Copyright © 1998, American Society for Microbiology
Relative Rates of Retroviral Reverse Transcriptase Template
Switching during RNA- and DNA-Dependent DNA Synthesis
ROBERT R. BOWMAN,1WEI-SHAU HU,2,3ANDVINAY K. PATHAK2,4*
Department of Genetics and Developmental Biology,1Mary Babb Randolph Cancer Center,2Department of
Microbiology and Immunology,3and Department of Biochemistry,4West Virginia University,
Morgantown, West Virginia 26506 Received 3 November 1997/Accepted 11 February 1998
Retroviral reverse transcriptases (RTs) frequently switch templates during DNA synthesis, which can result in mutations and recombination. The relative rates of in vivo RT template switching during RNA- and DNA-dependent DNA synthesis are unknown. To determine the relative rates of RT template switching during copying of RNA and DNA templates, we constructed spleen necrosis virus-based retroviral vectors containing a 400-bp direct repeat. The directly repeated sequences were upstream of the polypurine tract (PPT) in the RB-LLP vector; the same direct repeats flanked the PPT and attachment site (att) in the RB-LPL vector. RT template switching events could occur during either RNA- or DNA-dependent DNA synthesis and delete one copy of the direct repeat plus the intervening sequences. RB-LLP vectors that underwent direct repeat deletions during RNA- and DNA-dependent DNA synthesis generated viral DNA that could integrate into the host genome. However, any deletion of the direct repeats in the RB-LPL vector that occurred during RNA-dependent DNA synthesis resulted in deletion of the essential PPT and att site and generated a dead-end viral DNA product. Thus, only RB-LPL vectors that underwent direct repeat deletions during DNA-dependent DNA synthesis could integrate to form proviruses. The RB-LLP and RB-LPL vectors were permitted to undergo a single replication cycle, and the frequencies of direct repeat deletions were determined by PCR and Southern analysis of the resulting proviruses. A comparison of the frequency of direct repeat deletions in the RB-LLP and RB-LPL vectors indicated that the in vivo rates of RT template switching during RNA- and DNA-dependent DNA synthesis are nearly identical.
Reverse transcription is an essential step of the retroviral life cycle (1, 40, 41). During this process, the virally encoded re-verse transcriptase (RT) copies viral RNA into a double-stranded DNA (5). RT has a propensity to undergo frequent template switching events by dissociating and reassociating with the template (42). Two such template switching events, minus-strand strong-stop and plus-strand strong-stop DNA transfers, are necessary for completion of reverse transcription (17). Because these template switching events are obligatory to viral replication, it has been hypothesized that RT evolved to frequently switch templates (42). In addition to the two strong-stop DNA transfers, which occur at the ends of the template, RT can also undergo additional internal template switching events (12, 21, 23, 27, 28, 30, 31, 49). The internal template switching events can be either intramolecular (same template) or intermolecular (copackaged template). Intramolecular tem-plate switches can result in deletions, deletions with insertions, insertions, and duplications, whereas intermolecular template switches can result in homologous or nonhomologous recom-bination (21). Thus, the template switching property of RT plays an important role in generating variation in retroviral populations.
Another consequence of RT template switching is that di-rectly repeated sequences in viral genomes are deleted at very high frequencies. Direct repeat instability in retroviral vectors and in viral genomes has been previously observed (6, 22, 29, 33, 43). The rates of direct repeat deletions were recently determined for a single replication cycle of spleen necrosis
virus (SNV) and murine leukemia virus (MLV) (8, 23, 30). It was shown that direct repeats of 110, 383, 788, and 1,333 bp in SNV-based vectors were deleted at high rates of 41, 40, 85, and 93%, respectively (23, 30). In addition, a 701-bp direct repeat in an MLV-based vector was shown to be deleted at rates of 57% when the repeated sequences were in tandem and 89% when the repeats were separated by the MLV packaging se-quence (8). We recently determined that the RT template switching events that lead to deletions of the direct repeat are primarily intramolecular (21). We also observed that retroviral recombination exhibits high negative interference, whereby vi-ruses that exhibit one intermolecular RT template switch have a higher probability of exhibiting another intermolecular RT template switch (21).
RT template switching events that lead to deletions of the direct repeat can potentially occur either during RNA-depen-dent (minus-strand) DNA synthesis or during DNA-depenRNA-depen-dent (plus-strand) DNA synthesis. Although previous in vitro stud-ies have suggested that RT template switching occurs at a higher rate during RNA-dependent DNA synthesis than dur-ing DNA-dependent DNA synthesis (16, 26), the relative in vivo rates of RT template switching during RNA- and DNA-dependent DNA synthesis are unknown. Since direct repeat deletions occur at a high rate and can be easily identified, they provide a good model system for studying RT template switch-ing events. Previously, we proposed that the high frequency of direct repeat deletions occurred primarily during RNA-depen-dent DNA synthesis (23). It was postulated that the RNase H activity of RT continually degrades the RNA template 18 to 20 nucleotides (nt) behind the site of polymerization. Therefore, the nascent DNA strand and the template RNA would be expected to be held together by only a few hydrogen bonds, which would in turn be expected to promote dissociation of the * Corresponding author. Mailing address: Mary Babb Randolph
Cancer Center, West Virginia University, Morgantown, WV 26506. Phone: (304) 293-0495. Fax: (304) 293-4667. E-mail: VPATHAK @WVUMBRCC1.hsc.wvu.edu.
5198
on November 9, 2019 by guest
http://jvi.asm.org/
nascent DNA and RT from the template. Once dissociated, the nascent DNA and the RT may reassociate with the point of dissociation or with the homologous point in the 59 direct repeat. Reassociation with the 59 direct repeat would be fa-vored by increased base pairing, leading to deletion of the direct repeat and intervening sequence.
In an effort to shed light on the relative rates with which RT switches templates during RNA- and DNA-dependent DNA synthesis, we designed a strategy to distinguish the two tem-plate switching events in vivo. Our results indicate that RT template switching events occur at nearly equal rates during RNA- and DNA-dependent DNA synthesis.
MATERIALS AND METHODS
Definitions.pRB-LLP, pRB-LPL, pVP212, pRB-PPT, and pWH342 refer to plasmids, and RB-LLP, RB-LPL, VP212, RB-PPT, and WH342 refer to the viruses derived from these plasmids.
Plasmid construction.SNV-based retroviral vectors pRB-LLP and pRB-LPL were derived from the previously described retroviral vector pVP212 (30). Stan-dard molecular cloning procedures were used (36). To construct pRB-LLP, pVP212 was first partially digested with XmnI and treated with calf intestinal phosphatase (CIP; Boehringer Mannheim Biochemicals [BMB]) to dephosphor-ylate the ends. A 70-bp blunt-ended polylinker was then ligated into the XmnI site to generate pRB-RFE. The polylinker was designed to contain ClaI, XhoI,
NdeI, EcoRV, and EcoRI restriction sites. The sequence of the polylinker was
59ATCGATTATTATCCTGCTCGAGCCTGATAGCCCATATGTTAGTCCG ACGATATCCGCCGATGGTGAATTC39. pRB-RFE was cut with EcoRI and then treated with CIP followed by Klenow fragment (BMB) to generate dephos-phorylated blunt ends. pUC-Nco was derived from pUC19 by replacement of the pUC19 polylinker with an 8-bp NcoI linker. A 400-bp fragment of the lacZa peptide gene was generated by digestion of pUC-Nco with HaeII, followed by treatment with T4 DNA polymerase to generate blunt ends. The exact size of the
lacZafragment is 399 bp; this fragment will be referred to as the 400-bp frag-ment for the sake of simplicity. The HaeII fragfrag-ment was isolated by gel elution and ligated into the EcoRI site of pRB-RFE to generate pRB-LLP.
pRB-LPL was derived by digestion of pVP212 with NotI followed by treatment with CIP and Klenow fragment to generate dephosphorylated blunt ends. The same HaeII fragment isolated from pUC-Nco was inserted into the NotI site to generate pRB-LPL.
pRB-PPT was derived by partial digestion of pRB-LPL with NcoI followed by self-ligation. pRB-PPT contains one copy of the lacZafragment and lacks the polypurine tract (PPT) and att site.
pWH342 was derived from the previously described vector pEB232F (3). pEB232F was digested with HindIII and XbaI, followed by treatment with Kle-now fragment to generate blunt ends. The resulting DNA fragment was self-ligated to generate pWH342, a vector that contains the promoter region of the
lacZagene but lacks the PPT and att site.
Cells, transfections, and infections.D17 and C3A2 cells (obtained from the American Type Culture Collection) were maintained in Dulbecco’s modified Eagle’s medium (ICN) supplemented with 6% bovine calf serum (HyClone Laboratory), penicillin (50 U/ml) (Gibco), and streptomycin (50mg/ml) (Gibco). D17 is a dog osteosarcoma cell line that can be infected with SNV (34). C3A2 is a D17-derived reticuloendotheliosis virus-based helper cell line that can effi-ciently package RNAs containing SNV encapsidation sequence (46). The selec-tive drugs puromycin (Sigma) and G418 (an analog of neomycin; Gibco) were present at final concentrations of 1.75mg/ml (3.2mM) and 400mg/ml (0.58 mM), respectively.
Cells were transfected by the previously described dimethyl sulfoxide-Poly-brene method (24). For virus infection, D17 cells were plated at a density of 23 105cells on 60-mm-diameter plates or 106cells on 100-mm-diameter dishes. Twenty-four hours later, cells were infected with 0.2 ml of virus (60-mm-diameter dishes) or 1 ml of virus (100-mm-diameter dishes) in the presence of Polybrene (50mg/ml [final concentration]) as previously described (20). Transfected or infected cells were placed on puromycin selection 48 h later or G418 selection 24 h later.
Viral transfections, infections, and titer comparisons.The titers of retroviral vectors pRB-LLP, pRB-LPL, pVP212, pRB-PPT, and pWH342 were compared by the following method. The DNA of each retroviral vector was cotransfected into C3A2 helper cells with pBSpac at a ratio of 10mg to 1mg of plasmid DNAs. Plasmid pBSpac contains the puromycin N-acetyltransferase gene (7, 45). The transfected C3A2 cells were selected for puromycin resistance, pooled, ex-panded, and replated at a density of 23106cells per 100-mm-diameter dish. The culture medium was changed on day 1. On day 2, the culture medium containing virus was harvested and used to infect D17 cells. The average virus titers were determined from two to nine independent experiments.
PCR analysis of proviral DNAs.Proviral DNAs were amplified by PCR (19, 35) in an automated thermal cycler (OmniGene) with two primers that annealed to RB-LLP and RB-LPL proviruses. The 59primer annealed to the region of
each provirus containing the pBR origin of replication and was comprised of the sequence 59-GGACAGGTATCCGGTAAGCGGCAGGGTC-39. The 39primer annealed to the U3 region of each provirus and was comprised of the sequence 59-GCTTCTCGAATCGGCTGCATTTCTCGGCATC-39. DNA purification, PCR amplification, and restriction enzyme digestions were performed by stan-dard techniques (36). A 5% polyacrylamide gel was used for separation of DNA fragments generated by restriction digestion.
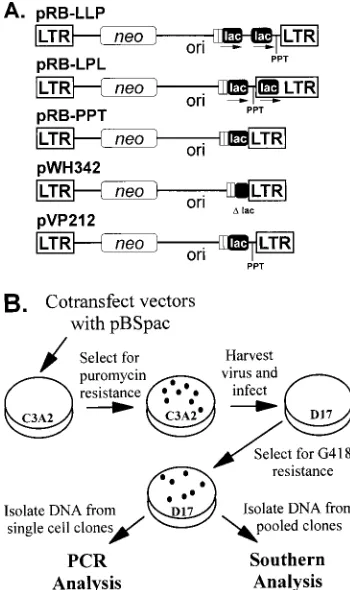
Southern analysis of proviral DNAs.Genomic DNAs were isolated and pro-viral structures were analyzed by Southern blot hybridizations using standard procedures (36). A 1.9-kb fragment of pVP212 containing the pBR and F1 origins of replication (ori) and a 0.4-kb lacZafragment were separately used to generate probes with the random-priming method (13) (Random Hexamer kit; BMB) and [a-32P]dCTP (ICN Pharmaceuticals, Inc.). The specific activities of these probes were greater than 109cpm/mg. Genomic DNAs were digested with restriction enzymes, separated by agarose gel electrophoresis, and transferred to nylon membranes as specified by the manufacturer (GeneScreen; Dupont NEN Research Products). The probes were hybridized to the nylon membranes and subsequently exposed to autoradiography film (Kodak) and/or a Phosphor-FIG. 1. Structures of SNV-based retroviral vectors and experimental proto-col to determine the relative rates of direct repeat deletions during minus-strand and plus-strand DNA synthesis. (A) All vectors contain a neomycin phospho-transferase gene (neo) and the pBR and F1 origins of replication (ori). The vectors pRB-LLP, pRB-LPL, pRB-PPT, and pVP212 contain a 400-bp lacZa fragment (lac). pWH342 contains the promoter region of the lacZafragment. pRB-LLP contains a direct repeat of the lacZafragment 59of the PPT. pRB-LPL contains one copy of lacZa59of the PPT and a second copy of lacZa39of the PPT within the U3 region of the 39long terminal repeat (LTR). The dis-tances between the two copies of the repeated sequences are the same in pRB-LLP and pRB-LPL. All vectors also contain a 110-bp direct repeat (open boxes). The PPT and att site were deleted from both pRB-PPT and pWH342. pVP212 contains one copy of lacZafragment 59of the PPT and att site. (B) Experimental protocol. C3A2 helper cells were cotransfected with pRB-LLP or pRB-LPL in the presence of pBSpac, an expression vector which confers resis-tance to puromycin. Pools of puromycin-resistant helper cells were expanded; virus was harvested from the helper cells and used to infect D17 target cells. The infected D17 cells were selected for resistance to G418. Genomic DNAs were isolated from either single-cell clones or pools of G418-resistant cells. The DNAs from single-cell clones were analyzed by PCR, and the DNAs from pools of cells were analyzed by Southern hybridization. Some of the single-cell clones were also analyzed by Southern hybridization.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.338.513.69.364.2]Imager cassette (Molecular Dynamics). Quantitation of bands was accomplished with the ImageQuant program (Molecular Dynamics).
RESULTS
Construction of retroviral vectors with direct repeats.Two SNV-based retroviral vectors, pRB-LLP and pRB-LPL, were constructed to determine the relative rates of in vivo RT tem-plate switching during RNA- and DNA-dependent DNA syn-thesis. Both vectors contained a neomycin phosphotransferase gene which was expressed from the long terminal repeat pro-moter (Fig. 1A). Both vectors also contained two copies of a 400-bp lacZagene fragment. The pRB-LLP vector contained both copies of the lacZafragment 59of the PPT. In contrast, the pRB-LPL vector contained one copy of the lacZafragment 59of the PPT and another copy 39of the PPT and att site. Both vectors were designed so that the distance between the two lacZa fragments was 70 bp. Deletion of the 400-bp direct repeat in the pRB-LLP vector during either RNA- or DNA-dependent DNA synthesis was expected to result in viral DNA that could integrate to form a provirus. On the other hand, deletion of the 400-bp direct repeat in the pRB-LPL vector during RNA-dependent DNA synthesis was expected to result in deletion of the intervening PPT and att site. Deletion of the PPT would be expected to prevent initiation of plus-strand DNA synthesis and generate a dead-end product. Deletion of the direct repeat in the pRB-LPL vector during DNA-depen-dent DNA synthesis was expected to generate heteroduplex viral DNA that could integrate to form a provirus. In addition to the 400-bp direct repeat, the pRB-LLP and pRB-LPL vec-tors also contained a 110-bp direct repeat 59 of the 400-bp direct repeat that has previously been shown to delete at a rate of 30 to 41% per replication cycle (21, 30). The 110-bp direct repeat was included in the vectors to determine whether RT template switching events that lead to deletion of the 400-bp direct repeat affect the frequency of deletion of the 110-bp direct repeat. To experimentally determine whether the dele-tion of the PPT and att site from the pRB-LPL vector would result in failure to complete reverse transcription, we also constructed pRB-PPT and pWH342 (Fig. 1A). These vectors were similar to pRB-LPL except that one copy of the lacZa fragment plus the PPT and att site were deleted from both of these vectors. Because of these deletions, pRB-PPT and pWH342 were not expected to complete reverse transcription and form proviruses. Finally, the relative efficiencies of viral replication of the pRB-LLP, pRB-LPL, pRB-PPT, and pWH342 vectors were determined by comparison to the con-trol vector pVP212, which is very similar to pRB-LLP except that it contains only one copy of the lacZagene (Fig. 1A).
Protocol for analysis of template switching and virus titers. The experimental approach taken to directly compare in vivo RT template switching during RNA- and DNA-dependent DNA synthesis in one replication cycle is outlined in Fig. 1B.
One replication cycle is defined as the steps of viral replication necessary for the generation of a provirus in the target cells from the vector DNA in the helper cells. This process includes RNA transcription in the helper cells and reverse transcription in the target cells. The vectors pRB-LLP and pRB-LPL were separately cotransfected into C3A2 helper cells along with pBSpac, a plasmid that contains the puromycin N-acetyltrans-ferase gene. The transfected cells were selected for puromycin resistance, and the resulting colonies were separately pooled and expanded. Viruses produced from the cells were used to infect D17 target cells, and the infected cells were selected for G418 resistance. Genomic DNAs were isolated from single-cell clones and from pools of the D17 target single-cells. The fre-quencies of direct repeat deletions were quantified by PCR amplification and restriction analysis of single-cell clones. These frequencies were then verified by examining a much larger number of proviruses by Southern analysis of the target cell pools.
Virus titers were determined by quantitation of G418-resis-tant D17 colonies after infection. The results of four indepen-dent experiments performed with both LLP and
[image:3.612.49.292.80.162.2]pRB-FIG. 2. Analysis of target cell clones for direct repeat deletions. (A) Loca-tions of primers used for PCR amplification and expected fragments after diges-tion with BglI. Structures of the 39ends of RB-LPL and RB-LLP proviruses without (above) and with (below) direct repeat deletion are shown. The large arrowheads indicate the approximate locations and directions of primers used for PCR amplification. The locations of BglI restriction sites (Bgl) are shown above the proviruses without direct repeat deletions. All other abbreviations are as for Fig. 1A. BglI digestion of PCR products derived from RB-LPL proviruses is expected to generate 1.1-kb (or 1.0-kb) and 0.56-kb fragments from proviruses without or with deletion of the 400-bp direct repeat. A 0.47-kb fragment is expected only from RB-LPL proviruses without deletion of the 400-bp direct repeat. Similarly, BglI digestion of PCR products derived from RB-LLP provi-ruses is expected to generate 1.1-kb (or 1.0-kb) and 0.62-kb fragments from proviruses without or with deletion of the 400-bp direct repeat. A 0.47-kb frag-ment is expected only from RB-LLP proviruses without deletion of the 400-bp direct repeat. (B) Representative restriction digestion analysis of PCR products derived from RB-LPL and RB-LLP proviruses. Lanes 1 and 2, RB-LLP provi-ruses with and without, respectively, direct repeat deletion; lanes 3 and 4, RB-LPL proviruses with and without, respectively, direct repeat deletion. The ap-proximate sizes of the restriction fragments are shown. Lane M, molecular weight marker.
TABLE 1. Titers in infected D17 cell pools
Expt Virus titer (CFU/ml)
a
RB-LLP RB-LPL
1 680 380
2 360 190
3 870 170
4 880 360
Avg 700 275
a23106helper cells were plated on day 0, the culture medium was changed on day 1, and virus was harvested on day 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.330.522.298.553.2]LPL vectors are summarized in Table 1. The average virus titers of pRB-LPL (275 CFU/ml) were approximately 2.5-fold lower than the virus titers of pRB-LLP (700 CFU/ml). Dele-tion of the direct repeat in the pRB-LPL vector during RNA-dependent DNA synthesis was expected to delete the interven-ing PPT and att site and generate a dead-end product. Therefore, the virus titers obtained were consistent with the expectation that the viral titers of pRB-LPL would be lower than the viral titers of pRB-LLP. Four additional infections were also performed in order to obtain more single-cell clones for PCR analysis (data not shown).
PCR amplification and restriction analysis of target cell clones to determine the frequencies of direct repeat deletions. To determine the frequencies of direct repeat deletions, the infected D17 target cell clones were individually propagated and 39regions of the proviral genomes were amplified by PCR. The locations of the PCR primers and the sizes of DNA frag-ments expected after digestion with BglI are shown in Fig. 2A. Digestion with BglI of PCR products derived from RB-LLP proviruses without deletion was expected to yield 1.1-, 0.47-, and 0.62-kb fragments. In contrast, digestion with BglI of PCR products derived from RB-LLP proviruses with deletion was expected to yield only a 1.1- and a 0.62-kb fragment. Thus, for pRB-LLP the presence of the 0.47-kb fragment was indicative of a provirus that did not undergo a direct repeat deletion.
Similar PCR amplification and restriction digestion analysis of proviruses derived from pRB-LPL were also performed (Fig. 2A). Digestion with BglI of PCR products derived from RB-LPL proviruses without deletion was expected to yield 1.1-, 0.47-, and 0.56-kb fragments. In contrast, digestion with BglI of PCR products derived from RB-LPL proviruses with deletion was expected to yield only a 1.1- and a 0.56-kb fragment. Thus, the presence of the 0.47-kb fragment was indicative of a RB-LPL provirus that did not undergo a direct repeat deletion.
The 1.1-kb fragment contained another 110-bp direct repeat that was previously shown to delete at a frequency of 30 to 41% per replication cycle (21, 30). The 110-bp direct repeat was located 59 of the 400-bp direct repeat in LLP and pRB-LPL, and deletion of this direct repeat was not expected to interfere with virus replication. Based on previous studies, approximately 30 to 40% of the proviruses analyzed were ex-pected to generate a 1.0-kb fragment rather than a 1.1-kb fragment.
Representative restriction analysis of a provirus with dele-tion of the 400-bp direct repeat and a provirus without deledele-tion of the 400-bp direct repeat for both RB-LLP and RB-LPL are shown in Fig. 2B. Digestion with BglI of the PCR product derived from a RB-LLP provirus generated a 1.1-kb and a 0.62-kb fragment indicating that the provirus had deleted one copy of the 400-bp direct repeat (lane 1). Similar analysis of
another RB-LLP provirus generated 1.1-, 0.62-, and 0.47-kb fragments (lane 2). The presence of the 0.47-kb fragment in-dicated that this provirus had retained the 400-bp direct re-peat.
Identical restriction analysis of PCR products derived from a RB-LPL provirus with deletion of the 400-bp direct repeat and another RB-LPL provirus without deletion of the 400-bp direct repeat are shown in lanes 3 and 4, respectively. A 1.1-and a 0.56-kb fragment were generated from the PCR product shown in lane 3; however a 1.0-, a 0.56-, and a 0.47-kb fragment were generated from the PCR product shown in lane 4. These analyses indicate that the RB-LPL provirus in lane 3 under-went deletion of the 400-bp direct repeat whereas the RB-LPL provirus in lane 4 did not undergo deletion of the 400-bp direct repeat. The presence of the 1.0-kb band in lane 4 indicated that this provirus also underwent deletion of the 110-bp direct re-peat. The parental RB-LPL and RB-LLP vectors containing both copies of the direct repeats were used as positive controls for all PCRs. PCR amplification of the vector DNAs always yielded only products that contained both copies of the direct repeats, indicating that under the conditions used, direct re-peat deletions did not occur during PCR (data not shown).
The PCR amplification and restriction analysis illustrated in Fig. 2 were performed on 43 cell clones infected with RB-LLP and 40 cell clones infected with RB-LPL (Table 2). These clones were derived from eight independent infections with each vector and represent separate infection events. Most of these clones were isolated from separate plates of infected cells (34 of 43 for RB-LLP and 36 of 40 for RB-LPL). In the few cases where two clones were derived from the same plate, one clone generated a 1.1-kb band and another generated a 1.0-kb band, indicating that the two cell clones were generated from independent infection events (data not shown).
The data indicated that deletion of the 400-bp direct repeat occurred more frequently in LLP proviruses than in RB-LPL proviruses. Approximately 86% of the RB-LLP proviruses (37 of 43) underwent deletion of the 400-bp direct repeat either during transfection of the helper cells or during reverse transcription. In contrast, approximately 62% of the RB-LPL proviruses (25 of 40) underwent deletion of the 400-bp direct repeat. The difference between these frequencies of direct repeat deletions was statistically significant (P 5 0.02, two-proportions test). The lower frequency of direct repeat dele-tions observed for the RB-LPL vector than for the RB-LLP vector is consistent with the expectation that only deletions occurring during DNA-dependent DNA synthesis can be ob-served in this population.
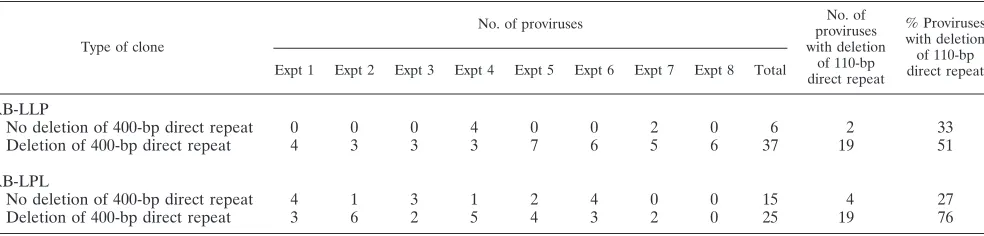
[image:4.612.55.547.81.199.2]We also compared the frequencies of 110-bp direct repeat deletions in RB-LLP and RB-LPL proviruses with and without deletion of the 400-bp direct repeat. Approximately 33% of the TABLE 2. PCR analysis of infected cell clones for deletions of direct repeats
Type of clone
No. of proviruses provirusesNo. of
with deletion of 110-bp direct repeat
% Proviruses with deletion of 110-bp direct repeat Expt 1 Expt 2 Expt 3 Expt 4 Expt 5 Expt 6 Expt 7 Expt 8 Total
RB-LLP
No deletion of 400-bp direct repeat 0 0 0 4 0 0 2 0 6 2 33
Deletion of 400-bp direct repeat 4 3 3 3 7 6 5 6 37 19 51
RB-LPL
No deletion of 400-bp direct repeat 4 1 3 1 2 4 0 0 15 4 27
Deletion of 400-bp direct repeat 3 6 2 5 4 3 2 0 25 19 76
on November 9, 2019 by guest
http://jvi.asm.org/
RB-LLP proviruses (2 of 6) and 27% of the RB-LPL provi-ruses (4 of 15) that did not have deletions of the 400-bp direct repeat had deletions of the 110-bp direct repeat. In contrast, 51% of the RB-LLP proviruses (19 of 37) and 76% of the RB-LPL proviruses (19 of 25) in which the 400-bp direct re-peat was deleted also had deletions of the 110-bp direct rere-peat. Thus, proviruses that were deleted of the 400-bp direct repeat had deletions of the 110-bp direct repeat at a higher frequency than proviruses that were not deleted of the 400-bp direct repeat (38 of 62 versus 6 of 21, P50.02, two-proportions test). Southern analysis of pools of transfected and infected cells to determine the frequencies of direct repeat deletions. Dele-tion frequencies in pRB-LLP and pRB-LPL were also
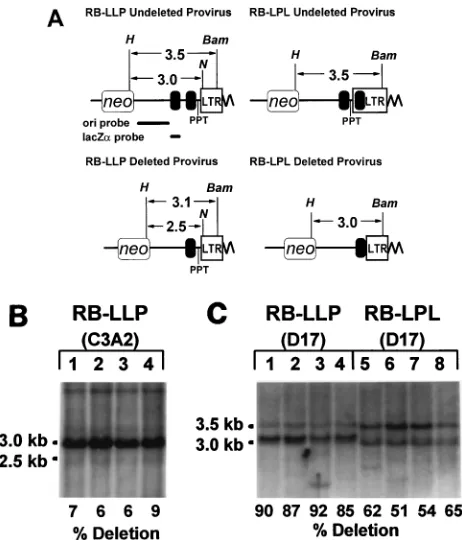
deter-mined by Southern blotting analysis (Fig. 3). Genomic DNAs from four pools of cells infected with RB-LLP and four pools of cells infected with RB-LPL were analyzed (the sizes of the pools ranged from 1,700 to 8,800 colonies per pool). In addi-tion, four pools of C3A2 helper cells transfected with pRB-LLP DNA were analyzed to determine the extent to which direct repeat deletions occurred during transfection and selec-tion of the helper cells.
The restriction sites used and the sizes of the resulting frag-ments are shown in Fig. 3A. Genomic DNAs from pools of C3A2 cells transfected with pRB-LLP were digested with HindIII plus NotI and hybridized to a 0.4-kb lacZaprobe (Fig. 3A, upper left panel). It was necessary to use the lacZaprobe rather than the ori probe because the genome of the C3A2 helper cells contains several copies of plasmid sequences that are expected to hybridize to the ori probe and complicate analysis. Viral DNAs without deletion of the 400-bp direct repeat were expected to generate a 3.0- or 2.9-kb fragment and DNAs with direct repeat deletions were expected to generate a 2.5- or 2.4-kb fragment (Fig. 3A, left panels). In this Southern analysis, the 3.0- and 2.9-kb fragments were not separated and are referred to as 3.0-kb fragments, and the 2.5- and 2.4-kb fragments were not separated and are referred to as 2.5-kb fragments. Southern analysis indicated that the 3.0-kb frag-ments were clearly evident in DNAs from the pools of cells transfected with pRB-LLP, while the 2.5-kb fragments were detectable at low levels in all four of the pools analyzed (Fig. 3B, left panel). Additional faint bands larger than 3.0 kb, which most likely represent nonspecific binding to the genomic DNA, were visible. Quantitation of the 3.0- and 2.5-kb bands indi-cated that the signal intensity of the 3.0-kb bands was approx-imately 28 fold higher than that of the 2.5-kb bands. The undeleted 3.0-kb fragments were expected to have a twofold-higher signal intensity, since these bands were expected to contain two copies of the lacZafragment to hybridize to twice as much of the lacZa probe. Since the 3.0-kb band was ex-pected to have a twofold-higher signal intensity, the molar ratio of the 3.0-kb band to the 2.5-kb band was 14:1. Therefore, approximately 7% of the pRB-LLP DNAs underwent direct repeat deletions during the process of transfection and selec-tion of helper cell clones.
[image:5.612.54.285.67.337.2]Southern hybridization analysis of four independent RB-LLP- and RB-LPL-infected D17 cell pools was also performed (Fig. 3B, right panel). Genomic DNAs from the pools were separately isolated and digested with HindIII plus BamHI. RB-LLP proviruses without deletion of the 400-bp direct re-peat were expected to generate a 3.5- or 3.4-kb fragment, and RB-LLP proviruses with deletion of the 400-bp direct repeat were expected to generate a 3.1- or 3.0-kb fragment (Fig. 3A, left panels). In this Southern analysis, the 3.5- and 3.4-kb fragments were not separated and are referred to as 3.5-kb fragments, and the 3.1- and 3.0-kb fragments were not sepa-rated and are referred to as 3.1-kb fragments. Similarly, RB-LPL proviruses without deletion of the 400-bp direct repeat FIG. 3. Southern analysis of C3A2 helper cells transfected with pRB-LLP
and pools of D17 cells infected with RB-LLP or RB-LPL. (A) Structures of the 39ends of RB-LLP and RB-LPL proviruses without deletion of the 400-bp direct repeat (above) and with deletion of the 400-bp direct repeat (below) are shown. Restriction digestion with HindIII (H) plus NotI (N) of genomic DNAs from the C3A2 helper cells transfected with RB-LLP is expected to generate a 3.0-kb band from the undeleted proviruses and a 2.5-kb band from the deleted proviruses. The black bars below the RB-LLP undeleted provirus represent the 1.9-kb fragment used to generate the ori probe and the 0.4-kb lacZafragment used to generate the lacZa probe for Southern analysis. Restriction digestion with
HindIII (H) plus BamHI (Bam) of genomic DNAs from pools of D17 cells
infected with RB-LLP is expected to generate a 3.5-kb band from the undeleted proviruses and a 3.1-kb band from the deleted proviruses. Similarly, restriction digestion with HindIII (H) plus BamHI (Bam) of genomic DNAs from pools of D17 cells infected with RB-LPL is expected to generate a 3.5-kb band from the undeleted proviruses and a 3.0-kb band from the deleted proviruses. All other abbreviations are as for Fig. 1A. (B) HindIII-plus-NotI digestion of DNAs from four pools of pRB-LLP-transfected C3A2 cells followed by hybridization to the
lacZa probe. The intensities of the 3.0-kb band representing the undeleted vectors and the 2.5-kb bands representing the deleted vectors were quantitated by PhosphorImager analysis. Frequencies of deletions of the 400-bp direct repeat based on the ratios of the intensities of the 2.5-kb band to the 3.0-kb band are shown below the lanes. (C) HindIII-plus-BamHI digestion of DNAs from four pools of RB-LLP-infected D17 cells (lanes 1 to 4) and four pools of RB-LPL-infected D17 cells followed by hybridization to the ori probe (lanes 5 to 8). The intensities of the 3.5-kb band representing the undeleted proviruses and the 3.1-or 3.0-kb bands representing the deleted proviruses f3.1-or RB-LLP and RB-LPL, respectively, were also quantitated by PhosphorImager analysis. The estimated frequencies of deletion of the 400-bp direct repeat are shown below the lanes.
TABLE 3. Effects of PPT and att deletion on viral titers
Expt Virus titer (CFU/ml)
a
RB-LLP VP212 RB-PPT WH342
1 390 400 10 10
2 450 10 10
Avg 390 425 10 10
a23106helper cells were plated on day 0, the culture medium was changed on day 1, and virus was harvested on day 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.308.547.646.708.2]were expected to generate a 3.5- or a 3.4-kb fragment, and RB-LPL proviruses with deletion of the 400-bp direct repeat were expected to generate a 3.0- or 2.9-kb fragment (Fig. 3A, right panels). In this Southern analysis, the 3.5- and 3.4-kb fragments were not separated and are referred to as 3.5-kb fragments, and the 3.0- and 2.9-kb fragments were not sepa-rated and are referred to as 3.0-kb fragments. The digested DNAs were hybridized to a 1.9-kb probe fragment derived from the ori region of the RB-LPL vector. The results obtained from four pools of cells infected with RB-LLP indicated that most of the proviruses underwent deletion of the 400-bp direct repeat (Fig. 3C, lanes 1 to 4). In comparison, the results ob-tained from four pools of cells infected with pRB-LPL indi-cated that a lower proportion of the proviruses underwent deletion of the 400-bp direct repeat (Fig. 3C, lanes 5 to 8). Quantitation of the 3.5-kb undeleted bands and the 3.1- or 3.0-kb deleted bands was performed to determine the frequen-cies of direct repeat deletions. The results indicated that on average 88% (63%) of the RB-LLP proviruses underwent direct repeat deletions and 58% (67%) of the RB-LPL pro-viruses underwent direct repeat deletions. Thus, the direct repeat deletion frequencies of RB-LLP and RB-LPL, as de-termined by Southern analysis, were very similar to the fre-quencies determined by PCR amplification and restriction di-gestions (86% for RB-LLP and 62% for RB-LPL).
Deletion of the PPT and attachment sites reduces the effi-ciency of provirus formation. The deletion frequencies ob-tained from pRB-LPL suggested that direct repeat deletions occurred at a high rate during DNA-dependent DNA synthe-sis. This interpretation was based on the assumption that any direct repeat deletions that occurred during RNA-dependent DNA synthesis would result in the deletion of the PPT and att site and generate a dead-end product. To validate this assump-tion, we generated two vectors, pRB-PPT and pWH342, which were similar to pRB-LPL but lacked the PPT and att sites. Our rationale for using two independently constructed vectors was to rule out the possibility of cryptic PPT or att sites being inadvertently present in the vectors, which would permit com-pletion of viral replication. For both vectors, initiation of DNA-dependent DNA synthesis from the PPT primer and subsequent proviral formation were expected to be greatly decreased, leading to a severe reduction in virus titers. Inde-pendent and parallel transfections and infections were per-formed, and the virus titers obtained with pRB-PPT and pWH342 were compared to the virus titers obtained with the control vectors pVP212 and pRB-LLP. As shown in Table 3, the virus titers obtained from pVP212 and pRB-LLP vectors were approximately 400 CFU/ml. In contrast, the virus titers obtained from pRB-PPT and pWH342 were approximately 10 CFU/ml. Therefore, deletion of the PPT and att site from pRB-PPT and pWH342 resulted in a 40-fold reduction in the efficiency of provirus formation. The 40-fold reduction in the virus titers represents the minimum reduction in the virus titers as a result of PPT deletions, since it is conceivable that higher virus titers from the control vectors would further increase the difference between the virus titers.
Southern analysis of clones of infected cells indicates effi-cient DNA repair of heteroduplex viral DNAs.Deletion of the direct repeats during DNA-dependent DNA synthesis of the pRB-LPL vector is expected to generate a heteroduplex viral DNA (Fig. 4A). The host cell DNA repair machinery may resolve these heteroduplexes in three different ways. First, it is possible that the sequence of the plus strand will be used as a template to replace the sequence of the minus strand (Fig. 4A, Repair to plus-strand). If this occurs, each daughter cell will inherit a provirus with a direct repeat deletion following inte-gration of the viral DNA and cell division of the infected cells. The resulting cell clones will be homogeneous with respect to the provirus genotype. Second, it is possible that the sequence of the minus strand will be used as a template to replace the sequence of the plus strand (Fig. 4A, Repair to minus-strand). If so, then both of the daughter cells that form after cell division will inherit a provirus without direct repeat deletion, and the resulting cell clone will be homogeneous with respect to the provirus genotype. Third, if DNA repair does not take place, then the heteroduplex viral DNA will integrate into the host cell chromosome, with one strand containing a direct repeat deletion and the other strand containing both copies of the direct repeat (Fig. 4A, No repair). After cell division, one daughter cell will inherit a provirus with direct repeat deletion, and the other daughter cell will inherit a provirus without direct repeat deletion. The resulting cell clone will be hetero-geneous, containing both deleted and undeleted proviruses. Thus, the frequency of cell clones that are heterogeneous with respect to the provirus genotype can be used to determine whether efficient DNA repair of the heteroduplex DNAs oc-curred before integration and cell division.
[image:6.612.63.274.69.275.2]Because selective amplification of the RB-LPL proviruses with deletion of the 400-bp direct repeats (1.6- or 1.5-kb frag-ments) occurred under the amplification conditions used, PCR analysis of the cell clones could not be used to determine FIG. 4. Analysis of RB-LPL-infected single-cell clones for potential DNA
repair of heteroduplex viral DNA. (A) Potential effects of DNA repair of het-eroduplex viral DNA on colony phenotypes. Black ovals represent cells contain-ing a provirus with deletion of the 400-bp direct repeat; white ovals represent cells containing a provirus that was not deleted of the 400-bp direct repeat. All other abbreviations are as for Fig. 1A. Deletion of the 400-bp direct repeat during plus-strand synthesis of RB-LPL is expected to generate a heteroduplex viral DNA. If the heteroduplex DNA is repaired to the plus-strand, then homo-geneous colonies containing deleted proviruses will form (Repair to plus-strand). If the heteroduplex DNA is repaired to the minus strand, then homogeneous colonies containing undeleted proviruses will form (Repair to minus-strand). If the heteroduplex DNA is not repaired prior to viral DNA integration and cell division, a mixed colony containing both deleted and undeleted proviruses will form (No repair). LTR, long terminal repeat. (B) Southern analysis of RB-LPL single-cell clones. The genomic DNAs isolated from 12 single-cell clones of infected D17 cells were analyzed to determine the presence of mixed-colony phenotypes. Each DNA was digested with HindIII plus NotI and hybridized to radioactively labeled ori probe (Fig. 3A). All 12 clones underwent direct-repeat deletions as determined by PCR amplification and restriction digestion analysis. In all 12 clones, only the 3.0-kb fragment characteristic of a deleted provirus was present, indicating that these clones were derived from homogeneous colonies.
on November 9, 2019 by guest
http://jvi.asm.org/
heterogeneity with respect to the provirus genotype (data not shown). We therefore analyzed 12 RB-LPL cell clones by Southern analysis to determine whether any of the cell clones were heterogeneous and contained both undeleted and deleted proviruses. Since all direct repeat deletions in the RB-LPL vector occurred during DNA-dependent DNA synthesis, all of these clones were derived from heteroduplex viral DNAs. Genomic DNAs isolated from the cell clones were digested with HindIII plus BamHI and hybridized to the ori probe. The results are shown in Fig. 4B. Any cell clones heterogeneous with respect to the provirus genotype were expected to gener-ate a 3.5-kb band from the undeleted proviruses and a 3.0-kb band from the deleted proviruses. The results obtained clearly showed that only the 3.0-kb band was generated from all 12 cell clones (Fig. 4B), indicating that none of the cell clones were heterogeneous and contained only the deleted proviruses. These results suggested that efficient DNA repair of the het-eroduplex DNAs occurred. The DNA repair could have taken place either before integration of viral DNA into the chromo-some or after integration but before replication of the provirus through cell division.
DISCUSSION
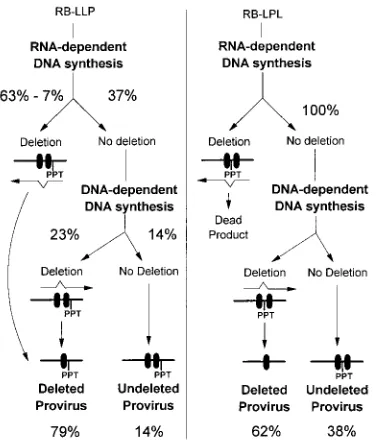
RT switches templates at similar rates during RNA- and DNA-dependent DNA synthesis.The frequencies of deletion of the 400-bp direct repeat observed for the LPL and pRB-LLP vectors can be used to estimate the frequencies of RT template switching during RNA- and DNA-dependent DNA synthesis. Data generated from PCR analysis provided a more quantitative measure of the frequencies of direct repeat dele-tions and were used for the analysis. The observed and esti-mated fractions of proviruses that underwent direct repeat deletions during RNA- and DNA-dependent DNA synthesis are shown in Fig. 5. As discussed earlier, deletions that are observed for the pRB-LPL vector represent only RT template switching events occurring during DNA-dependent DNA syn-thesis. Based on the PCR analysis, the frequency of RT tem-plate switching events that lead to direct repeat deletions dur-ing DNA-dependent DNA synthesis of RB-LPL was 62%, and the ratio of RB-LPL proviruses with and without direct repeat deletions was 62%/38%. It should be noted that approximately 7% of the RB-LPL vector DNAs are expected to undergo direct repeat deletions during the process of transfection and selection of helper cells. Since viruses derived from these DNAs with direct repeat deletions are expected to lack the PPT and att site, they are not expected to complete reverse transcription, integrate, and generate proviruses. Therefore, it is not necessary to adjust the 62% frequency of direct repeat deletions during DNA-dependent DNA synthesis.
[image:7.612.78.266.69.289.2]In contrast to pRB-LPL, the frequency of direct repeat de-letions observed for the pRB-LLP vector represents RT tem-plate switching events occurring during transfection, RNA-dependent DNA synthesis, and DNA-RNA-dependent DNA synthesis. Based on the PCR analysis, the frequency of RT template switching events that resulted in direct repeat dele-tions of RB-LLP was 86%. It was determined that approxi-mately 7% of the direct repeat deletions in RB-LLP vectors occurred during the process of transfection and selection of helper cells (Fig. 3B). Therefore, we estimate that approxi-mately 79% (86%27%) of RB-LLP proviruses underwent a direct repeat deletion either during RNA- or DNA-dependent DNA synthesis. The ratio of the RB-LLP proviruses with and without direct repeat deletions during reverse transcription was 79%/14%. It is important to note that only RB-LLP pro-viruses that retain the direct repeat either during RNA-depen-dent DNA synthesis or during transfection can serve as sub-strates for direct repeat deletions during DNA-dependent DNA synthesis. An unknown fraction of the RB-LLP provi-ruses (designated x) underwent deletions during DNA-depen-dent DNA synthesis, and the remaining fraction of the RB-LLP proviruses (79%2x) underwent deletions during RNA-dependent DNA synthesis. Therefore, the ratio of RB-LLP proviruses that did and did not undergo deletions during DNA-dependent DNA synthesis was x/14%. Assuming that the same fraction of RB-LLP and RB-LPL proviruses undergo deletions during DNA-dependent DNA synthesis, we can es-timate that approximately 23% of the RB-LLP proviruses de-leted the direct repeat during DNA-dependent DNA synthesis (if x/14%5 62%/38%, then x562% 314%/38% 523%). Since an estimated 23% of the RB-LLP proviruses deleted the direct repeat during DNA-dependent DNA synthesis, 56% of the proviruses deleted the direct repeat during RNA-depen-dent DNA synthesis (79%223%). Based on this analysis, an estimated 56% of the RB-LLP proviruses underwent direct repeat deletions during RNA-dependent DNA synthesis, and 62% of the RB-LPL proviruses underwent direct repeat dele-tions during DNA-dependent DNA synthesis. These results
FIG. 5. Illustration of the rationale used to extrapolate frequencies of RT template switching during RNA- and DNA-dependent DNA synthesis. Deletion of the direct repeat (black boxes) may occur during either transfection of the vectors into packaging cells, RNA-dependent DNA synthesis, or DNA-depen-dent DNA synthesis during replication of the RB-LLP or the RB-LPL vectors. Approximately 7% of the viral DNAs undergo deletion during transfection (Fig. 3B). Direct repeat deletion during transfection or RNA-dependent DNA syn-thesis of the RB-LPL vector results in the generation of a dead-end (Dead) product. Therefore, it is not necessary to adjust the rates of deletion during RNA-dependent DNA synthesis of the RB-LPL vector for the rate of deletion during transfection. The frequencies of RB-LPL deleted proviruses (62%) and undeleted proviruses (38%) are shown. The frequencies of RB-LLP undeleted proviruses (14%) is also shown. Approximately 86% of the RB-LLP vectors underwent direct repeat deletions during either transfection or RNA-dependent DNA synthesis. Since 7% of the viral DNAs were deleted during transfection, approximately 79% (86%27%) of the RB-LLP vectors underwent direct repeat deletion during RNA- and DNA-dependent DNA synthesis. The frequencies of RB-LLP proviruses that underwent direct repeat deletions during RNA-depen-dent DNA synthesis (56%) and DNA-depenRNA-depen-dent DNA synthesis (23% of total) were extrapolated from the observed frequencies of direct repeat deletions for the RB-LPL vector (see Discussion). Only 37% (100%2[56%17%]) of the RB-LLP proviruses that did not undergo direct repeat deletion during transfec-tion or RNA-dependent DNA synthesis were capable of deleting the direct repeat during DNA-dependent DNA synthesis.
on November 9, 2019 by guest
http://jvi.asm.org/
indicate that the rates of RT template switching during RNA-and DNA-dependent DNA synthesis are nearly identical.
On the surface, a comparison of the frequencies of deletions for RB-LLP during RNA-dependent and DNA-dependent DNA synthesis may suggest that direct repeat deletions occur at different rates during RNA- and DNA-dependent DNA synthesis (56% versus 23%). However, it is important to note that approximately 63% of the RB-LLP proviruses underwent direct repeat deletions during transfection or RNA-dependent DNA synthesis (56% during RNA-dependent DNA synthesis and 7% during transfection); therefore, only 37% of the vi-ruses could potentially delete the direct repeats during DNA-dependent DNA synthesis. We have extrapolated that 62% of the proviruses that could undergo direct repeat deletion during DNA-dependent DNA synthesis did so (37%30.62523%). Therefore, 56% of the RB-LLP viruses underwent direct re-peat deletion during RNA-dependent DNA synthesis, and 62% of the remaining viruses underwent direct repeat deletion during DNA-dependent DNA synthesis.
It is important to note that the rate of RT template switching during DNA-dependent DNA synthesis may represent an un-derestimate. Direct repeat deletions during DNA-dependent DNA synthesis are expected to generate heteroduplex DNAs, a fraction of which may be corrected to the undeleted minus strand by host DNA repair mechanisms (discussed below).
The previously proposed model for high-frequency deletion of direct repeats was based on other published studies indicat-ing that RNase H continually degrades the template RNA 18 to 20 nt behind the site of DNA polymerization (10, 11, 14, 15, 18, 48). In vitro studies have indicated that the RNase H activity of RT is important for RT template switching (16, 26). In the latter study, only template switching events that oc-curred when the polymerase reached the end of the template were analyzed (26). In addition, these studies have shown that the RNase H activity plays an important role in the minus-strand strong-stop DNA transfer. Although some in vitro stud-ies have suggested that the RNase H degradation of the tem-plate and DNA synthesis activities are coupled (11, 15, 18, 48), other in vitro studies have suggested that RNase H degrada-tion may not be coupled to the synthesis of nascent DNA (9, 37–39). The results of this study are consistent with the view that the RNase H degradation in vivo may not be coupled to DNA synthesis, since the frequency of direct repeat deletions during RNA-dependent DNA synthesis was not higher than the deletion frequency during DNA-dependent DNA synthe-sis. These results extend our previous studies, which indicate that substitutions and frameshift mutations occur at similar rates during RNA- and DNA-dependent DNA synthesis (25). Alternatively, it is possible that RNase H degradation of the template RNA facilitates RT template switching during RNA-dependent DNA synthesis, and other mechanisms promote RT template switching during DNA-dependent DNA synthesis. Mechanisms that may promote RT template switching during DNA-dependent DNA synthesis include strand displacement synthesis, secondary structure of the template DNA, and low affinity of the RT for the DNA template.
Heteroduplex viral DNAs are efficiently repaired by host cell DNA repair mechanisms.The results of this study indicate that heteroduplex viral DNAs, generated as a result of direct repeat deletions during DNA-dependent DNA synthesis of pRB-LPL, were efficiently repaired. The potential repair of the heteroduplex DNAs can result in three types of cell clones (Fig. 4). First, DNA repair of the viral DNAs may not occur. In this case, heteroduplex colonies would be expected. Since all of the cell clones analyzed were homogeneous, containing only proviruses with direct repeat deletions, we conclude that
effi-cient DNA repair of the viral DNAs occurred. Second, DNA repair may not exhibit strand specificity. If so, then 50% of the time, the deleted plus strand may get repaired by using the undeleted minus strand as a template. Consequently, the ob-served frequency of direct repeat deletions for the RB-LPL proviruses (62%) would be an underestimate, and the actual rate of direct repeat deletions during DNA-dependent DNA synthesis may approach 100%. Second, DNA repair may ex-hibit strand specificity and preferentially correct the plus strand by using the undeleted minus strand as a template; then the rate of RT template switching during DNA-dependent DNA synthesis may be much higher as well. However, since 62% of the proviruses exhibited direct repeat deletion, only 38% of the proviruses could have been corrected to the unde-leted form by DNA repair. Therefore, DNA repair mecha-nisms did not preferentially correct the plus strand by using the minus strand as a template. Third, if strand-specific repair occurred and preferentially corrected the undeleted minus strand by using the deleted plus strand as a template, then the observed frequency of direct repeat deletions for the RB-LPL proviruses (62%) would be an accurate estimate of the actual rate of RT template switching during DNA-dependent DNA synthesis.
Repair of viral DNAs containing small heteroduplexes has been previously described (2, 32). In these studies, the effi-ciency or potential strand specificity of DNA repair could not be determined. DNA repair of 16-nt loops in nonviral DNAs has been observed to occur efficiently (44); the repair of the loops exhibited strand specificity and required the presence of a nick in close proximity to the heteroduplex. Since the retro-viral integration process creates a gap at the 59 end of the minus strand, which would be approximately 600 bp from the heteroduplex, it is possible that strand-specific repair of the RB-LPL heteroduplex DNAs occurred and preferentially cor-rected the undeleted minus strand by using the deleted plus strand as a template.
In this study, the ability of retroviral vectors lacking a PPT and att site to complete reverse transcription and form provi-ruses was analyzed. Surprisingly, the vectors without PPT and att site were able to form proviruses at 2% efficiency relative to the wild-type vectors. The viral DNAs produced from these vectors are expected to lack the 59 att site and may have integrated into the target cells by aberrant integration events or by a mechanism not involving viral integrase.
Finally, proviruses that underwent deletion of the 400-bp direct repeat also were deleted of the 110-bp direct repeat at a higher than expected frequency, suggesting that direct repeat deletions exhibit high negative interference (4, 47). This ob-servation suggests that RTs that undergo one template switch event have a higher probability of undergoing another tem-plate switch event. We have previously shown that retroviral recombination exhibits high negative interference, whereby vi-ruses that exhibit one intermolecular template switch have a higher probability of exhibiting another intermolecular tem-plate switch. It should be noted that direct repeat deletions primarily occur by intramolecular template switching events whereas retroviral recombination requires intermolecular tem-plate switching events (21). Experiments to verify that intramo-lecular RT template switching events exhibit high negative interference are in progress.
ACKNOWLEDGMENTS
We thank Jeffery Anderson, Benjamin Beasley, Que Dang, Krista Delviks, Elias Halvas, John Julias, Evguenia Svarovskaia, Yegor Voronin, and Wenhui Zhang for critical reading of the manuscript.
This work was supported by Public Health Service grants CA58875
on November 9, 2019 by guest
http://jvi.asm.org/
to V.K.P. and CA58345 to W.-S.H. from the National Institutes of Health.
REFERENCES
1. Baltimore, D. 1970. Viral-dependent DNA polymerase. Nature 226:1209– 1211.
2. Berwin, B., and E. Barklis. 1993. Retrovirus-mediated insertion of expressed and non-expressed genes at identical chromosomal locations. Nucleic Acids Res. 21:2399–2407.
3. Bowman, E. H., V. K. Pathak, and W.-S. Hu. 1996. Efficient initiation and strand transfer of polypurine tract-primed plus-strand DNA prevents strand transfer of internally initiated plus-strand DNAs. J. Virol. 70:1687–1694. 4. Chase, M., and A. H. Doerman. 1958. High negative interference over short
segments of the genetic structure of bacteriophage T4. Genetics 43:332–353. 5. Coffin, J. M. 1996. Retroviridae: the viruses and their replication, p. 1767– 1848. In B. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Fields virology, vol. 3. Raven Press, New York, N.Y.
6. Czernilofsky, A. P., A. D. Levinson, H. E. Varmus, J. M. Bishop, E. Tisher, and H. M. Goodman.1980. Nucleotide sequence of an avian sarcoma virus oncogene (src) and proposed amino acid sequence of gene product. Nature 287:198–203.
7. de la Luna, S., I. Soria, D. Pulido, J. Ortin, and A. Jimenez. 1988. Efficient transformation of mammalian cells with constructs containing a puromycin-resistance-marker. Gene 62:121–126.
8. Delviks, K., W.-S. Hu, and V. K. Pathak. 1997.Cvectors: murine leukemia virus-based self-inactivating and self-activating retroviral vectors. J. Virol. 71:6128–6224.
9. Destefano, J. J., R. G. Buiser, L. M. Mallaber, T. W. Meyers, R. A. Bambara, and P. J. Fay.1991. Polymerization and RNase H activities of the reverse transcriptases from avian myeloblastosis, human immunodeficiency, and Moloney murine leukemia viruses are functionally uncoupled. J. Biol. Chem. 266:7423–7431.
10. Destefano, J. J., R. G. Buiser, L. M. Mallaber, R. A. Bambara, and P. J. Fay. 1991. Human immunodeficiency virus reverse transcriptase displays a par-tially processive 39to 59endonuclease activity. J. Biol. Chem. 266:24295– 24301.
11. Destefano, J. J., L. M. Mallaber, P. J. Fay, and R. A. Bambara. 1993. Determinants of the RNase H cleavage specificity of human immunodefi-ciency virus reverse transcriptase. Nucleic Acids Res. 21:4330–4338. 12. Dougherty, J. P., and H. M. Temin. 1988. Determination of the rate of
base-pair substitution and insertion mutations in retrovirus replication. J. Vi-rol. 62:2818–2822.
13. Feinberg, A. P., and B. Volgelstein. 1983. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal. Biochem. 132:6–13.
14. Fu, T. B., and J. Taylor. 1992. When retroviral reverse transcriptases reach the end of their RNA templates. J. Virol. 66:4271–4278.
15. Furfine, E. S., and J. E. Reardon. 1991. Reverse transcriptasezRNase H from the human immunodeficiency virus. J. Biol. Chem. 266:406–412. 16. Garces, J., and R. Wittek. 1991. Reverse-transcriptase-associated RNase H
activity mediates template switching during reverse transcription in vitro. Proc. R. Soc. Lond. 243:235–239.
17. Gilboa, E., S. W. Mitra, S. Goff, and D. Baltimore. 1979. A detailed model of reverse transcription and tests of crucial aspects. Cell 18:93–100. 18. Gopalakrishnan, V., J. A. Peliska, and S. J. Benkovic. 1992. Human
immu-nodeficiency virus type 1 reverse transcriptase: spatial and temporal rela-tionship between the polymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 89:10763–10767.
19. Higuchi, R. 1989. Simple and rapid preparation of samples for PCR, p. 31–38. In H. A. Erlich (ed.), PCR technology; principles and applications for DNA amplification. Stockton Press, New York, N.Y.
20. Hu, W.-S., and H. M. Temin. 1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of genetic recombination. Proc. Natl. Acad. Sci. USA 87:1556–1560. 21. Hu, W.-S., E. H. Bowman, K. Delviks, and V. K. Pathak. 1997. Homologous
recombination occurs in a distinct retroviral subpopulation and exhibits high negative interference. J. Virol. 71:6028–6036.
22. Hughes, S., and E. Kosik. 1984. Mutagenesis of the region between env and
src of the SR-A strain of Rous sarcoma virus for purpose of constructing
helper and independent vectors. Virology 136:89–99.
23. Julias, J. G., D. Hash, and V. K. Pathak. 1995. E2vectors: Development of novel self-inactivating and self-activating retroviral vectors for safer gene
therapy. J. Virol. 69:6839–6846.
24. Kawai, S., and M. Nishizawa. 1984. New procedures for DNA transfection with polycation and dimethyl sulfoxide. Mol. Cell. Biol. 4:1172–1174. 25. Kim, T., R. A. Mudry, Jr., C. A. Rexrode II, and V. K. Pathak. 1996.
Retroviral mutation rates and A-to-G hypermutations during different stages of retroviral replication. J. Virol. 70:7594–7602.
26. Luo, G., and J. Taylor. 1990. Template switching by reverse transcriptase during DNA synthesis. J. Virol. 64:4321–4328.
27. Mansky, L. M., and H. M. Temin. 1994. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J. Virol. 69:5087–5094.
28. Mansky, L. M., and H. M. Temin. 1995. Lower mutation rate of bovine leukemia virus relative to that of spleen necrosis virus. J. Virol. 68:494–499. 29. Omer, C. A., K. Pogue-Geile, R. Guntaka, K. A. Stakus, and A. J. Faras. 1983. Involvement of directly repeated sequences in the generation of dele-tions of the avian sarcoma virus src gene. J. Virol. 47:380–382.
30. Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: substitutions, frameshifts, and hyper-mutations. Proc. Natl. Acad. Sci. USA 87:6019–6023.
31. Pathak, V. K., and H. M. Temin. 1990. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: deletions and deletions with insertions. Proc. Natl. Acad. Sci. USA 87:6024–6028.
32. Pulsinelli, G. A., and H. M. Temin. 1994. High rate of mismatch extension during reverse transcription in a single round of retrovirus replication. Proc. Natl. Acad. Sci. USA 91:9490–9494.
33. Rhode, B. W., M. Emerman, and H. M. Temin. 1987. Instability of large direct repeats in retrovirus vectors. J. Virol. 61:925–927.
34. Riggs, J. L., R. M. McAllister, and E. H. Lennette. 1974. Immunofluorescent studies of RD-114 virus replication in cell culture. J. Gen. Virol. 25:21–29. 35. Saiki, R. K., D. H. Gelfand, S. Stoffel, S. J. Scharf, R. Higuchi, G. T. Horn, K. B. Mullis, and H. A. Erlich.1988. Primer-directed enzymatic amplifica-tion of DNA with a thermostable DNA polymerase. Science 239:487–491. 36. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a
laboratory manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
37. Schatz, O., J. Mous, and S. F. J. Le Grice. 1990. HIV-1 RT-associated ribonuclease H displays both endonuclease and 39359exonuclease activity. EMBO J. 9:1171–1176.
38. Schultz, S. J., S. H. Whiting, and J. J. Champoux. 1995. Cleavage specific-ities of Moloney murine leukemia virus RNase H implicated in the second strand transfer during reverse transcription. J. Biol. Chem. 270:24135–24145. 39. Telesnitsky, A., and S. P. Goff. 1993. Two defective forms of reverse tran-scriptase can complement to restore retroviral infectivity. EMBO J. 12:4433– 4438.
40. Temin, H. M., and S. Mizutani. 1970. RNA-dependent DNA polymerase in virions of Rous sarcoma virus. Nature 226:1211–1213.
41. Temin, H. M. 1976. The DNA provirus hypothesis: the establishment and implications of RNA-directed DNA synthesis. Science 192:1075–1080. 42. Temin, H. M. 1993. Retrovirus variation and reverse transcription: abnormal
strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci. USA 90:6900–6903.
43. Tikhonenko, A. T., and M. Linial. 1993. Transforming variants of the avian
myc-containing retrovirus FH3 arise prior to phenotypic selection. J. Virol.
67:3635–3638.
44. Umar, A., J. C. Boyer, and T. A. Kunkel. 1994. DNA loop repair by human cell extract. Science 266:814–816.
45. Vara, J., A. Portela, J. Ortin, and A. Jimenez. 1986. Expression in mamma-lian cells of a gene from Streptomyces alboniger conferring puromycin resis-tance. Nucleic Acids Res. 14:4617–4624.
46. Watanabe, S., and H. M. Temin. 1983. Construction of a helper cell line for avian reticuloendotheliosis virus cloning vectors. Mol. Cell. Biol. 3:2241. 47. White, R. L., and M. S. Fox. 1974. On the molecular basis of high negative
interference. Proc. Natl. Acad. Sci. USA 71:1544–1548.
48. Wohrl, B. M., and K. Moelling. 1990. Interaction of HIV-1 ribonuclease H with polypurine tract containing RNA-DNA hybrids. Biochemistry 29:10141–10147.
49. Zhang, J., and H. M. Temin. 1993. Rate and mechanism of nonhomologous recombination during a single cycle of retroviral replication. Science 259: 234–238.