JOURNALOFVIROLOGY,Dec. 1993, p. 7254-7263 0022-538X/93/127254-10$02.00/0
Copyright
© 1993, AmericanSociety forMicrobiology
Binding Sites for the
Herpes
Simplex Virus Immediate-Early
Protein
ICP4 Impose
an
Increased
Dependence
on
Viral DNA
Replication
on
Simple Model
Promoters
Located
in
the Viral Genome
KAREN E. KOOP,' JOANNE DUNCAN,2 ANDJAMES R.
SMILEYI'*
Molecular Virology and Immunology Programme, Departments of Biology' and Pathology,2McMaster
University,
1200 Main StreetWest,
Hamilton,
Ontario L8N3Z5,
Canlada
Received 5 May 1993/Accepted 15 August1993
We examined the ability of binding sites for the herpes simplex virus immediate-early protein ICP4toalter the regulation of closely linked promotersby placing strongICP4 binding sites upstream ordownstream of
simple TATApromotersin the intact viralgenome.Wefound that binding sitesstrongly reduced the levelsof expressionatearlytimes postinfection and that this effectwaspartiallyovercomeafter theonsetof viral DNA replication. These data confirm that DNA-bound ICP4caninhibittheactivity ofaclosely linkedpromoterand
raise the possibility that ICP4 binding sites contributeto temporalregulation during infection.
The 70genes of herpes simplex virus type 1 (HSV-1) (37)
aresequentially expressed in athree-tiered regulatory cascade
during lytic infection (26, 27; seereferences 14, 53, and 71 for reviews). Five immediate-early (IE) genes are expressed first,
and theseare the only HSVgenesthatare transcribed in the absence of de novo viral protein synthesis. The IE proteins then induce expression of the early (E) and late (L) genes.E genes areactivatedpriortotheonsetof viral DNAreplication, while L genes require DNA replication for maximal expres-sion. Lgenes canbe further subclassifiedasleaky Lortrue L, according to the stringency of the replication requirement. Considerable evidence indicates thatIEproteins regulate viral geneexpression atthe both thetranscriptional (22, 48, 60, 73) and theposttranscriptional (56) levels and that the kinetics of expression of individual HSVgenes are greatly influenced by sequencesintheirpromoterregions (24, 25, 29, 32, 35, 48, 60). However, the precise mechanisms underlying temporal regu-lation ofgene expression remain unknown.
The IE protein ICP4 (infected-cell protein 4 [26]) plays a critical role during infection: itis essentialfor activation of E
and L transcription and serves to downregulate IE gene expression. Thus, ICP4-deficient temperature-sensitive and deletion mutants fail to express most E and L genes and overproduceIEgeneproducts (7, 12, 49, 72). These regulatory responsescanbereproduced in transient cotransfectionassays,
inwhich ICP4 activatesanumber of E and Lpromoters(8, 13, 19, 41, 50) and represses the promoters of the IE ICP4 and ICP0genes (8, 20, 21, 42, 51, 52). These results demonstrate that ICP4 acts directly as both a positive and a negative
regulator of HSV geneexpression.
ICP4 is asite-specific DNA-binding protein, andabinding
consensus sequence, 5'-ATCGTCnnYnCCGRCnnCRYCR-3', has been described (16, 17, 31, 40), although binding to nonconsensus sites alsooccurs (28, 33, 39, 68). The results of
extensive mutational studies of ICP4 suggestthat DNA bind-ing is required for its transregulatory activities (9, 10, 45, 46, 59; butsee alsoreferences 47 and 58). However, theextent to
*Correspondingauthor.
which specific ICP4 binding sites modulate the activity of nearbypromotersis less clear. For example, ICP4 binding sites inthe vicinity of the glycoprotein D (gD) promoterappearto contribute to the activation ofgene expression in vitro, and
multimerization of these sites increased induction by ICP4 in transient transfection assays (3, 68, 69); however, mutations that eliminate these sites have no detectable effect on gD expression during lytic virus infection (63). Similarly, muta-tions of ICP4 that eliminatebindingtolow-affinitysites inthe
thymidine kinase (tk) gene have no effect on TKexpression (28). Incontrast, mutation ofan ICP4binding site in the UL
49.5 gene was reported to reduce expression during lytic infection(54). There isstrongerevidence foraroleof individ-ual ICP4binding sites in the negative regulation of IE gene
expression. High-affinity ICP4 binding sites are located over the ICP4 transcription start site and upstream of the ICPO
TATAelement (17, 33, 40). In bothcases, these sitesserve as targetsfor ICP4-mediatedrepressionintransient transfection assays (20, 51, 52). Consistent with these data, Michael and
Roizman (38) found that the ICP4 binding site locatedatthe ICP4 transcription start site serves as a target for negative regulation during lytic HSV infection. Incontrast,Everettand Orr (15) found that a mutation that eliminates the ICP4
binding sitein the ICPOpromoterhad nodetectable effecton ICP0 expression after transfer intothe viralgenome.
We reasoned that the contribution of individual ICP4
bind-ing sites might be more readily apparent in the context of
simple promoters lackingresponse elements for other regula-tors.Tothisend,weintroduced ICP4bindingsites into model promoters consisting of an isolated TATA box, reproducing the same spacing as is found in the native ICP4 and ICP0 genes.Thesestructureswerethenincorporated into the intact
HSVgenome and tested foractivity during lytic infection of Vero cells. We found that a single ICP4 binding site placed
upstream ordownstream of the TATAbox strongly reduced
the levelsoftranscripts producedatearlytimespostinfection. Furthermore, this inhibition was partially reversed after the onset of viral DNA replication, leading to a shift to later kinetics ofexpression. These results confirmthat ICP4binding sites in the viral genome can serve as targets for negative 7254
Vol.67, No. 12
on November 9, 2019 by guest
http://jvi.asm.org/
ICP4 BINDING SITES INHIBIT ACTIVITY OF MODEL PROMOTERS 7255
regulation
and
demonstrate that
they
canalso influence
thetemporal regulation
ofaclosely
linked promoter.MATERLALS
AND METHODSViruses and cells.
HSV-1 strain KOS
PAANS
(23)
wasusedthroughout this study. Virus stocks
weregrownand their titers
weredetermined
onmonolayers
of
Verocells.
Verocells
weremaintained
in a minimalessential medium
(GIBCO)
supple-mented with
5% fetal calf
serum,2mM
L-glutamine,
100 Uof
penicillin
G
perml, and
100p.g
of
streptomycin
sulfate
perml. Insertion ofICP4binding
site into model promoters.Syn-thetic
oligonucleotides bearing
ICP4
binding
sites
(purchased
from the Central
Facility
of the
Institutefor
Molecular
Biology
and
Biotechnology,
McMasterUniversity)
wereinserted into
pTKSB
derivatives
containing previously
described
TATA promoters(32) by using
standard methods
(55). pTKSB (70)
contains the HSV-1
tk
genebearing
a200-nucleotide
(nt)
deletion
extending
from +480
to+680;
this deletion
elimi-nates apromoterfor the
overlapping
and
diverging
UL24
gene andis
spanned by
aBam
HIlinker. Constructs
11T
and MLPT(previously
referred
to asUSII
TATA and MLP TATA[32])
bear the
TATAbox
regions
of the
HSV-1
US1
1 and adenovi-rus type 2(Ad2) major
late
promoter(MLP),
respectively,
inserted into the
BamHI site of
pTKSB in the UL24
orienta-tion
(32).
Theseinsertions
regeneratethe
BamHI site
down-stream
of
the TATAbox
andintroduce
aunique
XhoI
site
upstream
of the
TATAbox.
Promoters
bearing
downstream ICP4
binding
sites
wereconstructed
by inserting
adouble-stranded
35-mer,
consisting
of
31 ntspanning
the ICP4
mRNA capsite
and5'-GATC
protruding ends,
into the
unique
BamHI sites
on constructs11T
andMLPT,
regenerating
the
BamHI site downstream of
the insert. The
sequenceof
the topstrand of the
oligonucleo-tide
used was5'-gatcACGCCCCGATCGTCCACACGGA
GCGCGGCTG-3'. The
resulting
constructs weredesignated
1
IT/4
andMLPT/4. Control
promotersbearing
a2-ntdeletion
within the
ICP4
binding
site
were alsomade
(constructs
1
IT/mut4
andMLPT/mut4).
The sequenceof the
topstrandof
the
oligonucleotides
used
toinsert
the mutantsite
was5'-gatcACGCCCCGCGTCCACACGGAGCGCGGCTG-3'.
ICP4
binding
sites
werealso introduced into the
XhoI site
upstream
of the
US1
I TATA boxby using
double-stranded
40-mers
consisting
of
sequencesspanning
the ICP4
mRNA capsite and 5'-TCGA
protruding
ends. The
XhoI site
wasregen-erated
upstreamof the
insert. The
sequencesof
the
topstrands
of the
oligonucleotides
used
were asfollows:
(i)
distal
site,
5'-tcgagGACGCCCCGATCGTCCACACGGAGCGCGGCT
GCCg-3',
and(ii) proximal site, 5'-tcgagCCGAGGACGCCC
CGATCGTCCACACGGAGCGCGGg-3'.
The
resulting
con-structs weredesignated
D4/1
IT and
P4/i
iT.
Control promoters
bearing
a 2-ntdeletion within
theup-stream
ICP4
binding
sites
were also constructedin
asimilar
manner(Dmut4/1
IT
andPmut4/I
IT).
The sequencesof the
top
strands of
theoligonucleotides
used
were asfollows:
(i)
distal
site, 5'-tcgagGACGCCCCGCGTCCACACGGAGCGC
GGCTGCCg-3',
and(ii) proximal site, 5'-tcgagCCGAGGAC
GCCCCGCGTCCACACGGAGCGCGGg-3'.
The sequencesof
allinsertions
wereverified
by
themethod of
Sanger
et al.(57).
Transfer of mutations into the viral genome.
Viral
recom-binantsbearing
inserts of
thetestpromotersweregenerated by
in vivo recombination
following
cotransfection ofinfectious
HSV-1
PAAr5
DNA andlinearized
plasmids containing
theappropriate
sequences(61, 62).
TK-deficient
progeny wereisolated
by plaque
purification
in the presenceof
100pLg
of
5-bromodeoxycytidine
perml,
and recombinantsbearing
the desired insert were identifiedby Southern blot hybridization(66).
RNA extraction. Verocells were infected at a
multiplicity
of 10PFU percell;
whereindicated,
aphidicolin was added at 10,ug/ml
at the time of infection. Total cellular RNA wasextracted by
using
amodified version
of the RNAzol B method(TEL-TEST,
Inc.,Friendswood,
Tex.),
based on themethod
ofChomczynski
and Sacchi(6). Briefly,
1 ml of RNAzol B wasadded
toeach 10-cm-diameter culture dish containing
approx-imately
5 x 106 cells.The
lysate
waspassed through
apipette
several times
tosolubilize
RNAand transferred
to amicro-centrifuge tube.
After the addition of 200 ,ul ofchloroform,
thesuspension
wasincubated for 5min on ice.Samples
werethencentrifuged
at12,000
x gfor
15 min(4°C). Following
anextraction with chloroform, the
RNA wasprecipitated from
the
aqueousphase
with
isopropanol. After
asecond
precipita-tion with
ethanol, the pellet
waswashedthree times with 1:3
diethylpyrocarbonate-treated
0.4 Msodium acetate-ethanol,
once
with
70% ethanol,
and oncewith
95%
ethanol.Pellets
were
dried
under vacuumfor
10 to 15 min andresuspended
indiethylpyrocarbonate-treated
water.Primer extension analysis.
Primer extension
analysis
wasperformed
aspreviously described (64).
Thefollowing
syn-thetic
primers
werepurchased from the Central
Facility
of theInstitute for Molecular
Biology and
Biotechnology,
McMasterUniversity: (i) UL24b, 5'-ACACAACACCGCCTCGACCA
GGGTG-3'; (ii) gD, 5'-CCCCATACCGGAACGCACCACA
CAA-3'
(43); and (iii) 7SL RNA, 5'-AACTTAGTGCGGACA
CCCGATCGGC-3'
(44).
Preparation
of an ICP4-GST fusion protein. Afusion
pro-tein containing the DNA-binding domain of
ICP4(74) fused
toglutathione S-transferase (GST) from Schistosoma
japonicum
(65)
wasprepared
asfollows.
ASacII
fragment
ofpGX58
(67)
containing codons
262 to 490 ofthe
ICP4 gene was insertedinto the
BamHI site in
themultiple cloning site of pGEX-2T
(Pharmacia) by using
aBamHI-Saclladaptor (5'-GATCCTC
CGCGGAG-3', purchased
fromthe Central
Facility of
theInstitute for Molecular Biology
andBiotechnology,
McMasterUniversity)
tomaintain the
properreading frame; the
termi-nation codon
wasprovided by
vectorsequencesdownstream of
the insert. The
sequencespanning the junction between the
GST
andICP4
coding
sequences wasconfirmed by
Sangersequencing (57). The resulting plasmid
wasdesignated
pIGF-17.
Purified ICP4-GST fusion protein
wasprepared
asfollows.
Escherichia
coli HB1OI transformed with pIGF-17
was grown to anoptical density
at 600 nmof
0.9 in 1liter
of TB(55)
at37°C. Cultures
werethen induced
by
theaddition
ofisopropyl-13-D-thiogalactopyranoside
(IPTG)
to afinal concentration of 0.1 mMand incubated
for 2 h at37°C.
Allsubsequent
steps wereperformed
at4°C. Cells were harvested bycentrifugation
at
11,000
xgfor 15min,
washed in 50 mM Trishydrochloride
(pH 8.0),
andthen
lysed
in 25 ml oflysis
buffer[13%
sucrose, 100 mM EDTA(pH 8.0), 20
mMethylene
glycol-bis(,B-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA; pH 8.0),
50 mMTris
hydrochloride (pH 8.0),
250 mMsodiumchloride,
1 mM
phenylmethylsulfonyl fluoride,
and 20 mgof lysozyme
per
ml].
After
15min onice, Nonidet
P-40wasaddedto afinal
concentration of
0.1%,
and thelysate
wasincubated for
afurther
15 min. Cellular debriswas thenpelleted by
centrifu-gation
at150,000
x gfor 1 h in a BeckmanTi5O.2
rotor, and the fusionprotein
waspurified
from the supernatantby
affinity
chromatography
onglutathione-Sepharose
4B(Pharmacia)
perthe
manufacturer's
instructions. Eluted material
wasthen
dialyzed against
2liters
ofdialysis
buffer(100
mMKCl,
20 mMVOL.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
7256 KOOP ET AL.
N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid
[HEPES;
pH
7.9], 10%
glycerol, 0.5 mMphenylmethylsulfonyl fluoride,
and 0.5 mM
dithiothreitol
[DTT]).
DNA affinity chromatography. DNA
affinity
chromatogra-phy
wasperformed by themethod of
Kadonagaand Tjian
(30),
with
the exception that the column was constructed by usingcommercially
preparedCNBr-activated Sepharose
4B(Phar-macia). Oligomers
of ICP4binding sites
wereconstructed by
using
double-stranded30-mers prepared from the
following
oligonucleotides:
5'-GATCCCGATCGTCCACACGGAGC
GCGGCTA-3'
and5'-GATCTAGCCGCGCTCCGTGTGGA
CGATCGG-3'
(purchased
from theCentral
Facility of
theInstitute for
MolecularBiology
andBiotechnology,
McMasterUniversity).
GST-Sepharose-purified
ICP4-GST
wassubjected
to
dialysis (see above)
prior
toapplication
tothe
DNAaffinity
column.
DNA-bound
protein
waseluted
fromthe column with
buffer
Z(25
mM HEPES[pH 7.8],
12.5 mMMgCl,,
1 mM DTT,20%
glycerol,0.1% Nonidet
P-40) containing
1 MKCl
and
dialyzed against
2liters of dialysis
buffer.
Southwestern (DNA-protein) blotting.
Protein
samples
weresubjected
toelectrophoresis through
a13%
N,N'-diallytartar-diamide-cross-linked sodium
dodecyl sulfate
(SDS)-polyacryl-amide
gel
(34)
andtransferred
to anitrocellulose
membraneby electroblotting
at15
Vfor
12 min.The
gel, which retained
some
of the
protein,
wasstained
with Coomassiebrilliant
blue.The
protein affixed
tothe nitrocellulose
wasrenatured
andprobed with the duplex ICP4
binding site
used to generate constructs IIT/4 and MLPT/4, by
themethod of
Michaeletal.
(39). The
DNAprobe
was 3' endlabelled with the
Klenowfragment of
E. coli DNApolymerase
I and[cx_-32P]dCTP.
DNase I footprinting assays. DNase I
footprinting
assayswere
conductedby
using XhoI-PvuII fragments
from theplasmids
used to constructviral recombinants
MLPT/4
and MLPT/mut4, which were 3' end labelled at theXhol site
withthe
Klenowfragment of
E. coli DNApolymerase
I and[0-32P]dCTP.
The
footprinting
procedure
used wasbased
onthat of Galas
andSchmitz
(18)
withsomeof the
modifications
of Brenowitz
etal.
(4).
Binding
reactions
wereperformed
in50-pl1
mixtures
containing
25 mMTris
hydrochloride (pH 7.9),
2 mM
MgCl,, 10% glycerol,
0.5 mMEDTA,
0.5 mMDTT,
0.5pLg
of sheared
herring
spermDNA,
and 0.5 to 20VLg
of the
ICP4-GST fusion
protein. After
preincubation
at roomtem-peraturefor 5
min,
20,000
cpmoflabelled
DNAwasadded
and was incubated at30°C
for 20 min. Theprotein-bound
DNA was thendigested
for 60 s at room temperaturewith
0.2 U of DNase I(Pharmacia) freshly diluted
in DNase Idilution buffer
(50
mM Trishydrochloride
[pH
7.2],
10 mMMgSO4,
1 mM DTT,50% glycerol,
25 mMCaCl,).
DNase Idigestion
wasterminated
by
theaddition
of 100 I of DNase I stop buffer(1% SDS,
100[tg
of tRNA perml,
200 mMNaCl,
20 mMEDTA,
200p.g
of
proteinase
Kperml.After incubation for
10 min at50°C, the
DNA wasethanol
precipitated
and
then loaded on a 7 M urea-8%polyacrylamide
sequencing
gel.
RESULTS
Experimental design. We wishedtodetermine the
effects
ofa strong ICP4
binding site
on theactivity
andtemporal
regulation
of asimple
model promoter located in the HSV genome. To thisend,
wegenerated
two sets of artificial promotersin which
theICP4
binding site from the
ICP4 genetranscription
startsite
waslinked
to a TATAbox(Fig. 1);
ascontrols,
we alsogenerated
corresponding
constructscontain-ing
a mutantICP4binding site lacking
the first two residues of the ATCGTC consensus(52).
One set of constructs contained the ICP4binding site
downstream of the HSV- 1 US11 orAd2MLP TATA
boxes, reproducing
the
arrangementfound
in the nativeICP4
promoter(constructs
IIT/4
andMLPT/4
andtheir
mutant derivatives1lT/mut4
andMLPT/mut4,
respectively).
The other setcontained the
binding
site
ateither
of twopositions
upstreamof the USII
TATAelement;
in onecase, thesite
wasplaced
tomimic the
spacing
inthe
ICPO
promoter(D4/1
IT
andDmut4/1
IT),
while
inthe other
case,thesite
waslocated
5 ntcloser
tothe TATA box(P4/1
IT andPmut4/l
IT).
Wethen used
apreviously
described
system to linkthese
artificial
promoterstothe UL24
genein
the HSV
genome(32).
Each
promoterwasinserted
acrossthe
endpoints
of
a200-nt
deletion
(ASB)
that extends from
-172to +24relative
tothetranscription
startsite of the
largest
UL24transcript
(UL24b),
thereby
placing
UL24b
sequences underthe
control of the
inserted
promoter(32).
The
resulting
structures werethen
transferred into
theUL24
locusof
HSV-1strain
KOSPAAN5
by
DNA-mediated marker rescue and thenselected for
TK-deficient
recombinants
(Materials
andMethods).
Interaction with ICP4. The
oligonucleotides
usedtoassem-ble the ICP4
binding
sites
were chosen to span theminimal
region required
forICP4
binding (52).
Weconfirmed the
ability
of
ICP4tobindtothe site present inconstructMLPT/4
by
DNase Ifootprinting.
Inthis
assay,weused
anICP4-GST
fusion
protein bearing
the
DNA-binding
domain of ICP4
(residues
262 to 490[74])
linked
toGST from
S.japonicum
(65).
Gel
electrophoretic
analysis
of fusion
protein
isolated
by
affinity chromatography
onglutathione-Sepharose
demon-strated
amajor product
withanapparentmolecular
massof
ca. 50kDa,
thepredicted
sizeof
thefull-length
fusion
protein
(Fig. 2A).
Inaddition,
twoless-abundant
larger products
andaprominent
25-kDa band(corresponding
totheexpected
size ofGST)
were observed.Only
the50-kDa
protein
bound alabelled
DNAprobe bearing
anICP4binding
site ina South-westernblotting
assay(Fig. 2B)
andwasselectively
retained
on aDNAaffinity
column
bearing
tandem
arraysof
ICP4binding
sites
(Fig.
2).
In DNase Ifootprinting
assaysusing
a DNAfragment
spanning
theMLPT/4
site,
theICP4-GST fusion
protein
protected
a ca.23-ntregion (Fig. 3);
theposition
ofthe
protected
region
wassimilar
tothat
produced by
intactICP4
on
the native ICP4 promoter
(17),
although
the
sizeof
the
footprint
wasmarginally
smaller(ca.
23
versus ca. 28nt).
Asexpected
(52),
nofootprint
wasobserved
onthecorresponding
construct
bearing
the mutantICP4
binding
site(MLPT/mut4
[Fig.
3]).
Effects ofan ICP4
binding
site located downstream of the TATA box. Vero cellswere infectedwith recombinant
virusesbearing
inserts of theUSI
I TATA element(lIT),
theUS1I
TATAbox linkedto a downstream ICP4binding
site(llT/4),
orthe
USI
I TATA boxlinked to amutant ICP4binding
site
(1I
T/mut4).
Whereindicated,
viral DNAreplication
wasblocked
by
the
addition of
aphidicolin
atthe time of
infection.
Total cellular
RNAwasthen extracted
at 6and
12 hpostin-fection and examined
by
primer
extension with aprimer
complementary
toresidues +91 to +67 of thenative UL24b
transcript (Fig. 4).
Ascontrols,
the RNAsamples
werealso
scored for HSV
gD
mRNAandcellular
7SL RNAby
primer
extension.
Inagreement
with
previous
work(32),
constructlIT
gave rise to a ca. 53-ntprimer
extension
product,
thepredicted
size fortranscripts
initiating
30ntdownstreamof the
inserted TATA element. As
expected,
constructs I1T/4
and IIT/mut4
gaverise
tolarger
extensionproducts
(ca.
85nt),
reflecting
theinsertion
ofoligonucleotides bearing
the ICP4binding
sites.Comparison
ofconstructs11T/4
and11
T/mut4
indicated
that thefunctional
ICP4binding
sitepresent
in 1IT/4
alteredUL24b
expression
in three ways.First,
the levels ofUL24b
transcripts
inconstruct 1IT/4
were reducedmore thanJ VlIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
ICP4 BINDING SITES INHIBIT ACTIVITY OF MODEL PROMOTERS 7257
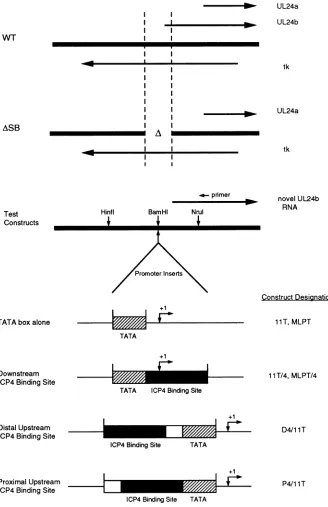
I I
I
-L
I I
I WT
UL24a UL24b
i
I I
I I
I I
I I
I I I
I I
mAnm
I I
I I
I I
-
prmer
Hinf BamHl Nrul
4f
i
J,
romoterInse
tk
UL24a
tk
novelUL24b RNA
Construct Designation
TATA box alone
Downstream ICP4Binding Site
DistalUpstream ICP4BindingSite
TATA
+1
TATA ICP4BindingSite
ICP4Binding Site TATA
ProximalUpstream
ICP4Binding Site
Km
L
P4/11 TICP4Binding Site TATA
FIG. 1. Experimental design. The ASB deletion removes 200 nt of tk coding sequences and the promoter for the larger UL24 transcript (UL24b)and isspanned byaBamHI linker.Oligonucleotides bearingtest promotersequencesconsisting ofanICP4binding site locatedupstream
or downstream of a TATA box were inserted into the intact HSV-1 genome across the ASB deletion, oriented to drive the UL24 gene.
Correspondingconstructsbearingmutantbinding siteswerealsomade.Theactivity ofeachtestpromoterwasthenscoredbyassayingthe levels
of novel UL24btranscripts by primer extension.Thediagrams oftestpromotersshown in the bottom portionare notdrawnpreciselyto scale.
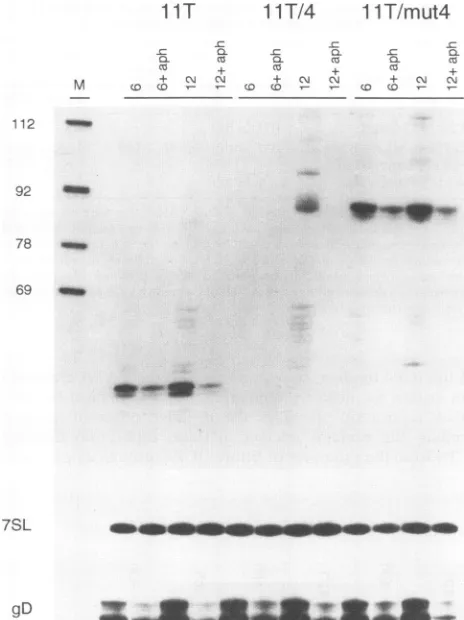
100-fold at 6 hpostinfection relative to 1lT/mut4 (Fig. 4and
Table 1). Second, this negative effectwaslargelyovercome by
12 h postinfection, such that IT/4 displayed only a three to
fourfold reduction relativeto 1lT/mut4.
Thus,
the ICP4bind-ing site shifted the kinetics oftranscript accumulationtolater
timespostinfection. Third, the binding site conferredagreatly
increased dependenceonviral DNAreplication. Thus,at 12 h
postinfection,
transcripts arising from construct IT/4 werereduced more than 100-foldwhen infections werecarried out
in the presence of aphidicolin; in contrast, lT/mut4
tran-scripts were reduced only 4 to 7-fold (Table 2). Taken in
combination, these findings demonstrate that addition of the
ICP4 binding site changed the kinetics of UL24b expression fromleaky late to true late.
Todeterminewhether these effectsdepended onthe nature
of the TATA element, we analyzed analogous constructs ASB
Test Constructs
11T, MLPT
1 1T/4,MLPT/4
D4/1 1T I
Vol.. 67, 1993
I+1
Ion November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.138.466.77.584.2]7258 KOOP ET AL.
A
M
1
2
3
B
M
1
2_>40P4
"-I.._.
_. .. Ag
-30ip
FIG. 2. Analysis ofthe ICP4-GST fusion protein produced from plasmidpIGF-17. Protein samples were subjected to electrophoresis
through an SDS-13% polyacrylamide gel and then transferred to nitrocellulose. (A) Coomassie brilliant blue staining pattern of pro-teins retained in the gel after transfer; (B) propro-teins bound to nitrocel-lulose reacted with a 32P-labelled oligonucleotide duplex bearing an ICP4 binding site. Lanes 1, ICP4-GST purified by DNA affinity chromatography; lanes 2, ICP4-GST purified by chromatography on glutathione-Sepharose; lanes 3, GST purified by chromatography on glutathione-Sepharose; lanes M, protein markers (Amersham); sizes in kilodaltons are shown on the left.
bearing the much stronger Ad2 MLP TATA box
(constructs
MLPT, MLPT/4,
and MLPT/mut4).
In this case aswell,
addition of the ICP4 binding site led to greatly
reduced
transcript levels at 6 hpostinfection, and the imposition ofan
increased
dependence on viral DNA replication (Fig. 5;Tables
1 and 2). Therefore, we conclude
that
the
observedeffects
observed are independent of the sequence of TATA
box.
In particular, these data establish that the effects observed with construct 11T/4 are not due to the weak promoteractivity
of the USI1 TATA box (construct MLPT is at least as active as the native UL24b promoter [32]).Effects of upstream ICP4 binding sites. The
preceding
results demonstrated
anegative effect
of an ICP4 bindingsite
placed over the transcription start site-the arrangement found in the native ICP4 promoter. We next examined con-structsbearing the bindingsitelocated upstream of the TATA box to mimic the spacing found in
the ICPO
promoter(con-structs D4/llT and its mutant
derivative, Dmut4/1lT
[33]).
The results demonstrated
that the upstream binding site had asimilar,
but lesspronounced,
effect (Fig. 6). Thus, levelsof
UL24b
RNAarising
fromD4/1
iT werereduced
more thansixfold
at 6h
postinfection, and
two- to threefold at 12 hpostinfection,
relative to the construct bearing the mutatedbinding site (Table 1).
In addition, at 12 h postinfection,ptg
ofICP4-GST igofICP4-GST00t}
++ LOCMJL) 0 0 CD C) LC
+ + LOOC\x,LOs° CD
iD<~ ,
WI_ _ _
_ll ._rm .z....
...WW
lo.jr ... .. ....
W_
- x_m_-40
.._ _~~Eu
Wildtype
Mutant
FIG. 3. DNaseIfootprintingof the ICP4 binding site inconstructMLPT/4.XhoI-PvuIIfragments spanning the ICP4 binding site inconstructs
MLPT/4 and MLPT/mut4 (3' end labelled atthe XhoI end)were incubated with 0, 0.5, 1.25, 2.5, 5, 10, or20 p.g of ICP4-GST (purifiedby
chromatographyonglutathione-Sepharose)and then treated briefly withDNaseI.Digestion productswereseparated by electrophoresis through
a7 Murea-8% polyacrylamidegel. The region of the fragment protected by theICP4-GSTfusion product is indicated by the bracketonthe left.
Wildtype, MLPT/4; mutant, MLPT/mut4. Lanes G, A+G, T+C, and C, base-specific chemical cleavage products of the same end-labelled
fragments (36).
200
97.4
69
46
30
L.
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.64.304.75.202.2] [image:5.612.151.475.344.665.2]ICP4 BINDING SITES INHIBIT ACTIVITY OF MODEL PROMOTERS 7259
1
1T/4
1 1 T/mut4
c-a.o=a 0
= CL _E nL -E a
CL OLn CLQ C;s
(b + C + 0 +
+ \ C'J + ('I N + C\U (N
M E(C0 c0 (0 (0(0
1 12
-MLPT
_f-C- aco
CZ +
M eD QO C c
MLPT/4
MLPT/mut4
cl CIO El C
CZ + 0 + + C\M CMN + (N\ c\l
CO (d - - Cf (D0
112
92
0
VWV
78
69
-rn--
n----gD W5
FIG. 4. Effects ofan ICP4bindingsiteplaced downstreamof the US11TATAelement. Vero cellswereinfected with 10 PFUpercellof the indicated viralrecombinants,andtotal cellular RNAwasharvested
at6 and 12 h postinfection. Samples (20
jig)
were then assayed forUL24transcripts by primerextension withaprimercomplementaryto
residues +91to +67of thenative UL24btranscript.Sampleswerealso
scored for7SLandgDRNAsby primer extension,by using0.5 and5 jigofRNA,respectively. Whereindicated, 10,ugofaphidicolin(aph)
permlwasaddedatthe time of infection andmaintainedcontinuously. Molecularweightmarkers (M)were3-end-labelled HpaIIfragments
ofpBR322. Markerfragmentsizes(in nucleotides) are shownon the left.
transcripts arising from D4/1ITwere reducedover20-foldby blocking DNA replication, while those arising from Dmut4/
lITwere reduced only 5-to7-fold (Table 2).
DiDonato and Muller (1) found that ICP4 and TFIID are
locatedon opposite helical faces of the DNA when bound at theirrespective sitesinboth theICP4 and the ICPOpromoters;
furthermore,
chemical probing suggested that ICP4 binding alteredthe helicalgeometryofthe TATAregion. On the basisof these observations, these investigators proposed that the stereospecificorientation of ICP4 andTFIID might be impor-tant to ICP4-induced repression of IE gene expression. To examine the importance of the helical orientation of bound ICP4 with respect toTFIID,we constructed an additionalset of viral recombinants in which the ICP4 binding site was
positioned 5 bp (approximately 1/2 helical turn) closer tothe TATAbox(P4/liT and
Pmut4/1lT).
We found that this shiftdid not alter the effect of the ICP4 binding site relative to construct D4/1IT(Fig. 6; Tables I and 2), suggestingthatthe
helical orientation ofICP4with respect tothe TATAbinding protein is notcritical for the effects thatwehave observed.
7SL
[image:6.612.58.290.74.384.2]gD _
.a
FIG. 5. Effectsofan ICP4 bindingsite placeddownstream of the Ad2 MLP TATA element. Vero cellswere infected with 10 PFUper
cell ofthe indicated viralrecombinants,and RNAsamplesextracted6 and 12 hpostinfectionwerescored forUL24b, 7SL,andgDRNAsas
described in thelegendtoFig.4.Whereindicated,10 p.gofaphidicolin (aph) per ml was added at the time of infection and maintained continuously. Molecular weight markers (M) were 3-end-labelled
HpaII fragments ofpBR322. Marker fragmentsizes (in nucleotides)
areshown onthe left.
DISCUSSION
Theexperiments describedinthispaperdemonstrate thatan ICP4binding site placed eitherupstreamor downstream ofa
simpleTATA boxpromotercan alterboththe levels and the
kinetics ofgene expressionduring HSV infection. Thus,
addi-tion ofan intact binding site reduced expression relative to control constructs, and this inhibitory effect wassubstantially
more pronounced at early times postinfection. In addition,
inhibition was exaggerated when viral DNA replication was
blocked withaphidicolin. Wetake these resultstoindicatethat
ICP4 bound in closeproximitytothe TATAbox blocksgene
expression by interferingwith some aspect of promoter func-tionand that DNAreplicationpartially bypassesthiseffect;the netoutcome istoshift thekinetics ofexpressiontolatertimes
postinfection. How does DNA-bound ICP4 inhibit promoter
function?Although it has beenproposed that repressionmay
involvesterichindranceorconformationalchangesinduced in
11T
92 _m
78 _
69 _
7SL
36
-VOL.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.317.552.74.436.2]7260 KOOP ET AL.
TABLE 1. Inhibition conferred by ICP4 binding site
Inhibition (fold) relative to mutant site" Virus pair
6h 12 h
1lT/4, IIT/mut4 >100, >100 3.8, 3.0 MLPT/4, MLPT/mut4 >100, >100, >100, 43 3.7, 1.9, 4.4,8 D4/1 IT, Dmut4/11T 22, 6.0, 7.6 2.6, 2.2, 2.3 P4/lIT, Pmut4/1lT 10, 6.5, 11 2.9, 2.2, 2.8
aGels were exposed to preflashed KodakXAR-5 film with an intensifier screen,and relative signal intensities were quantified by microdensitometry. The degree of inhibition conferred by theICP4 bindingsite relative to the mutant site wascalculatedbydividing the signal obtained for the mutant site by that obtained for the intact site. In some cases, the signal from the construct bearing the wild-type site couldnot be detected, representing an inhibition of more than
100-fold relative to the control construct. For eachvirus pair, the results of severalexperiments are listed.
the TATA region (11, 40), the
existence of a mutant form of
ICP4
that binds DNA but fails to autoregulate (47) suggests
that
anadditional function of ICP4 other than
simple
DNAbinding is required. It seems unlikely that this additional
function involves
adirect interaction
with the
TATAbinding
protein, because
ourresults
show that the
helical orientation
11T
D4/11T
-~ ~ ~ ~ ~ ~
Q ~C Q U
m + U +
+ CU CM + CM CU
[image:7.612.66.305.84.155.2]M (00 (0(
TABLE 2. Inhibitionby aphidicolin observedat 12 hwith ICP4
bindingsite andmutantbindingsite
Inhibition(fold) by aphidicolin'atfollowing
Viruspair binding site:
ICP4 Mutant ICP4
I1T/4, 1I T/mut4 >100, > 100 6.8, 4.1 MLPT/4,MLPT/mut4 >100, >100, >100, >100 24, 8.5, 16,69
D4/1lT, Dmut4/IIT 44,21,24 5.8, 5.3,6.7
P4/lIT, Pmut4/11T 34, 57, 20 3.8,7.7, 6.4 "Gels were exposed to preflashed Kodak XAR-5 film with an intensifier
screen,and relativesignalintensities werequantified by microdensitometry.Fold inhibitionby aphidicolinwascalculated by dividingthesignal obtainedat12h withoutaphidicolin bythat obtained at 12 h in the presence of aphidicolin. In some cases signals could not be detected in the presence of aphidicolin, representingadecreaseofmorethan100-fold.Foreachvirus pair, the resultsof severalexperimentsareshown.
of the
ICP4
binding site with
respect tothe TATA element is
not
critical
fornegative
regulation.
Themechanism
by which
DNA
replication alleviates the negative
effect of anICP4
binding site remains
unclear:perhaps replication dislodges
ICP4 from
thepromoter
ortitrates
ICP4through amplification
Dmut4/1
1TP4/1
l T Pmut4/1 1 TQ.Z5 O. .C Q.
CU + X + X +
+ (V (V + (VJ N + N CU
( ( C (0D - CD(0
78
69
p
__r.
FIG. 6. Effects of ICP4 binding sites placed upstream of the
USlI
TATAelement. Vero cellswere infected with 10 PFU per cell of theindicatedviralrecombinants, andRNAsamplesextracted 6 and12hpostinfection were scoredfor UL24b,7SL, and gD RNAsasdescribed inthe
legendtoFig. 4. Where indicated, 10
jig
of aphidicolin(aph) per ml was added at the time of infection and maintainedcontinuously.Molecularweightmarkers (M) were
3-end-labelled HpaII
fragments of pBR322. Marker fragment sizes(in nucleotides)areshownonthe left.1w
36
_
7SL
gD
*j-
4000.1
w~~~~~~~Ado _- _NMM
-J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.321.562.94.171.2]ICP4 BINDING SITES INHIBIT ACTIVITY OF MODEL PROMOTERS 7261
of
binding sites; alternatively, replication-induced
conforma-tional
changes
in
the
promotermight
allow
transcription
evenin the presence
of bound ICP4. Further
experiments arerequired
todistinguish
betweenthese
possibilities.
The
ability
of anICP4
binding site
tostrongly
reduceexpression
from asimple
TATA promoter atearly times
postinfection
is consistent with
findings
that the ICP4binding
sites
inthe native
ICP4 and ICP0 promoters arerequired for
ICP4-mediated
repression
intransfection
assays(20, 51, 52)
and the
observation that the site
atthe ICP4
genetranscription
start
site
serves as a targetfor
negativeregulation during
infection
(38).
However,
Everettand Orr
(15) found that
deleting
the ICP4
binding
site from the native ICP0
promoterhad
noeffect
onICP0
expression during
lytic
HSV
infection.
We suggest
that three factors
might
contribute
tothe minimal
effect
ofICP4
binding
tothenative
ICP0
promoter.First,
wefound that
abinding
site
placed
upstreamof the
TATAbox
tomimic the
arrangementin the
ICPO
promoterhad
aless
pronounced
inhibitory
effect
than a siteplaced
overthe
transcription start site, as in the
ICP4
promoter.Second,
transactivation
by
Vmw65strongly boosts
IE genetranscrip-tion
(2, 5, 48)
before
theaccumulation of
ICP4,
and it is
possible
that these
preexisting transcription complexes
par-tially
inhibit
binding
of
ICP4 tonative
IE promoters.Third,
ourdata demonstrate that the
negative
effects of ICP4
binding
sites
arepartially
reversed after the
onsetof viral
DNAreplication.
These considerations
suggestthat
downregulation
of
IE promotersthrough
ICP4
binding
sites
might
assumegreater
importance
under conditions of
limiting
Vmw65,
par-ticularly prior
to the onsetof
viral
DNAreplication,
for
example, during
theearly
stagesof
infection
atlow
multiplicity
or
following
reactivation from
latency.
Asimilar
argumentmight
alsoexplain why mutating
the
ICP4binding
sites in
thegD
promoter had noobvious effect
on thelevels of
gD
expression during
infection(63). Thus,
the ICP4
binding
site
mostproximal
to thegD
transcription
startsite
begins
at ca.-
90,
perhaps
toofarfrom the
corepromotertoexert astronginhibitory effect
in the presenceof
viralactivators and
DNAreplication.
Our
finding
thatICP4
binding
sites
canconfer
agreatly
increased
requirement
for viral
DNAreplication
onsimple
TATA promoters
raises the
possibility
thatthis mechanism
contributes
to thereplication dependence
of atleast
some bonafide HSV
L promoters.According
tothis
hypothesis,
ICP4
binding
sites located
inthe
vicinity of
L promotersmight
act asnegative
controlelements that restrict
expression
atearly
times
postinfection.
Inthis context,it is
interesting
to notethat
our data demonstrate that themagnitude
of the
replication
requirement imposed by
anICP4binding
sitecanvarywith the
position
of
thesite, illustrating
that this
relatively simple
mechanism
could
give
rise
to thewide
range ofreplication
requirements
that
isobserved
amongHSV
L promoters. Thehypothesis
thatDNA-bound
ICP4serves as anegative
regula-torof
nearby
L promotersis consistent with the observation of
Arsenakis
etal.
(1)
that ICP4
caninhibit
expression
of the
E/L
gD
genein
stably
transfected cell
lines;
in
addition,
earlier
work fromour
laboratory
has shownthe
trueLUS1I
promoter contains sequences that are able toimpose
anincreased
requirement
for
DNAreplication
on asimple
TATApromoter(32).
However, this
hypothesis
leads
to aninteresting
paradox:
ICP4
is
alsoarequired
activator of
Lgeneexpression,
andthe available evidence indicates that itmustbind DNA in orderto servethis function(9, 10, 45, 46, 59). Perhaps
DNAreplication
converts DNA-bound ICP4 froma local repressor intoalocalactivator; alternatively,
it ispossible
that L promoters can beinduced
only by
ICP4 molecules thatareboundtodistal sites.According to the latter hypothesis, DNA binding may serve to
concentrate ICP4
in thevicinity
of the viralgenome,
ratherthan
directly targeting individual
Lpromoters for activation.
We
currently
seek todirectly
assess theregulatory
role of ICP4binding sites in HSV
Lpromoters.
ACKNOWLEDGMENTS
This research was funded by the Medical Research Council of Canada. J.R.S. isaTerry Fox Senior Scientist of the NCI(C).
REFERENCES
1. Arsenakis, M., G. Campadelli-Fiume, and B. Roizman. 1988. Regulation of glycoprotein D synthesis: does c4, the major regulatory protein of herpes simplex virus 1, regulate late genes bothpositively and negatively? J. Virol. 62:148-158.
2. Batterson, W., and B. Roizman. 1983. Characterization of the herpes simplex virion-associated factor responsible for the induc-tion of oc genes. J.Virol. 46:371-377.
3. Beard, P., S. Faber,K. W.Wilcox,and L. I.Pizer. 1986. Herpes simplex virus immediate early infected cell polypeptide 4 bindsto DNA and promotes transcription. Proc. Natl. Acad. Sci. USA
3:4016-4020.
4. Brenowitz, M., D. F. Senear, andR. E. Kingston. 1989. DNaseI footprinting analysisofprotein-DNA binding.In F. M.Ausubel,
R.Brent, R. E.Kingston, D. D. Moore, J. G.Seidman,J. A.Smith,
and K. Struhl (ed.), Current protocols in molecular biology.
GreenePublishing Associates andWiley-Interscience, NewYork. 5. Campbell,M. E.M., J.W.Palfreyman,andC.M.Preston. 1985. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsibleforstimulation of immediate earlytranscription. J. Mol. Biol. 180:1-19.
6. Chomczynski, P., andN.Sacchi. 1987.Single-step methodofRNA isolation by acidguanidinium thiocyanate-phenol-chloroform ex-traction. Anal. Biochem. 162:156-159.
7. DeLuca,N.A.,A. M.McCarthy,and P. A.Schaffer. 1985.Isolation and characterization ofdeletion mutantsofherpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4.J. Virol. 56:558-570.
8. DeLuca,N.A.,and P.A. Schaffer. 1985.Activation of
immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes simplex virus type I protein ICP4. Mol. Cell. Biol. 5:1997-2008.
9. DeLuca, N. A., and P. A. Schaffer. 1987. Activities of herpes
simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense
peptides. Nucleic Acids Res. 15:4491-4511.
10. DeLuca, N.A., and P. A. Schaffer. 1988. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4.J. Virol.62:732-743.
11. DiDonato, J. A.,and M. T. Muller. 1989. DNAbinding and gene
regulation by the herpes simplex virus type 1 protein ICP4 and involvement of theTATAelement.J.Virol.63:3737-3747. 12. Dixon,R. A.F.,and P. A.Schaffer. 1980. Fine-structure mapping
and functional analysis of temperature-sensitive mutants in the gene encoding the herpes simplex virus type 1 immediate-early
protein VP175. J. Virol. 36:189-203.
13. Everett,R.D.1984.Transactivation oftranscription by herpesvirus products: requirement for two HSVI immediate-early polypep-tides formaximumactivity. EMBO J.3:3135-3141.
14. Everett,R. D. 1987. The regulation of transcription ofviral and cellular genes by herpesvirus immediate-early gene products. Anticancer Res.7:589-604.
15. Everett,R. D.,andA. Orr. 1991.TheVmwl75bindingsitein the IE-1promoterhasnoapparentrole intheexpression of Vmwl 10 during herpes simplexvirus type 1 infection. Virology 180:509-517.
16. Faber, S.W.,and K. W.Wilcox. 1986. Association of theherpes simplex virus regulatory protein ICP4 with specific nucleotide sequences inDNA.Nucleic AcidsRes. 14:6067-6083.
Vol.67, 1993
on November 9, 2019 by guest
http://jvi.asm.org/
7262 KOOP ET AL.
17. Faber, S. W., and K. W. Wilcox. 1988. Association of herpes simplex virus regulatory protein ICP4 withsequencesspanning the ICP4genetranscription initiation site. Nucleic AcidsRes.
14:555-570.
18. Galas, D., and A. Schmitz. 1978. DNase footprinting: a simple
method for the detection of protein-DNA binding specificity. Nucleic Acids Res. 5:3157-3170.
19. Gelman,I. H., and S.Silverstein. 1985. Identification of
immedi-ate earlygenes from herpes simplexvirus that transactivate the virus thymidine kinasegene.Proc.Natl.Acad. Sci.USA 82:5265-5269.
2(0. Gelman,I.H., andS. Silverstein. 1987. Dissection of
immediate-early genc promoters from herpes simplexvirus: sequences that respond to the virus transcriptional activators. J. Virol. 61:3167-3172.
21. Gelman, I. H., and S. Silverstein. 1987. Herpes simplex virus
immediate-earlypromoters areresponsive tovirus and cell trans-acting factors.J. Virol. 61:2286-2296.
22. Godowski,P. J., and D. M.Knipe.1986. Transcriptional control of herpesvirusgene expression:genefunctions required for positive
and negative regulation. Proc. Natl. Acad. Sci. USA 83:256-260. 23. Hall, L. M., K. G. Draper, R. J. Frink, R. H.Costa, and E. K.
Wagner. 1982. Herpes simplex virus mRNA species mapping in EcoRI fragment 1. J.Virol. 43:594-607.
24. Homa,F. L., J.C. Glorioso, and M.Levine. 1988. Aspecific 15-bp TATA box promoter element is required for expression of a
herpes simplex virustype I lategene. Genes Dev. 2:40-53. 25. Homa, F. L., T. M. Otal,J. C. Glorioso, and M. Levine. 1986.
Transcriptionalcontrolsignalsofaherpes simplexvirustype1late
(y2)genelie within bases -34to+124 relativetothe5'terminus
of the mRNA. Mol. Cell. Biol. 6:3652-3666.
26. Honess, R. W., and B. Roizman. 1974. Regulationofherpesvirus macromolecularsynthesis.I.Cascaderegulationof thesynthesisof
threegroupsof viralproteins.J. Virol. 14:8-19.
27. Honess,R.W., and B.Roizman. 1975. Regulationofherpesvirus macromolecular synthesis: sequential transition of polypeptide synthesis requiresfunctional viralpolypeptides.Proc. Natl.Acad. Sci. USA 72:1276-1280.
28. Imbalzano, A. N., A. A. Shepard, and N. A. DeLuca. 1990. Functional relevance ofspecificinteractions betweenherpes sim-plexvirustype 1 ICP4 andsequencesfrom the
promoter-regula-torydomain of the viralthymidinekinasegene.J. Virol. 64:2620-2631.
29. Johnson, P.A., and R. D. Everett. 1986. The control ofherpes simplexvirus type 1 late gene expression: aTATA box/cap-site region is sufficient for fully efficient regulated activity. Nucleic Acids Res. 14:8247-8264.
30. Kadonaga, J. T., and R. Tjian. 1986. Affinity purification of
sequence-specific DNA binding proteins. Proc. Natl. Acad. Sci.
USA 83:5889-5893.
31. Kattar-Cooley, P.,and K. W. Wilcox. 1989. Characterization of the DNA-binding properties ofherpes simplexvirusregulatory
pro-tein ICP4. J. Virol. 63:696-704.
32. Kibler,P.K., J. Duncan,B. D.Keith,T.Hupel,andJ.R.Smiley. 1991.Regulationofherpes simplexvirustruelategeneexpression:
sequencesdownstream from theUS] TATA box inhibit expres-sion fromanunreplicated template. J. Virol. 65:6749-6760. 33. Kristie, T. M., and B. Roizman. 1986. (4, the major regulatory
proteinofherpes simplextype 1,isstablyandspecifically associ-ated withpromoter-regulatorydomains ofagenesand of selected other viralgenes.Proc. Natl. Acad. Sci. USA 83:3218-3222. 34. Laemmli, U. K. 1970.Cleavage ofstructuralproteins during the
assembly of the head of bacteriophage T4. Nature (London) 227:680-685.
35. Mavromara-Nazos, P., and B. Roizman. 1989. Delineation of regulatory domains ofearly (,B) and late(y2) genesby construc-tion of chimeric genes expressed in herpes simplex virus 1 ge-nomes. Proc. NatI. Acad. Sci. USA 86:4071-4075.
36. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labelled DNA with base-specific chemical cleavages. Methods Enzymol. 65:499-525.
37. McGeoch, D.J.,M.A.Dalrymple,A.J.Davison,A.Dolan,M. C. Frame,D.McNab,L.J. Perry, J.E.Scott,and P.Taylor.1988. The
complete nucleotide sequence of the
long unique region
in the genome ofherpes simplexvirustype 1. J. Gen. Virol.69:1531-1574.
38. Michael, N., and B. Roizman. 1993. Repression of the herpes
simplex
virus I cx4 geneby
its geneproduct
occurswithin thecontextof the viralgenomeand is associated with all three cognate sites. Proc.Natl.Acad. Sci. USA 90:2286-2290.
39. Michael, N.,D.Spector,P.Mavromara-Nazos,T. M.Kristie,and B. Roizman. 1988. The
DNA-binding properties
of themajor
regulatory
protein
4 ofherpes simplex
virus. Science 239:1531-1534.40. Muller,M. T. 1987. Bindingof the herpes simplexvirus
immedi-ate-earlygene
product
ICP4to itsowntranscription
startsite. J. Virol. 61:858-865.41. O'Hare, P.,andG. S.Hayward.1985.Evidence foradirect role for both the 175,000-and
110,000-molecular-weight immediate-early
proteinsofherpessimplexvirus in the transactivation of
delayed-earlypromoters.J.Virol. 53:751-760.
42. O'Hare, P.,andG. S. Hayward. 1985.Three
trans-acting
regula-toryproteins of herpes
simplex
virus modulateimmediate-early
gene
expression
in apathway
involving positive
andnegative
feedbackregulation.J.Virol. 56:723-733.43. Panning, B.,andJ.R. Smiley. 1989.
Regulation
of cellulargenes transducedbyherpes simplex
virus. J. Virol. 63:1929-1937. 44. Panning, B., and J. R. Smiley. 1993. Activation of RNApoly-merase III
transcription
of human Alurepetitive
elementsby
adenovirustype5:requirement
for the Elb58-kilodaltonprotein
andtheproducts
ofE4 openreading
frames 3 and6.Mol. Cell. Biol. 13:3231-3244.45. Paterson,T.,andR. D.Everett. 1988.Mutational dissection of the HSV-1
immediate-early protein
Vmwl75 involved in transcrip-tional transactivation andrepression.
Virology
166:186-196. 46. Paterson,T.,and R. D. Everett. 1988.Theregions
of theherpes
simplex
virustype1immediateearly
protein
Vmwl75required
for sitespecific
DNAbinding closely
correspond
tothose involved intranscriptional regulation.
Nucleic Acids Res. 16:11005-11025. 47. Paterson, T.,V.G.Preston,and R.D. Everett. 1990.Amutantofherpes
simplex
virustype 1immediateearly
polypeptide
Vmw175 which bindstothecapsite of itsownpromoterin vitrobut failstoautoregulate
invivo. J. Gen. Virol. 71:851-861.48. Post,L. E.,S.
Mackem,
andB.Roizman. 1981.Regulation
of ce genes ofherpes
simplex
virus:expression
of chimeric genesproduced by
fusion ofthymidine
kinase with x gene promoters. Cell 24:555-565.49. Preston,C. M. 1979.Control of
herpes
simplex
virus type 1 mRNAsynthesis
incells infected withwild-type
virusorthe temperature-sensitive mutanttsK. J.Virol. 29:275-284.50. Quinlan,M.P.,andD. M.Knipe. 1985. Stimulation of
expression
of aherpes simplex
virusDNA-binding
protein by
two viral functions. Mol. Cell. Biol. 5:957-963.51. Resnick, J.,B. A.Boyd,and M. L.
Haffey.
1989. DNAbindingbythe
herpes simplex
virus type 1 ICP4protein
isnecessaryfor the efficient downregulation
of theICPO promoter.J.Virol. 63:2497-2503.52. Roberts, M. S., A. Boundy, P.
O'Hare,
M. C.Pizzorno,
D. M.Ciufo,
and G. S.Hayward.
1988. Direct correlation between anegative autoregulatory
response element at the cap siteof theherpes
simplex
virus type 1 IE175(x4)
promoter and aspecific
binding
site for theIE175(ICP4)
protein.
J. Virol.62:4307-4320. 53. Roizman, B.,and A. E.Sears.1990.Herpes
simplex
viruses and theirreplication.
InB. N.Fields and D. M.Knipe (ed.), Virology,
2nd ed.Raven
Press,
NewYork.54. Romanelli, M.
G.,
P.Mavromara-Nazos,
D.Spector,
and B. Roizman. 1992. Mutationalanalysis
ofthe ICP4binding
sites in the 5' transcribednoncoding
domains of theherpes
simplex
virus1
UL49.5
y2gene. J. Virol. 66:4855-4863.55. Sambrook, J., E. F.
Fritsch,
and T. Maniatis. 1989. Molecularcloning:
alaboratory
manual,2nd ed. ColdSpring
Harbor Labo-ratory,ColdSpring Harbor,
N.Y.56. Sandri-Goldin, R. M., andG. E. Mendoza. 1992. A
herpesvirus
regulatory
protein
appearsto actposttranscriptionally by
affecting
mRNA
processing.
GenesDev.6:848-863.57. Sanger, F.,S.
Nicklen,
andA. R.Coulson. 1977.DNAsequencing
J. VIROL.on November 9, 2019 by guest
http://jvi.asm.org/
ICP4 BINDING SITES INHIBIT ACTIVITY OF MODEL PROMOTERS 7263 with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA
74:5463-5467.
58. Shepard,A.A., and N. A. DeLuca.1991. A second-siterevertantof a defective herpes simplex virus ICP4 protein with restored regulatory activities and impaired DNA-binding properties. J. Virol. 65:787-795.
59. Shepard, A. A., A. N. Imbalzano, and N. A. DeLuca. 1989. Separation of primary structuralcomponentsconferring autoreg-ulation, transactivation, andDNA-bindingproperties tothe her-pessimplex virus transcriptional regulatory protein ICP4. J. Virol. 63:3714-3728.
60. Silver, S.,and B. Roizman. 1985. -y2-thymidine kinase chimeras are identically transcribed but regulated as y2 genes in herpes simplex virusgenomesandas , genesin cellgenomes.Mol. Cell. Biol. 5:518-528.
61. Smiley, J.R.1980. Construction in vitro andrescueofathymidine kinase-deficient deletion mutation of herpes simplex virus. Nature
(London)285:333-335.
62. Smiley, J. R.,B.S. Fong,andW.-C.Leung. 1981. Construction of a double-jointed herpes simplex virus DNA molecule: inverted repeats are required for segment inversion and direct repeats promotedeletions. Virology 113:345-362.
63. Smiley, J. R.,D.C. Johnson, L. I. Pizer, and R. D. Everett. 1992. The ICP4binding sites in the herpes simplex virustype1 glycop-rotein D (gD) promoter are not essential for efficient gD tran-scription during virus infection. J. Virol. 66:623-631.
64. Smiley, J. R., C. Smibert, and R.D.Everett.1987.Expression ofa cellulargenecloned in herpes simplex virus: rabbit beta-globin is regulated as anearly viral gene in infected fibroblasts. J.Virol. 61:2368-2377.
65. Smith,D.B.,K. M.Davern,P.G.Board,W. U.Tiu, E. G.Garcia, and G. F. Mitchell. 1986. Mr 26,000 antigen of Schistosoma
japonicumrecognized by resistant WEHI 129/J mice isaparasite glutathione S-transferase. Proc. Natl. Acad. Sci. USA 83:8703-8707.
66. Southern, E. 1975. Detection ofspecific DNAsequences among DNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
67. Stow,N.D.,and E. C.McMonagle.1982.Eukaryotic viralvectors, p. 199-204. Cold Spring Harbor Laboratory Publications, Cold Spring Harbor, N.Y.
68. Tedder, D. G., R. D. Everett,K.W.Wilcox, P. Beard, and L. I. Pizer. 1989.ICP4-binding sitesinthe promoterandcodingregions ofthe herpes simplexvirus gDgenecontributetoactivation ofin vitrotranscription by ICP4.J. Virol. 63:2510-2520.
69. Tedder, D. G., and L. I. Pizer. 1988. Role for DNA-protein interactioninactivation of the herpes simplex virus glycoproteinD gene.J. Virol. 62:4661-4672.
70. Varmuza, S. L., and J. R. Smiley. 1985. Signals for site-specific cleavageofHSV DNA:maturation involvestwo separatecleavage events at sites distal to therecognitionsequences.Cell41:793-802. 71. Wagner, E. K. 1985.Individual HSV transcripts: characterization ofspecificgenes, p.45-104. In B. Roizman (ed.), The herpesvi-ruses, vol. 3.Plenum Press,NewYork.
72. Watson, R.J., andJ. B.Clements. 1980.A herpessimplexvirus type1 functioncontinuously required for early and late virus RNA synthesis. Nature (London) 285:329-330.
73. Weinheimer, S. P., and S. L. McKnight. 1987.Transcriptional and post-transcriptional controls establish the cascade ofherpes sim-plex virus protein synthesis. J. Mol. Biol. 195:819-833.
74. Wu, C. L., and K. W.Wilcox. 1990.Codons 262to490from the herpes simplex virus ICP4 gene are sufficient to encode a se-quence-specific DNA bindingprotein.Nucleic AcidsRes. 18:531-538.
VOL.67, 1993