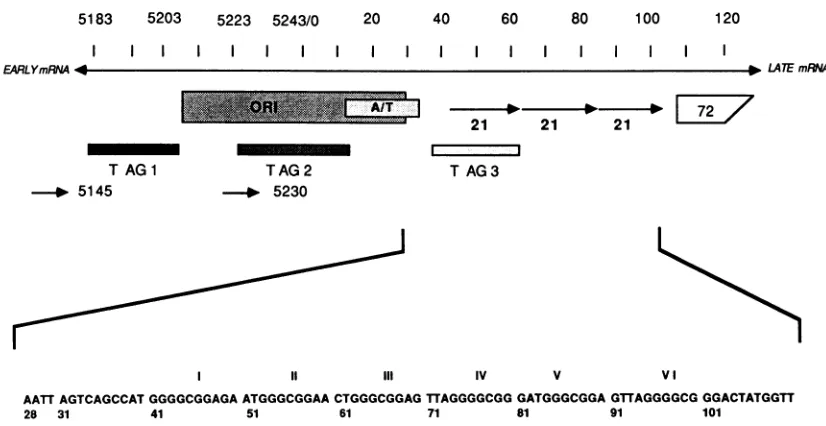
5183 5203 5223 5243/0 20 40 60 80 100 120
I I
EARLYmRNA
4-T AG 1 TAG 2
* 5145 -* 5230
.-
*
L
21 21 21
TAG3
11 III IV v v I
AATTAGTCAGCCATGGGGCGGAGAATGGGCGGAACTGGGCGGAGTTAGGGGCGGGATGGGCGGAGTTAGGGGCG GGACTATGGTT
28 31 41 51 61 71 81 91 101
FIG. 1. TheSV40controlregion witholigonucleotide primersused infootprinting experiments.The bidirectional arrowdesignates the linear SV40 DNAsequencewith nucleotide numbers accordingtoTooze(45). The directions ofearlyand latetranscriptionareindicated.
Relevant SV40controlregionsarealsoshown, includingthe centers of threeT-antigen (T AG)-bindingdomains. Three arrowslabeled21
depictthe six GC-boxregulatoryelements.Thewedgelabeled 72 is theorigin-proximal portionof the72-base-pairviral enhancerelement.
Oligonucleotideprimersareshownas arrowswith numbers(5145and5230) indicatingthe 3' ends of theprimers.At thebottom,theSV40 GC-boxregionisexpandedtoshow thenucleotidesequencefromnts28to111. Within thisexpanded region,the GC boxesarenumbered with romannumerals. ORI, Originofreplication.
to65 hpostinfectionwerelinearized andelectrophoresedon
1%agarosegels after dilutionstypically intherangeof 1:10 to 1:1,000.DNAwas visualizedby ethidium bromide stain-ing andphotographed withKodak Tri-X film. Theamountof DNA ineach lanewasdeterminedbydensitometry.Arange ofexposures weretaken toassure linearresponse.
RESULTS
Thelatephase of theSV40lifecycleinvolvesprogrammed production of viral DNA and protein with the goal of assembling infectiousvirus(45). As the latephase proceeds,
more RNA is produced fromthe expanding pool ofDNA,
butthe averagetranscriptional activity ofSV40 DNA tem-plates remainsconstant(18). However, theaverage
replica-tion activity ofSV40 DNAdecreases during this time (24, 33). Thus, instead of the exponential production of DNAthat would accompany uncontrolled replication, the amount of SV40 DNA simply increases linearly. This relationship be-tween replication and transcription apparently ensures a
balanced ratio of protein to DNA. The purpose of these
initialexperiments wastoprobe thein situ interactions with DNA elements nearand within the replication core during
thetimewhentheprogrammeddecrease inaveragetemplate
activity occurs.
The method of probing DNA-protein interactions (6, 20) involves adding DNase I to nuclei isolated from SV40-infected CV-1 cells to lightly nick the endogenous SV40 DNA. The extrachromosomal SV40 DNA is then isolated anddenaturedto produce strands whoseendswere formed
byDNase Inicking insitu.Theattackpatternisrevealed by hybridizing5'-end 32P-labeledDNA primers adjacenttothe region ofinterest and extending them in vitrotorevealstop
sites correspondingtonicks(20).Primer 5145 is designedto probe thecore replication elements, and primer5230 reads throughtheaccessoryGC-box element (Fig. 1). Previously, probing with primer 5230 showed that the GC-box region
was partially protectedfrom DNase Idigestionbyabound factorat40 hpostinfection (6). Thetechniquedetects only the interaction of abundant factors with nonencapsulated DNA;the approximately90% ofSV40 DNAin virionsand previrions is relatively DNase I resistant (see reference 6) and does not contribute significantly to the signal in this assay. Factorbindingtoless thanhalf of the remaining10% of the DNA is difficult to detect with this method(6).
Probingthereplicationcoreregionin vivo.Figure2shows the results of in situ probing of the replication core
se-quences and the adjacent T-antigen-binding site I. Factor binding is assessed by comparing digestion of DNA com-plexedwith protein (nuclei, lanes 2 to 4)with digestion of naked DNA(in vitro, lane1). Therewassignificant
protec-tion ofsequences overT-antigen site I at 32, 40, and 48 h postinfection. However,strongprotectionofsequences
cor-responding to the core elements for replication which are
adjacenttothis sitewasnotobserved(comparelanes 2to4 withlane 1).
These footprinting results are in excellent accord with
long-standingobservations in the literature. At this time in the lytic cycle, the dominant interaction in this region is expected to involve T antigen bound at site I acting as a
repressorofearly transcription (34). Immunological studies indicated thatapproximately 10% of SV40 DNA in vivo is boundby T antigen (42), and this agrees well with strong protection ofapproximately 10% of the templates in these nuclear footprints. The limits of protection seen in these
nuclearexperimentsaresimilarto limits of T-antigen binding to site I in vitro (34, 44). Replicating templates constitute only 1% of SV40 DNA (42), and thus, protection of the replicationcoreorigininthisproportion oftemplates would be well below thelevel ofdetection;noextensiveprotection
wasseen,evenwithlighterexposuresof theautoradiogram. The timecourseofprobingrevealed no changesthat could easily account for the programmed decrease in template
ILAEmRNA
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.101.514.77.290.2]PROGRAMMED GC-BOX BINDING AND DNA REPLICATION 349
C 24 3 2
I
*
WF
..
2:
M.F Om Om
[image:3.612.121.244.78.482.2]1 2 3 4
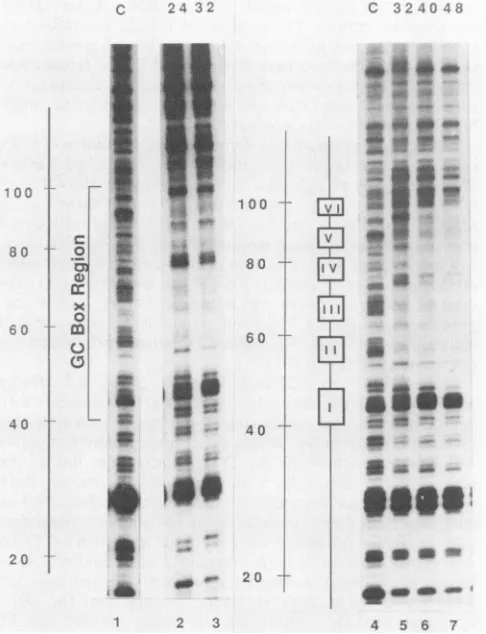
FIG. 2. Nuclear protection ofthe region near the replication
origin (ORI)was probed onthe late mRNA template strand with
primer 5145 at 32, 40, and 48 h postinfection. The minimum replication origin, which is approximatelycoincident with T-antigen (T Ag)-binding siteII, isindicated, asis siteI. Lane 1 contained
SV40nicked in vitroandprobed with primer 5145as acontrol(C),
while lanes2, 3, and 4 contained samples nicked in nucleiat32, 40,
or48 hpostinfection.
replication activity during late times. The only changeseen
wasthat theinternal A+T-richspacerregion within T-antigen
site Iwasslightlymoreprotectedastheinfection proceeded. Programmed bindingtomultiple GCboxesin vivo.Aquite differentresultwas seen when theaccessoryGC-box DNA elementwasprobed (Fig. 3). When DNA samples from 48 h postinfection wereprobed, strongprotectionwas seen over
the entire GC-box regulatory element. When this pattern (lane 7)wascompared with that of the in vitro control (lane
4), it was found that all bands between approximately nucleotides (nts) 50 and 100 were stronglyprotected from
DNase I digestion in SV40-infected nuclei. The region be-tween nts30 and 50waspartially protected, andonlyweak
[image:3.612.320.563.85.402.2]protectionwas evidentwithinthe enhancer (seealso refer-ence6). Attheearlier timeof40hpostinfection (lane 6),the patternwassimilar, exceptthattheprotectionwasnotquite
FIG. 3. Nuclear footprintsproducedonthe latemRNAtemplate
orCstrand in theGC-box region.Nuclei from SV40-infected cells were prepared at various times postinfection and nicked with
approximately0.01,ug ofDNase Iper,ulfor2min(lanes 2, 3, and
5to7).NuclearDNAswerepurified, denatured, andusedastemplates
forhybridization witholigonucleotide primer 5230. Lane C depicts controlSV40DNAnickedinvitrowithapproximately 0.0001 p.gof
DNase Iper,ul (lanes1 and4).The numbers above lanes 2, 3, and
5to7indicatethetimes (hours) postinfectionatwhich nucleiwere
probed. SV40 nucleotide numbersareshowntothe left of each gel, along with the locationsof theGC-box 21-base-pairrepeatelements (left panel)andindividual GC boxes (rightpanel).
as strong; this weakening occurredprincipally betweennts 70and 100. Evenearlier,at32hpostinfection (lane 5), this regionbetweennts70and 100wasmuch less wellprotected, andat24hpostinfection, the weak protection of this region
wasdiminishedfurther(lane 2). This loss ofprotectionwas
accompanied bytheappearanceoftwopositions of DNaseI hyperreactivity near nts 75 and 95. As discussed above, strongly protected regions inthisassay aretakentoreflect factorbindingtoabout 10%of the nuclearSV40molecules. This is an order ofmagnitude higher than the fraction of templates actively engagedineitherreplicationor transcrip-tion(15, 42).
Thus, ourviewof thisprogram canbegin at24 h postin-fection,whenreplication activityis stillhigh (24, 33).At that time, only the region surrounding GC boxes 2 and 3 is stronglyprotected. As thetemporalprogram proceeds, GC boxes 4 to 6 are bound progressivelywith the factor. This orderedbindinghas notbeenreportedforanyof thecellular factors known to bind the GC-box region. As reported previously (see Fig.3 in reference6), this factorprotectsthe earlymRNAorGstrand lessstronglyin theGC-boxregion,
C J24-0 4
IO
O
! i,
I.
100
8 0
;£0
40
C v!2 4 G2-.
*.a
*a
aS
-*._
45 7
100
t
80
-60'
E$
40
,tr
2I
20
20
700.il
VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
350 BUCHANAN AND GRALLA
another property not noted with isolated GC-box DNA transcription factors. This preference for the late mRNAor Cstrand wasmaintainedthroughout the latephase(datanot shown). We conclude that programmed factor binding ex-ists;duringthe time when the pool of templatesbecomesless active in producing DNA, the GC-boxDNAis increasingly boundby anabundant nuclearfactor.
Inhibition of programmed factor binding inhibition of DNA
replication. We beganexperiments toprobe the mechanism underlying this progressive factor binding to the GC-box
DNA region. A key issue involving genetic programs in
SV40 and in cells is whether DNA replication playsadirect role in influencingfactor binding (5, 45, 47, 48). This issue
canbe explored in this system because there is continuous production ofnew templates while the SV40 program
pro-ceeds. Thus, replication can be inhibited early in the pro-gram and it can be observed whether the programmed increase in bindingis also inhibited or whether the patternis otherwise perturbed.
Thepossibility that DNA replication is requiredforfactor bindingwastested initially by inhibiting DNA synthesiswith aphidicolin (29) and monitoring alterations in factorbinding by nuclear footprinting. In this experiment, a series ofplates
was infected with SV40. At 32 h postinfection, half of the
plates were treated with 5 ,g ofaphidicolin per ml. Both
control plates and drug-treatedplates were incubated for an
additional8 or 16 h. At these times (40 and 48 h
postinfec-tion,respectively), nuclei were prepared and the SV40 DNA was footprinted in situ to estimate factor binding. As a
further comparison, control samples from 32 h postinfection were processed in parallel. Thus, the effect of the DNA
synthesis inhibitor aphidicolin on factor binding can be
deducedfrom this collection of data (Fig. 4A).
The results showed that inhibition of DNA replication
with aphidicolin inhibited binding of factors to additional
sites within the GC-box region. When DNA synthesis was
inhibitedat 32 hpostinfection, continuedincubation until 40 or 48 h postinfection did not lead to significant additional
factor binding; that is, the 32-h pattern was nearly frozen
(Fig. 4A, compare lanes 1, 4, and 5). In parallel samples in which DNA synthesis was not inhibited, the expected in-crease in factor binding occurred during this time (lanes 2
and 3). These results also indicate that previously bound
GC-box interactions were stable and that nonreplicating DNA remained bound by factors for at least 16 h in vivo
(compare lanes 1 and 5). Similar results were obtained with
cycloheximide, which is known to inhibit protein synthesis and SV40DNAreplication (data not shown).
Sinceaphidicolin inhibitsbothSV40 (9) and cellular DNA synthesis,theeffect oftemplate replicationonfactor binding was assessed by using a viral mutant in which cellular
processes are not affected. In this experiment, parallel
cultures were infected with either tsA58 virus or
VA45-54,
the wild-type parent of mutant tsA58 (43). tsA58 is a virus with atemperature-sensitive T antigen, and thus, it cannot
replicateat42°C. The twoviruses were grown at 32°C for 40 h to allow replication and partial factor binding to
occur,
both ofwhich occur rather slowly at 32°C. TheVA45-54-and tsA58-infected cultures were then shifted to the nonpermis-sive temperature of41.5°C and incubated for 10 h longer.
Viral DNA replication during the 10 h occurred only in
VA45-54-infected cultures. After this incubation, nuclei were preparedfrom bothculturesandfootprintedtoassess
factor binding (Fig. 4B). This comparison showed that
additional factor binding at 41.5°C occurred when viral
replication
proceeded (VA45-54in lane 1B)and notwhen it32 C 4'O 4 O 4;a
vA&g
cr~~~~~~~~c
139 11 t
..
..
(5... z , ..
3i
.
yv. a>t :e:#
*.. #.
..
,s,...,.
:g '.;
F
3sFrc
4:A. ., M
z <'h
*<}s
:-. XS
r.s;.
.t.
FIG. 4. Inhibition of GC-box factor binding by inhibition of DNA replication. (A) Nuclei were harvested at 32, 40, and 48 h postin-fection, as indicated above the lanes. A plus or minus indicates whether aphidicolin (5 j±g/ml) was added to infected plates at 32 h postinfection. At the indicated times, nuclei were prepared, nicked withDNaseI,and probed with primer 5230. (B) Cells infected with VA45-54(lane 1) or tsA58 (lane 2) for 40 h at32°C.At 40 h, infected cells were shifted to41.5°C(nonpermissive for tsA58 viral replica-tion) and incubated for an additional 10 h at41.5°C.Nuclei were the prepared, nicked with DNaseI,and probed with primer 5230. The amount of viral DNA present at 40 h corresponded approximately to the amount that appeared in a 37°C wild-type infection at 24 h postinfection.
was blocked(tsA58in lane 2B). That is, the GC boxes were much more protected (lane 1B) when T-antigen-dependent viral replication was allowed to continue. The experiment demonstrated that SV40 T antigen is required for GC-box filling. Taken together with the inhibition of binding by aphidicolin, the data indicate that T-antigen-dependent viral DNAreplication is required for programmed factor binding. Effects of the region containing GC boxes on replication timing and rate. Previous studies have shown that deletions within the GC-box region lead to lowered levels of replicated SV40 DNA (23, 24, 32, 50). Since the data indicate that binding to the GC boxes depends on functional T antigen, it seemed possible that the GC boxes could also influence how J.VIROL.
.n
4-)
t-,
'IT
:1
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.353.522.79.467.2]PROGRAMMED GC-BOX BINDING AND DNA REPLICATION 351
. 1.. r,tr ii 3 1r . Jhr 2 4i-r.
[image:5.612.316.555.83.454.2].. _o -- -- -_
FIG. 5. Southernblot analysis of DNAs produced bywild-type SV40 (lanes 1, 4, 7, 10, 13, 16, and 19), SV-P7 (lanes 2, 5, 8, 11, 14, 17, and 20), and PXS7(lanes 3, 6, 9, 12, 15, 18, and 21) from 12 to 24 h postinfection. CV-1 cells were infected, and DNAs were harvested at the indicated times postinfection. Purified DNA was linearized and diluted before electrophoresis on 1% agarose gels. Gels were blotted with nitrocellulose and probed with whole nick-translated SV40 as described in Materials and Methods. Only the major SV40 band is shown.
Tantigenturns on replication. The footprinting procedures arenotsensitiveenough to probe the onset of replication at
earlytimes directly. Instead, we used mutants to investigate
the role of the GC-box region in replication timing.
Trans-fected plasmids are unsuitable for this purpose, since the
time at which they begin replication is altered (8). Thus, mutants built into viable virus must be used. We had
available two such mutantviruses, each ofwhich deletes a
different halfof the GC-box region. Virus SV-P7 lacks GC
boxes 1 to 3 and nine adjacent nucleotides (nt 32 to 73),
approaching but not impinging upon the replication core
(Buchanan and Gralla, unpublished data), and virus PXS7
(16) lacksGC boxes4to 6 (nt 73 to 100). Takentogether with
wild-type SV40, the setof viruses allows assessment of the
function ofthe40-base-pair regionadjacent to the replication coreand the30-base-pair region between this regionand the
enhancer.
In thefirstseries of experiments, the time of the onset of
replicationwasmeasuredinviruses lacking eitherearly-side
(SV-P7) orlate-side(PXS7) GCboxes. DNA was harvested every 2hfrom12to 24 hpostinfection, anditsamount was
estimated by probing Southern blots (Fig. 5). The two mutantviruses begantoreplicate atgrossly different times. SV-P7 DNA was visible at 12 h postinfection (lane 2) and
gradually increased inamount overthe time assayed (lanes 2, 5, 8, etc.). Bycontrast,PXS7DNA(lanes3, 6, 9,etc.)was notvisible until22hpostinfection (lane18), whenit beganto
accumulate. Thus, deletion of thetwohalves of this region hadvery different effectsonthe timing ofDNA accumula-tion. This differential effectonreplication timing is unlikely
to be dueto altered early accumulation ofTantigen, since
deletion of the late-side GC boxes should not be more
deleterioustoT-antigenexpressionthanis deletion of early-side GC boxes.
These sameblotsshow thatwild-typeDNA(lanes1,4, 7, etc.)wasbarelyvisible at 12 hpostinfectionandincreasedin amountby 16 hpostinfection, asexpected (45). During this time interval, the hierarchy in the amounts ofDNA was maintainedasSV-P7>wildtype>PXS7.Weconclude that
thePXS7mutationdrasticallyslows theonsetof
replication,
even
though
thePXS7early
promoter appears tobenearly
asefficient aswild-typeSV40 in vivo(21).Theearlier timeat which SV-P7DNAbegantoaccumulatewas
unexpected
for two reasons. (i) The region deleted is critical forearly
transcription (see above references), and thus, one
might
haveexpectedadelayduetoslowerproduction
of Tantigen.
(ii)Asdiscussedabove, other GC-box deletionshave ledto lower,nothigher, totallevels ofSV40
DNA,
andthislower ultimate levelofDNAwasalso truefor SV-P7(seebelow).
Ourbest
quantitative
estimate is that PXS7 isdelayed
for 6to8 hand SV-P7accelerated
modestly,
by2orperhaps
3 h.10
WtSV40
z 6
SV-P7
0
E
4c
4
2
PXS7
0
10 20 30 40 50 60 70 80
TimePost-infection(hours)
FIG. 6. Accumulation of wild-type (wt)SV40, SV-P7, and PXS7 from 24 to 65 h postinfection. DNAs from infected plates were harvested andquantifiedattheindicated timespostinfection. More generally, the
70-base-pair
region
coveredby
thesetwodeletions acts as a
timing
elementinfluencing
the onsetofSV40DNA
replication.
The amounts ofDNAmade by these three viruses were also monitored past 24 hpostinfection to observe the
long-term effect on rate ofDNA
production.
All three virusesaccumulatedDNAfrom24 to 65 hpostinfection
(Fig.
6)but atdifferentrates.During thistime, wild-typevirusproduced,
on average, 0.26 p,g ofDNA perh, SV-P7produced
0.18,ug/h,
and PXS7produced
0.05,ug/h.
Thesedata agree withthose of various studies
showing
lowered DNAyields
causedby GC-box deletions (16, 24, 32,50)butare
interest-ing
intwo respects.(i) SV-P7,
whichproduced
moreDNAthan did the wild type before 24 h
postinfection
(Fig. 5),
produced less by 65 h
postinfection (Fig. 6).
(ii)
Although
SV-P7 and PXS7, which delete separatesubregions,
hadopposite
effects on the onset ofreplication, they
had thesame
qualitative
effectonsteady-state
replication
rates;i.e.,
both reduced the rate ofSV40 DNA
synthesis. Thus,
theregion contains separatecontrolsfor
replication
timing
andsteady-state
replication
rate(Fig. 7).
Both halves of theregionare required for
optimal
DNAaccumulation,
consis-tent with the idea that when
they
are occludedby
pro-VOL.64, 1990on November 10, 2019 by guest
http://jvi.asm.org/
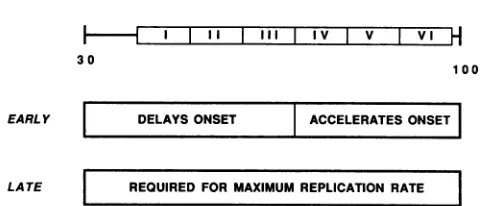
l I I I I I III IV v vI
H
30EARL Y DELAYSONSET ACCELERATESONSET
LATE REQUIRED FOR MAXIMUM REPLICATION RATE
FIG. 7. Effect(s) ofSV40GC boxes on viral DNA replication.
SV40 GC boxes are schematically shown as six adjacent boxes.
Belowaredepicted the effects oftwoGC-box subregionsonDNA
replication during the early and late phasesof the SV40 lyticcycle. grammedabundantfactorbindingorwhentheyaredeleted,
replication is inhibited. The two subregions encompassing the six GC boxes affect thetime ofreplication onset differ-ently, implying that before replication onset, an interesting
but unknown genetic program exists which controls the
initiation of DNAreplication. DISCUSSION
These experiments have described a temporal program
involving interactions at the repetitive SV40 GC-box
regu-latory elements. T-antigen-dependent DNA replication is requiredfor assembly ofa complex that protectsthe entire region from DNase digestion in nuclei. This assembly
pro-ceeds by filling the GC boxes in order, from the origin-proximal toward the origin-distal elements. It is not known whether therequirementforT-antigen-dependentreplication implies that this genetic program requires specific
compo-nents of the active SV40 replication complex, which is positioned nearby, or simplynewly replicated DNA.
The GC-box region consists of repeated sequences, and
repetitive elements areknowntobe associatedwith
tempo-ral control by diverse proteins, such as X repressor and T
antigen (34, 37). In X, the bound sequences have a dual
function and serve as activator sites at low factor
concen-trations but mediate repression when occluded at high
pro-teinconcentrations. These datasuggestthattheinteractions with the SV40 GC-box elements are analogous; i.e., the
elements serve as activation sites exceptwhen occludedby factors. TheamountofSV40DNA bound by factorsat late times is much greater than the amount involved in replica-tionortranscription; thus, atthesetimes, the factorappears
not to activate the templates but rather to occlude them. Recall that SV40 GC boxes are not required to activate transcription atlatetimes (24) and that no net activation of transcriptionoccursduringthistime(18).Onthebasisof this
circumstantial evidence, wesuggestthatprogrammedfactor binding at late times progressively occludes the GC-box replication element to program the gradual decrease in
template replication activity.
Inthe GC box and X examples, repeatunits arebound in a specified direction as a program proceeds. Since the
GC-box units are of very similar DNA sequences but are
bound inaspecified order, cooperative interactions maybe
involved. However, the mechanism underlying directional GC-box factor bindingremains unknown, since none of the
factors known to bind this region in vitro were reported as
having this property of directional binding (26). Neverthe-less, the ability of prebound factors to specify the nearby positioning ofnew factors only on templates competent to replicate could have an important influence on
reprogram-ming expression in newly made cells (5, 46, 47).
A second property of the bound complex may also have
interesting implications, since thefactorprotectsoneof the DNA strands more stronglythan theother
(6);
upon strandseparation during
replication,
it ispossible
that thisprefer-ence forone of the strands would be
maintained,
allowing
preferential transmission to one of the two
daughter
DNAmolecules (see also references 41and49).
Several different factorshave beenisolated
(17, 28,
35,36)
thatbind SV40 GC-boxDNAinvitrosince thediscoveryof SP-1 (11). The abundant GC-box factor bound inSV40-infected cells is believed to be ofcellular rather than viral
origin (7),
although
a role for late viral proteins cannotbeexcluded. At least two
previously
characterized GC-box-binding factors share the unusual property of strand prefer-ence. (i)Achickenglobin
genefactorbindsG-stringregula-tory sequenceswithastrandpreference similartothat of the SV40 GC-box factor (12). (ii) It has recently been demon-strated that uninfected CV-1 cells contain a factor that preferentiallybinds one strand ofanSV40 GCbox,although
it does not appear to bind double-stranded DNA (17). It is not known whether the monkey cell factordetected in this
studyisanalogoustothesefactors, SP-1,oranyof theother isolated human factors; GC-box-binding proteins have not beenpurified frommonkey cells.Inbothhumanandmonkey cells, GC-box elements are common (27) and are often repeated when present, as in SV40.Thus, cellscontain both components ofthe DNA-factor interaction described here,
and therefore, the interesting properties ofthe SV40 com-plex are likely to be shared by certain cellular genes.
These experiments have confirmed that the SV40GC-box repeat unit influences the SV40 replication rate (see refer-ences in the introduction) and have also shown that subre-gions control the timing of replication onset (Fig. 7). Since mammalian cellularorigins of replication arepoorly charac-terized, it is not known whether some origins are associated with GC boxes, G strings, or other repeat elements. How-ever, it is known that thetiming of cellular DNAreplication
is precisely controlled. The earliest genes to be replicated
aregenerallythe so-calledhousekeeping genes (25). GC-box repeat elements are highly concentrated in this gene type (19, 25). Since some proposals link origins ofreplicationwith transcriptional activation (22), it is possible that these cellu-lar GC boxes influence replication as well asgeneexpression;
recallthat SV40GCboxes have this dual effect (10). The data presented here are most easily interpreted as implying that the onset of viral replication requires removal of a repressor fromearly-region-proximal GC boxes I, II, and III (Fig. 7). This observation could have significant impact on the eval-uation and extension of models for control ofDNA
replica-tion onset ineucaryotic cells (2, 22, 30, 39, 40). ACKNOWLEDGMENTS
This research was supportedby grants from theNational Science Foundationand the American CancerSociety.
LITERATURE CITED
1. Barrera-Saldana,H.,K.Takahashi, M.Vigneron, A.Wildeman, I. Davidson, and P. Chambon. 1985. All six GC motifs of the SV40earlyupstream element contributetopromoter activity in vivo and in vitro. EMBO J. 4:3839-3849.
2. Berg, L., M. Lasky, A. Stenlund, and M. R. Botchan. 1986. Repressionof bovinepapillomavirusreplication ismediatedby a virally encoded transacting factor. Cell 46:753-762.
3. Borowiec, J. A., L. Zhang, S. Sasse-Dwight, and J. D. Gralla. 1987. DNA supercoiling promotes formation of a bent repres-sion loop in lac DNA. J. Mol. Biol. 195:101-111.
4. Brady, J., M. Radonovich, M. Thoren, G. Das, and N. P.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.60.301.70.173.2]PROGRAMMED GC-BOX BINDING AND DNA REPLICATION 353
Salzman. 1984. Simian virus 40 major late promoter: an up-stream DNA sequencerequired forefficient in vitro transcrip-tion. Mol. Cell. Biol. 4:133-141.
5. Brown, D. D. 1984. The role of stable complexes thatrepress and activate eukaryotic genes. Cell 37:359-365.
6. Buchanan, R. L., and J. D. Gralla. 1987. Factor interactionsat simian virus 40 GC-box promoter elements in intact nuclei. Mol. Cell. Biol. 7:1554-1558.
7. Cereghini, S., and M. Yaniv. 1984. Assembly of transfected DNAinto chromatin: structural changes inthe origin-promoter-enhancer region upon replication. EMBO J. 3:1243-1253. 8. Chou, J. Y., and R. G. Martin. 1975. DNAinfectivity and the
induction of host DNA synthesis with temperature-sensitive mutantsof simian virus 40. J. Virol. 15:145-150.
9. Decker,R.S., M. Yamaguchi,R.Possenti, andM.DePamphilis. 1986. Initiation of simian virus 40 DNA replication in vitro: aphidicolincausesaccumulation ofearly-replicating intermedi-atesand allows determination of the initial direction ofDNA synthesis. Mol. Cell. Biol. 6:3815-3825.
10. DePamphilis, M. L. 1988. Transcriptional elements as compo-nentsof eukaryotic origins of DNA replication. Cell 52:635-638. 11. Dynan, W. S., and R. Tjian. 1983. The promoter specific transcription factor Sp-1 binds to upstream sequences in the SV40early promoter.Cell 35:79-87.
12. Emerson,B.M., C.D. Lewis, and G. Felsenfeld.1985. Interac-tion ofspecific nuclear factors with the nuclease-hypersensitive region of the chicken adultP-globingene: natureof the binding domain. Cell 41:21-30.
13. Ernoult-Lange, M.,F.Omilli,and E.May. 1987. Contribution of different GC-motifs to the control of simian virus 40 late promoteractivity. Nucleic Acids Res. 15:8177-8193.
14. Fairman, M. P., andB.Stillman. 1988.Cellularfactorsrequired for multiple stages of DNA replication in vitro. EMBO J. 7:1211-1218.
15. Fernandez-Munoz, R., M. Coca-Prados, and M. T. Hsu. 1979. Intracellular forms of simian virus40nucleoprotein complexes. J. Virol. 29:612-623.
16. Fromm, M., and P. Berg. 1982. Deletion mapping of DNA regions required for SV40 early region promoter function in vivo.J. Mol.Appl. Gen. 1:457-481.
17. Gaillard, C., M. Weber, and F. Strauss. 1988. A sequence-specific single-strand-binding proteinfor thelate-codingstrand of the simian virus40controlregion.J.Virol. 62:2380-2385. 18. Gariglio, P.,R. Llopis,P. Oudet, andP. Chambon. 1979. The
template of the isolated native simian virus 40transcriptional complexesisaminichromosome. J. Mol. Biol. 131:75-105. 19. Goldman, M.A., G. P. Holmquist,M. C. Gray,L. A.Caston,
and A. Nag. 1984. Replication timing of genes and middle repetitivesequences. Science 224:686-692.
20. Grafla,J. D.1985. Rapid"footprinting" onsupercoiledDNA. Proc. Natl.Acad. Sci. USA82:3078-3081.
21. Hansen, U., and P.Sharp. 1983. Sequencescontrollingin vitro transcriptionofSV40 promoters. EMBO J. 2:2293-2303. 22. Harland,R.M.,and R. A.Lasky. 1980.Regulated replicationof
DNA microinjected into eggs of Xenopus laevis. Cell 21: 761-771.
23. Hartzell, S. W., B. J. Byrne, and K. N. Subramanian. 1984. Mapping of the late promoter of simian virus 40. Proc. Natl. Acad. Sci. USA 81:23-27.
24. Hertz, G. Z., and J. E. Mertz. 1988. The enhancer and GGGCGG boxes ofSV40providesimilarfunctions in bidirec-tionally promotingtranscription. Virology 163:579-590. 25. Holmquist,G. P.1987. Role ofreplicationtime in the controlof
tissue-specific gene expression. Am. J. Hum. Genet. 40:151-173.
26. Jones,N.C.,P. W.J. Rigby,and E. B. Ziff.1988.Trans-acting protein factors and the regulation ofeukaryotictranscription: lessons from studies on DNA tumor viruses. Genes Dev. 2:267-281.
27. Kadonaga, J. T., K. A. Jones, and R. Tjian. 1986. Promoter-specificactivation ofRNApolymeraseIItranscription bySP-1. Trends Biol. Sci. 11:20-23.
28. Kim, C.H., C. Heath,A.Bertuch,andU. Hansen. 1987.Specific stimulation of simian virus 40late transcription in vitroby a
cellular factor bindingthe simian virus 40 21-base-pairrepeat promoterelement. Proc. Natl.Acad. Sci. USA 84:6025-6029. 29. Krokan, H., P. Schaffer, andM. DePamphilis. 1979.
Involve-mentofeucaryotic deoxyribonucleicacidpolymerasesaand1B in the replication of cellular and viral deoxyribonucleic acid. Biochemistry18:4431 4443.
30. LaBelia, F., H. L. Sive, R. G. Roeder, and N. Heinz. 1988. Cell-cycle regulationofahumanhistone H2bgeneismediated bythe H2b subtype specific consensus element. Genes Dev. 2:32-39.
31. Lee-Chen, G.,and M. Woodworth-Gutai. 1986.Simian virus40 DNA replication: functional organization of regulatory ele-ments. Mol.Cell. Biol. 6:3086-3093.
32. Li,J.J.,K. W.C.Peden,R.A. F.Dixon,and T.
Keiley.
1986. Functionalorganizationof the simian virus 40origin ofDNA replication.Mol. Cell. Biol.6:1117-1128.33. Manteuil, S., J. Pages, D. Stehelin, and M. Girard. 1973. Replicationofsimian virus 40deoxyribonucleicacid:analysisof theone-stepgrowth cycle. J.Virol. 11:98-106.
34. McKnight,S.,and R.Tjian.1986.Transcriptionalselectivityof viralgenesin mammalian cells. Cell46:795-805.
35. Mermod, N., T. J. Williams, and R. Tjian. 1988. Enhancer bindingfactorsAP-4andAP-1 actinconcert toactivate SV40 latetranscriptionin vitro. Nature(London)332:557-561. 36. Mitchell, P. J., C. Wang, and R. Tjian. 1987. Positive and
negative regulation oftranscriptionin vitro: enhancer-binding proteinAp-2is inhibitedby SV40Tantigen.Cell 50:847-861. 37. Ptashne,M. 1986.Ageneticswitch. Blackwell Scientific
Publi-cations, Ltd., Oxford.
38. Queen, C.,S. T.Lord,T. F. McCutchan,and M.Singer. 1981. Three segments from the monkey genome that hybridize to simianvirus 40common structural elements. Mol. Cell. Biol. 1:1061-1068.
39. Roberts, J. M., and G. D'Ursu. 1988. An origin unwinding activity regulatesinitiation ofDNAreplication during mamma-lian cellcycle. Science 241:1486-1489.
40. Roberts, J. M., and H. Weintraub. 1988. Cis-acting negative control of DNAreplicationineukaryoticcells. Cell 52:397-404. 41. Sakonju,S.,and D. D. Brown. 1982.Contactpointsbetweena
positive transcriptionfactor andXenopus 5S RNA gene. Cell 31:395-405.
42. Tack,L. C., and M. L.DePamphilis. 1983. Analysis of simian virus40 chromosome-T-antigen complexes: T-antigenis pref-erentiallyassociated withearlyreplicatingDNAintermediates. J.Virol. 48:281-295.
43. Tegtmeyer,P.,C.Dohan, Jr.,andC. Reznikoff.1970. Inactivat-ingandmutageniceffects of
nitrosoguanidine
on simian virus 40. Proc. Natl. Acad. Sci. USA 66:745-752.44. Tegtneyer, P.,B. A.Lewton,A. L.DeLucia,V. G.Wilson,and K.Ryder.1983.Topographyof simianvirus 40 A
protein-DNA
complexes: arrangement of
protein
bound to theorigin
of replication.J. Virol. 46:151-161.45. Tooze,J.(ed.).1981. DNAtumorviruses,2nded. Cold
Spring
HarborLaboratory, ColdSpring Harbor,N.Y.
46. Wang, X. F., and K. Calame. 1986. SV40
enhancer-binding
factors are
required
at the establishment but not the mainte-nance step ofenhancer-dependent
transcriptional
activation. Cell47:241-247.47. Wolffe,A.P.,andD. D. Brown. 1986. DNA
replication
in vitroerases a Xenopus SS RNA gene
transcription
complex.
Cell 47:217-227.48.
Wolffe,
A.P.,and D. D.Brown.1988.Developmental
regulation
oftwo5SribosomalRNA genes. Science 241:1626-1632. 49. Wolffe,A. P., E. Jordan,and D. D. Brown. 1986. A
bacterio-phageRNApolymerasetranscribes
through
aXenopus 5SRNAgene
transcription
complex
withoutdisrupting
it. Cell44:381-389.
50. Yamaguchi,M.,andM.L.
DePamphilis.
1986. DNAbindingsite forafactor(s)required
toinitiate simian virus40DNAreplica-tion.Proc. Natl. Acad. Sci. USA 83:1646-1650.
VOL. 64,1990