Copyright © 1998, American Society for Microbiology
Activation of Heterologous Gene Expression by the Large
Isoform of Hepatitis Delta Antigen
YU WEI†
ANDDON GANEM*
Howard Hughes Medical Institute and Department of Microbiology and Immunology,
University of California Medical Center, San Francisco, California 94143-0414
Received 15 August 1997/Accepted 10 December 1997
Hepatitis delta virus (HDV) encodes two isoforms of its principal gene product, hepatitis delta antigen
(HDAg). These two forms play distinctive and complementary roles in viral replication. Here we report that the
large (LHDAg), but not the small (SHDAg), isoform of HDAg has the capacity to activate the expression of
cotransfected genes driven by a variety of promoters, including the pre-S, S, and C promoters of hepatitis B
virus. Mutational analysis of the C-terminal 19 amino acids unique to LHDAg shows that changing prolines
to alanines in the two PXXP motifs in this region specifically ablates the activation function without abolishing
another activity of LHDAg, namely, its ability to inhibit HDV RNA synthesis. However, C-terminal truncations
that also disrupt these PXXP motifs only slightly diminished the activation function, indicating that the proline
mutations were not acting by inactivating potential SH3 interactions that could be mediated by these motifs.
Mutation of the isoprenylated cysteine to serine decreases but does not abolish the activation activity, and
over-expression of SHDAg does not interfere with the transactivation function of LHDAg. Although the mechanism
and biological significance of this activity of LHDAg remain unknown, the presence of this activity serves as
yet another marker that functionally distinguishes this protein from the closely related isoform SHDAg.
Hepatitis delta virus (HDV) is an RNA virus that requires
coinfection with hepatitis B virus (HBV) to complete its life
cycle. The helper function supplied by HBV is limited to the
provision of envelope proteins (hepatitis B surface antigens)
for the completion of HDV assembly (28, 29, 31). HDV RNA
replication is independent of its HBV helper (19). In fact, the
presence of HDV suppresses HBV replication in vivo (30, 39).
Nonetheless, clinical studies have shown that HDV infection
can be associated with more severe hepatitis than HBV alone
and is often implicated in cases of fulminant hepatitis (4, 32).
The genome of HDV is a circular, single-stranded RNA of
about 1,700 nucleotides (nt), of which approximately 70% are
self-complementary (for a review, see references 20 and 21).
This self-complemetarity allows the genome to form an
un-branched rod-like structure. A unique functional protein,
hep-atitis delta antigen (HDAg), is encoded by the genome (3, 38),
and two isoforms of this protein are produced during infection.
The canonical small form of HDAg (SHDAg) is 195 amino
acids (aa) long; it harbors an N-terminal coiled-coil domain
responsible for oligomerization (37), a central domain
respon-sible for binding to the RNA genome (7, 23), a nuclear
local-ization signal (2, 7), and a C-terminal glycine- and proline-rich
region with an uncertain function. This form of HDAg is
es-sential for viral RNA replication, although it is not itself a
polymerase. Host RNA polymerase II is thought to supply the
polymerase function for replication (15, 26). During viral
rep-lication, an RNA editing event occurs at the UAG termination
codon of SHDAg, allowing readthrough of another 19 aa (Fig.
1) to generate the large isoform of the protein, LHDAg (25).
Since LHDAg contains all of the domains of SHDAg, it too
can form multimers with itself and with the SHDAg isoform,
bind HDV RNA (as a homo- or heteromultimer), and be
lo-calized to the nucleus.
Despite these similarities, the two HDAgs have very distinct
functions (22) and play complementary roles in HDV
replica-tion, which takes place largely in the nuclei of infected cells
(34). While SHDAg activates HDV RNA replication, LHDAg
is a trans-dominant inhibitor of this process (8). By contrast,
LHDAg, but not SHDAg, is capable of interacting with the
HBV envelope proteins to mediate envelopment of the HDV
ribonucleoprotein in viral assembly (6). This interaction has
been shown to require farnesylation of a cysteine residue found
in the C-terminal 19 aa unique to LHDAg (27, 16).
Further-more, it has been shown recently that only LHDAg is
phos-phorylated in cells (1).
In this report, we describe yet another activity of LHDAg
that further differentiates it from the related isoform SHDAg,
i.e., the ability to activate gene expression in trans.
MATERIALS AND METHODS
Plasmids.All wild-type (WT) and mutant HDAg coding regions were cloned into the KpnI and XbaI sites of pcDNA3 (Invitrogen). The complete LHDAg
open reading frame was first cloned into pBluescript II SK(2) (Stratagene) to
generate pHDL. All LHDAg mutants were prepared by PCR with pHDL as the template.
The plasmids expressing LHDAgP1m, LHDAgP2m, and LHDAgP1P2m were constructed by two rounds of PCR. In the first round, two independent PCRs were performed with two pairs of primers. The primers were designed such that
the 39-end primer of one pair has the complementary sequence of the 59-end
primer of another pair, and desired mutations were introduced into these two
primers. For LHDAgP1m, the 59-end primer HD1 (59-GACTTCTAGAGGAT
CCCCCGCTTTATTCACTGG-39[nt 944 to 960; the sequence and numbering
are according to reference 19]) was paired with the 39-end primer 59-GATATA
CTCTTCGCAGCCGATGCGCCCTTTTCTC-39, corresponding to nt 1010 to
977 (underlined nucleotides represent mutations), and the 59-end primer 59-G
AGAAAAGGGCGCATCGGCTGCGAAGAGTATATC-39 (nt 977 to 1010)
was paired with the 39-end primer HD2 (59-TTCGTCCCCAATCTGCAGGGA
GTC-39[nt 1100 to 1077]). For LHDAgP2m, primer HD1 was paired with 39-end
primer 59-CAGCCGATCCGGCCTTTTCTGCCCAGAGTTGTC-39(nt 997 to
965) and 59-end primer 59-GACAACTCTGGGCAGAAAAGGCCGGATCGG
CTG-39(nt 965 to 997) was paired with primer HD2. For LHDAgP1P2m, primer
HD1 was paired with the 39-end primer 59-GATATACTCTTCGCAGCCGAT
GCGGCCTTTTC-39(nt 1010 to 979) and primer 59-GAAAAGGCCGCATCG
* Corresponding author. Mailing address: Howard Hughes Medical
Institute and Department of Microbiology and Immunology,
Univer-sity of California Medical Center, San Francisco, CA 94143-0414.
Phone: (415) 476-2826. Fax: (415) 476-0939. E-mail: ganem@socrates
.ucsf.edu.
† Present address: U.R.E.G., Institut Pasteur, 75015 Paris, France.
2089
on November 9, 2019 by guest
http://jvi.asm.org/
GCTGCGAAGAGTATATC-39(nt 979 to 1010) was paired with primer HD2. pHDL was used as the template in all PCRs save in that for LHDAgP1P2m, in which LHDAgP2m was used as the template. After gel purification, the products from two PCRs were pooled. The presence of the complementary sequence in the primers allows annealing of part of the sequence between the PCR products. The resulting molecules were amplified in the second-round PCR by using primers HD1 and HD2. The final PCR products were then gel purified, digested with PstI and XbaI, and cloned into pHDL cut with PstI and XbaI. The resulting plasmids were digested with KpnI and XbaI and cloned into pcDNA3.
The deletion mutants were prepared by a PCR in which the 59- end primers
were 59-GACTCTAGACCCGCTTTATTCAATCGGCTGGGAAG-39(nt 990
to 1002) for the plasmid expressing S18, 59-GACTCTAGACCCGCTTTATTC
AGGGAGAAAAGGGC-39(nt 975 to 987) for the construct S113, 59-GACT
CTAGACCCGCTTTATTCAACAACTCTGGGGA-39 (nt 966 to 978) for
S116, and 59-GACTCTAGACCCGCTTTATTCAGGGTCGACAACTC-39(nt
959 to 972) for S118, and the 39-end primer was HD2. Lcys211 was also
gen-erated by a PCR in which the 59-end primer 59-GACTCTAGAGGATCCTAT
TCACTGGGGTCGAGAACTCTGGGGAG-39(nt 951 to 979) was paired with
primer HD2. pHDL was used as the template. The PCR products were gel purified, digested with PstI and XbaI, and cloned into pHDL cut with PstI and
XbaI. The inserts were prepared by digestion with KpnI and XbaI and cloned into
pcDNA3.
N-myc140CAT was constructed by a PCR in which primer N-myc59(59-CAG
TCTCGAGTTCCCAGCAGTCGCCTG-39) was paired with primer N-myc39
(59-TCGCCTGCCCGCC-39) (13). N-myc84CAT was also constructed by a PCR
in which the primer 59-CCCTAGGGAAAGGAAGCAC-39was paired with
N-myc39. N-myc2-CAT was used as the template (35). N-myc140Sp1(2)CAT was
constructed by two PCR rounds. In the first round, N-myc59paired with primer
59-GTGTGCGGAGAGGGGGGACAATAGCCA-39and primer 59-TGGCTA
TTGTCCCCCCTCTCCGCACAC-39 paired with N-myc39 were used in two
independent PCRs in which N-myc2-CAT was the template. The PCR products
were then pooled. The second PCR round was performed with N-myc59and
N-myc39as the primer. The final PCR products were cloned into the XhoI and
XbaI sites of pCAT, which was described previously (36). 3Sp1CAT was
gener-ated by annealing the sense oligonucleotide CAG CTC GAG ATT GCC CCC GCC CTC ATT GCC CCC GCC CTC ATT GCC CCC GCC CTC TAG ATA C to the antisense oligonucleotide GTA TCT AGA GGG CGG GGG CAA TGA GGG CGG GGG CAA TGA GGG CGG GGG CAA TCT CGA GCT G, which contains three Sp1 binding sites. The double-stranded oligonucleotide was cut with XhoI and XbaI and cloned into pCAT. 3Sp1mCAT was constructed in the same way with the sense oligonucleotide CAG CTC GAG ATT GTC CCC CCT CTC ATT GTC CCC CCT CTC ATT GTC CCC CCT CTC TAG ATA C and the antisense oligonucleotide GTA TCT AGA GAG GGG GGA CAA TGA GAG GGG GGA CAA TGA GAG GGG GGA CAA TCT CGA GCT G, which contain three mutated Sp1 binding sites.
The nucleotide sequence of the fragments from PCR was confirmed by con-ventional dideoxy sequencing.
Plasmid pDL452 was a gift from D. Lazinski (Tufts University) (unpublished data). The promoter constructs (hsp70 TATA series and simian virus 40 [SV40] early series) were the gift of D. Lukac and J. Alwine (University of Pennsylvania) and R. Kingston (Harvard Medical School) and have been described elsewhere (24, 33). pPreS1CAT, pSCAT, and pUCAT9 were the gift of T. S. B. Yen (University of California, San Francisco). pPreS1CAT and pUCAT9 have been described previously (17, 40). pSCAT was obtained by cloning the HBV fragment spanning nt 3125 to 33 in pCAT. Luciferase reporter vector pGL2-promoter (SV40 promoter) was obtained from Promega.
Cell transfection and CAT and luciferase assays.HepG2, 293, and CCL13 (Chang liver) cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented with 10% bovine serum and antibiotics. Cells were transfected by
calcium phosphate coprecipitation using 1mg of reporter plasmid and 4mg of
transactivator plasmids. (For the Western immunoblot assay, 10mg of DNA was
transfected.) At 12 h after transfection, cells were washed with phosphate-buffered saline and cultured for another 24 h, at which time cell extracts were assayed for chloramphenicol acetyltransferase (CAT) activity or HDAg (by im-munoblotting with anti-HDAg). For HDV RNA replication analyses, cells were harvested 4 to 6 days after transfection. The CAT assay method used is described in detail elsewhere (36). For the luciferase assay, cells were harvested 48 h after transfection and lysed in lysis buffer containing 25 mM Tris-HCl (pH 8.0), 8 mM
MgCl2, 1% Triton X-100, 1% bovine serum albumin, 15% glycerol, and 1 mM
dithiothreitol. Cellular extract was measured for luciferase activity in the lysis buffer in the presence of 0.4 mM ATP and 1 mM luciferin.
Northern blot analysis.Total RNA was isolated from cells with RNAzol B (TEL-TEST), electrophoresed through a standard 1% agarose–2.2 M formalde-hyde gel, and transferred to a positively charged nylon membrane (Boehringer
Mannheim) in 103SSC (13SSC is 0.15 M NaCl plus 0.015 M sodium citrate).
Membranes were hybridized at 72°C with an HDV antigenome-specific ribo-probe uniformly labeled with digoxigenin (5). After hybridization, blots were
washed at 72°C with 0.1% sodium dodecyl sulfate and 23, 0.53, and 0.13SSC,
successively. The probe was detected as previously described (12).
Western immunoblot analysis.Transfected cells were harvested and lysed in a solution containing 50 mM Tris-HCl (pH 7.5), 400 mM NaCl, 0.2% Nonidet P-40, and protease inhibitors. Lysates were fractionated by sodium dodecyl sulfate-polyacrylamide (12.5%) gel electrophoresis and transferred to Immo-bilon-P membranes (Millipore). The membranes were incubated with an anti-HDAg polyclonal antibody (1:10,000 dilution) for 1 h at room temperature. Immune complexes were detected with horseradish peroxidase-conjugated goat antibodies to rabbit immunoglobulin G (1:7,500 dilution) (Gibco BRL) and enhanced chemiluminescence (Amersham).
RESULTS
Evidence for transactivation activity of LHDAg.
The
possi-bility that LHDAg harbors an activation activity was initially
suggested by experiments aimed at identifying cellular factors
that might interact with the protein in the yeast two-hybrid
assay. In those experiments, a fusion protein containing the
DNA binding domain of Gal4p fused to intact LHDAg was
expressed in yeast cells. Surprisingly, this Gal4p-LHDAg
fu-sion protein was able to strongly activate transcription of a
lacZ reporter gene under the control of a promoter containing
multiple Gal4p binding sites, even in the absence of any other
interacting activator proteins. No comparable activation was
seen when a similar fusion protein containing SHDAg was
expressed in the same yeast strain (5). However, since many
proteins that do not activate transcription under normal
con-ditions can score in this assay, we decided to conduct further,
more rigorous tests of gene activation by LHDAg.
To examine the ability of LHDAg to activate transcription in
mamalian cells, plasmids expressing LHDAg or SHDAg were
cotransfected into HepG2 cells along with a CAT reporter
gene driven by the cellular N-myc2 promoter. Forty-eight
hours later, the cells were assayed for CAT activity; the results
are expressed as fold CAT activity above that induced in the
absence of LHDAg (each value represents the mean result of
at least three independent experiments). A plasmid expressing
human growth hormone was used as an internal control for
transfection efficiency. When the CAT gene was under control
of the N-myc2 promoter, CAT activity was increased more
than sixfold in the presence of LHDAg (Fig. 2A). In contrast,
no activation of the N-myc2 promoter by SHDAg was detected
(Fig. 2A). Since immunoblotting with anti-HDAg confirmed
the expression of both LHDAg and SHDAg in these
exper-iments (Fig. 2A), the absence of transactivation activity of
SHDAg is not due to the absence of the protein. By contrast,
a CAT gene under the control of a minimal promoter (TATA
box) from adenovirus E1B was not activated by either LHDAg
or SHADg (data not shown), suggesting that activation by
LHDAg likely operates through factors bound upstream of the
TATA box (see also Fig. 2D).
[image:2.612.64.278.70.192.2]The N-myc2 gene (originally cloned from woodchuck liver)
is a member of the myc gene family (14) whose promoter has
FIG. 1. Sequence of the 19 aa unique to the C terminus of LHDAg. ThePXXP motifs are underlined. Below are shown the amino acid changes present in the mutants employed in this study. The positions of the termination codons introduced into the truncation mutants are indicated by asterisks.
2090
WEI AND GANEM
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
been extensively studied in HepG2 cells (13). To map the sites
of LHDAg responsiveness, we constructed several deletions of
sequences in the N-myc2 regulatory region (cf. Fig. 2B and C).
Deletions to
2
140 (relative to the cap site of the mRNA)
maintained the transactivation activity of LHDAg, while
dele-tions to
2
84 reduced activation by 5.7-fold (Fig. 2C). A
com-puter search of the sequence from
2
140 to
2
84 revealed the
FIG. 2. LHDAg has transactivation activity. (A) One microgram of a plasmid containing a CAT gene under the control of the woodchuck N-myc2 promoter
was transfected with 4mg of the pcDNA3 vector or a plasmid expressing SHDAg
or LHDAg. CAT activity was measured, and that expressed in the cells trans-fected with the reporter plus the pcDNA3 vector was set arbitrarily at 1.0; other assay results were normalized to this value. At the bottom is a Western blot showing expression of SHDAg and LHDAg. (B) Part of the sequence of the woodchuck N-myc2 promoter. The TATA element is boxed. Sequences bearing putative binding sites for transcription factors are underlined. The transcription
start site is designated 11 (13). Mutations introduced into the Sp1 site in
33Sp1mCAT are shown separately below. (C) CAT assay of deletion mutants of
the N-myc2 promoter. One microgram of a CAT reporter plasmid under the control of the N-myc promoter containing 140 bp (N-myc140), 84 bp (N-myc84),
or 140 bp with a mutated Sp1 binding site [N-myc140Sp1(2)] regulatory
se-quence was transfected with 4mg of the pcDNA3 vector or with a plasmid
expressing SHDAg or LHDAg. A CAT assay was performed as described for A. (D) Activation of Sp1 by LHDAg in different cell lines. A CAT gene driven by the E1b TATA element with three WT or mutated Sp1 sites, as indicated, was cotransfected with either the pcDNA3 vector or a vector expressing SHDAg or LHDAg. Cells were assayed for CAT activity 48 h after transfection, and results were normalized to the level of CAT expressed from the pcDNA3 cotransfection, as described for A. (E) Detection of the activation activity of LHDAg by using the luciferase gene as a reporter. The luciferase gene driven by the SV40 early promoter was cotransfected with either the pcDNA3 vector or a vector express-ing SHDAg or LHDAg, and its activity was measured 48 h after transfection. The error bars show the standard deviation from the mean of at least three indepen-dent experiments.
2091
on November 9, 2019 by guest
presence of binding sites for Sp1 and for members of the
GATA family of transcription factors (Fig. 2B). To determine
if Sp1 might be a target of activation, we mutated the Sp1 site
in this promoter and tested the ability of the mutant to be
activated; this lesion reduced LHDAg induction twofold (Fig.
2C). From the fact that constructs with inactive Sp1 binding
sites continue to show a residual level of activation (Fig. 2C),
we infer that additional factors can also be up-regulated by
LHDAg (see below).
To provide a direct test of the ability of Sp1 to be activated
in trans (directly or indirectly) by LHDAg, we cotransfected
LHDAg or SHDAg into HepG2 cells together with a vector
containing three Sp1 binding sites upstream of the minimal
E1B TATA box-driven CAT gene (Fig. 2D). CAT activity was
measured 48 h after transfection. As expected, the presence of
SHDAg had no effect on CAT activity, whereas LHDAg
strongly activated the CAT gene linked to the three Sp1 sites
(over 20-fold) (Fig. 2D). The specificity of Sp1 activation by
LHDAg was further demonstrated by introducing mutations
into the Sp1 sites. Control experiments with nuclear extracts
showed that oligonucleotides bearing these mutant Sp1 sites
do not bind Sp1 in vitro (data not shown). Correspondingly,
transcription of a CAT gene linked to the mutant Sp1 sites was
not up-regulated by LHDAg (Fig. 2D). This result confirms
that Sp1 is one target of activation by LHDAg.
To explore the generality of this activation response, we
examined if it can be observed in cell lines other than HepG2.
Accordingly, we cotransfected the 3
3
Sp1E1B CAT reporter
gene with the plasmids expressing SHDAg and LHDAg into
293 (human embryonal kindey) and CCL13 (human liver) cells
and assayed for CAT activity. In both cell lines, LHDAg
acti-vated CAT gene expression as strongly as in HepG2 cells (Fig.
2D); as in HepG2 cells, SHDAg was inactive in this assay in
both lines.
The fact that CAT genes with differing 5
9
regions respond
differently to LHDAg makes it unlikely that the prime target of
this activation resides within CAT sequences. However, to
exclude the possibility of reporter-specific artifacts, we also
examined the ability of LHDAg to activate a luciferase
re-porter, in this case driven by an SV40 promoter. As shown in
Fig. 2E, this reporter was reproducibly up-regulated fourfold,
an amount similar to that observed with CAT reporters driven
by promoters containing SV40 components (Fig. 3). No effect
of SHDAg on luciferase expression was observed.
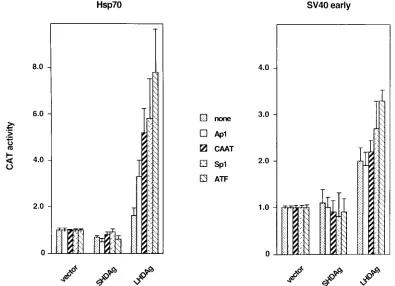
Effect of LHDAg on various promoters.
To assess the range
of potential transcription factor targets of LHDAg, we tested
two series of promoters in transient transfection assays. The
hsp70 minimal promoter and the TATA box of the SV40 early
promoter were each paired with a single copy of a variety of
upstream stimulatory elements (USEs), including CAAT, Ap1,
Sp1, ATF, and a nonsense sequence (none), all driving CAT
reporter genes (for detailed sequences, see references 24 and
33). Each of these plasmids was cotransfected into HepG2 cells
with constructs expressing either LHDAg or SHDAg, and the
transfected cells were assayed for CAT activity. As shown in
Fig. 3, the increase in CAT activity caused by LHDAg was
higher with the promoters based on the hsp70 minimal
pro-moter than those based on the SV40 early propro-moter for the
same USE. When different USEs were linked to the same
promoter and tested for response to LHDAg, there was a
hierarchy of responses, in the following order: ATF
.
Sp1
5
CAAT
.
AP1. These data affirm that Sp1 is not the only factor
that can be activated by LHDAg expression. By contrast,
pre-liminary experiments failed to show activation of Oct-1 and
HNF4 (data not shown), suggesting that LHDAg is not an
indiscriminate activator of all upstream activators.
Effect of LHDAg mutations on transactivation activity.
LHDAg and SHDAg are identical in sequence, save for the
additional 19 aa present at the C terminus of LHDAg. Since
SHDAg does not possess the transactivation activity displayed
by LHDAg, it seemed likely that this activity was endowed by
this 19-aa C-terminal extension. To explore this possibility, we
constructed several classes of LHDAg mutants, including both
truncations and amino acid substitutions. In a preliminary
in-spection of the sequence of this region, two particular features
that pointed to the potential for protein-protein interactions
attracted our attention. First, we noted the presence of two
PXXP motifs (Fig. 1); in some proteins, such motifs have been
implicated in interactions with other proteins harboring SH3
domains (9). The second and more well-established feature
was the CXXX motif responsible for the known farnesylation
of LHDAg at cysteine 211 (27) that is essential for interactions
with HBsAg (16). Special attention was paid to these two
features in the design of mutations in this region.
The mutations we constructed are summarized in Fig. 1.
Three lesions were specifically designed to target the PXXP
motifs. Mutations in LHDAgP1m and LHDAgP2m consist of
replacement of prolines with alanines in the first and second
PXXP motifs, respectively (Fig. 1), while LHDAgP1P2m bears
mutations in both PXXP motifs (Fig. 1). Mutant Lcys211
changes Cys 211 to serine, thereby ablating isoprenylation. The
remaining mutations introduce stop codons after codons 8, 13,
16, and 18 of this 19-codon region (Fig. 1) and are designated
S
1
8, S
1
13, S
1
16, and S
1
18, respectively. Each mutant was
transfected into HepG2 cells and tested for both
transactiva-tion of the 3
3
Sp1CAT reporter and for the expression of the
mutant proteins (as assayed by immunoblotting with
anti-HDAg). As shown in Fig. 4C, all of the mutants expressed
similar levels of LHDAg proteins and had electrophoretic
mo-bilities consonant with their predicted chain lengths.
While mutations in the individual PXXP motifs did not
significantly impair transactivation, the P1P2m double mutant
virtually ablated this activity (Fig. 4C) and was, in fact, the only
mutant in this series with this phenotype. Although this initially
suggested that SH3 interaction motifs might be important for
the activation activity, this simple interpretation is
counter-manded by the phenotype of mutant S
1
8. In this mutant, one
PXXP motif is deleted and the other is severely crippled (Fig.
1), yet transactivation is preserved at nearly WT levels (Fig.
4A). Thus, it appears that the phenotype of the P1P2m mutant
is due not to disruption of putative SH3 interaction but to
effects on another aspect of the structure or conformation of
this region. However, we note that the P1P2m lesion is highly
specific and its phenotype is not the trivial result of a global
disruption of protein structure. The protein stably accumulates
to WT levels, remains immunoreactive, and continues to
func-tion as an inhibitor of SHDAg-mediated HDV RNA
replica-tion. The latter result is shown in the experiment of Fig. 4A, in
which 10
m
g of the WT or mutant LHDAg expression vector
was cotransfected into 293 cells with 3
m
g of
replication-com-petent overlength HDV plasmid pDL452, which is designed to
express HDV genomic RNA. HDV RNA of antigenomic
po-larity was then assayed by Northern blotting; such RNA can
only be produced in these cells by authentic
(SHDAg-depen-dent) viral RNA replication (19). As can be seen in Fig. 4A, all
PXXP mutants, including P1P2m, retain the ability to
domi-nantly inhibit replication, implying that all of them retain the
ability to hetero-oligomerize with SHDAg. Thus, the P1P2m
lesion selectively impairs the transactivation function of LHDAg.
Mutant Lcys211, in which cysteine 211 was changed to a
serine, has previously been shown to be unprenylated (16), as
well as to be somewhat less active as an inhibitor of replication
2092
WEI AND GANEM
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
(18). As shown in Fig. 4C, the Lcys211 mutation reproducibly
diminished the activation activity (ca. twofold) but did not
abolish it. We conclude that farnesylation of LHDAg is not
essential for this activity. The transactivation results obtained
with the truncation mutants are also shown in Fig. 4C. Clearly,
all of the C-terminal truncations tested were stably expressed
and were able to activate expression of the reporter, with only
modest and variable reductions in activity. Figure 4B shows
that all were also still able to inhibit HDV RNA replication,
although there was clear variation in their potency in this
regard. There was little apparent correlation between this
in-hibitory activity and the strength of transactivation.
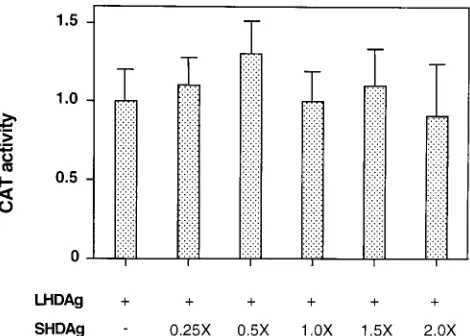
Effect of SHDAg on transactivation activity of LHDAg.
Dur-ing viral replication, SHDAg and LHDAg coexist in cells and
their relative stoichiometries are important in determining
function. For example, the inhibitory effects of LHDAg on
viral RNA replication can be overcome by overexpression of
the SHDAg isoform. To determine if expression of SHDAg
can influence the transactivation activity of LHDAg, HepG2
cells were transfected with a fixed quantity of the LHDAg
expression plasmid and increasing amounts of an
SHDAg-expressing construct, and the ability to activate CAT
expres-sion from a cotransfected reporter gene was assayed. Over an
eightfold range of relative SHDAg-to-LHDAg plasmid
con-centrations, no significant effect on the level of transactivation
was observed (Fig. 5).
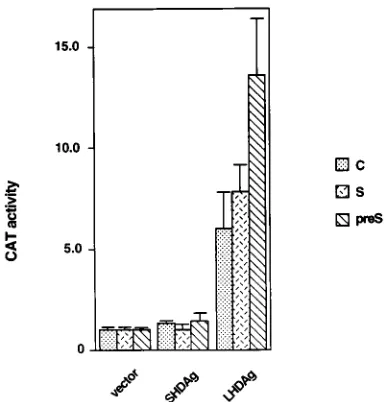
Effect of LHDAg on HBV promoters.
Since HBV is the
natural helper virus of HDV, we wanted to know what effect
expression of LHDAg would have on HBV promoters.
pPreS1CAT, pSCAT, and pUCAT9 are plasmids in which the
CAT gene is under the control of the HBV pre-S, S, and C
promoters, respectively (17, 40). Each of these plasmids was
cotransfected with the LHDAg expression vector into HepG2
cells, and extracts were assayed for CAT activity. As shown in
Fig. 6, all of the HBV promoters tested were activated by
LHDAg; again, SHDAg expression was without effect.
DISCUSSION
These studies document that, at least in acutely transfected
cell cultures, LHDAg, but not SHDAg, displays a novel
activ-ity, the ability to activate gene expression in trans. Activation is
independent of the reporter function used and occurs in
sev-eral cell types and on a wide variety of natural and synthetic
promoters but is not observed on minimal (basal) promoter
elements, suggesting that it operates principally on accessory
rather than basal transcription factors.
[image:5.612.100.495.71.357.2]We know little of the mechanism by which this activation
occurs. The yeast two-hybrid result that initially drew our
at-tention to this phenomenon could be interpreted to mean that
LHDAg can interact directly or indirectly with the basal
tran-scriptional machinery once targeted to DNA, and certainly
binding to accessory transcription factors might be one way to
achieve such targeting in vivo. However, since many proteins
with no transcriptional roles in vivo can score in two-hybrid
assays, we are reluctant to place too much emphasis on this
result as a clue to the mechanism. Many alternative
mecha-nisms can be envisioned, including much more indirect ones.
For example, LHDAg might function from a cytoplasmic
lo-cation (e.g., the cytosolic face of the endoplasmic reticulum) to
initiate a signal transduction cascade that ultimately converges
on nuclear transcription factors. Such a proposal would be
formally similar to certain models proposed for the activation
of gene expression by the HBV X protein (11). Additional in
FIG. 3. Activation of several upstream elements by LHDAg. CAT genes driven by various upstream stimulatory elements, including binding sites for ATF, CAAT, Sp1, and AP1, paired with either the hsp70 TATA element or the SV40 early promoter TATA element, were cotransfected into HepG2 cells with either the pcDNA3 vector or a vector expressing SHDAg or LHDAg. Cellular extracts were assayed for CAT activity, and the results were normalized as described in the legend to Fig. 2.on November 9, 2019 by guest
http://jvi.asm.org/
vivo and in vitro studies are required to address the
mechanis-tic issues raised here.
The potential biological significance of this activity is
like-wise a matter of conjecture. For example, we do not know if
activation occurs at levels of LHDAg achieved during infection
in vivo. Attempts to address this question in transfected cells
have been impeded by technical difficulties. Starting from
cloned HDV DNA, it takes days in cell culture before RNA
editing generates substantial LHDAg; by this time, most of the
input reporter CAT plasmid has disappeared. We have been
unable to examine rigorously whether native LHDAg
ex-pressed from edited RNA can activate a reporter gene,
al-though we think this highly likely. The larger question is what,
if any, functional consequences such transactivation might
have. It seems unlikely that such an activity would be required
for HDV RNA synthesis, since the latter is dependent upon
SHDAg and is achieved prior to peak LHDAg levels. By
con-trast, induction of HBV envelope protein expression by
LHDAg could well serve to enhance HDV assembly, and, as
shown in Fig. 6, the HBV pre-S and S promoters are clearly
responsive to LHDAg expression.
Irrespective of its role in HDV replication, the activation
FIG. 5. Expression of SHDAg does not affect the transactivation activity of
LHDAg. One microgram of 33Sp1CAT DNA was transfected with a fixed
amount (4mg) of a plasmid expressing LHDAg and increasing amounts of a
[image:6.612.312.547.490.658.2]plasmid expressing SHDAg into HepG2 cells. The molar ratio of SHDAg over LHDAg DNA is indicated; pcDNA3 plasmid DNA was added to each transfec-tion to bring the total amount of DNA used in each transfectransfec-tion to a constant level. At 48 h posttransfection, CAT activity was determined and normalized to the level produced in the presence of LHDAg alone, which was set at 1.0.
FIG. 4. Effect of mutations on the replication inhibition and transactivation activities of LHDAg. (A) Inhibition of HDV RNA replication by PXXP motif mutants. Three micrograms of HDV plasmid pDL452 DNA, which is capable of initiating viral replication upon transfection, was transfected into 293 cells with
10mg of plasmids expressing the mutant LHDAgs indicated above the gels (top).
Six days posttransfection, total RNA was extracted and 3mg of this RNA was
used for Northern blot analysis. Membranes were hybridized with a specific anti-genomic probe uniformly labeled with digoxigenin. At the bottom are the ethi-dium bromide-stained rRNAs used as a loading control. (B) Inhibition of HDV genome replication by truncation mutants. Three micrograms of pDL452 DNA
was transfected with 10mg of a plasmid expressing WT or truncated LHDAg into
293 cells. Northern blot analysis was performed as for B. The ethidium bromide-stained rRNAs shown were used as a loading control. (C) Effect of LHDAg
mutations on transactivation activity (top). One microgram of plasmid 33Sp1
CAT was transfected with 4mg of the indicated WT or mutant HDAg expression
vector into HepG2 cells, CAT assays were performed, and the results were normalized as described in the legend to Fig. 2. The immunoblot at the bottom shows expression of the indicated WT or mutant HDAgs in transfected cells.
2094
WEI AND GANEM
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
activity might influence other aspects of the infection, e.g.,
pathogenesis. Induction of host gene expression by LHDAg
could play a role in enhancing pathogenesis; for example, if
induction included class I major histocompatibility complex
genes, elevated liver cell cytotoxicity might result from
en-hanced cytotoxic T-lymphocyte action. Induction of HBV gene
expression (Fig. 6) could have similar effects in sensitized
in-fected hosts. This activity could contribute to the clear
associ-ation of HDV with fulminant hepatitis, which is thought to
result largely from exaggerated cytotoxic responses to viral
antigens. In this formulation, the reduction in HBV markers
typically observed in HDV coinfection (30) would be the result
of enhanced immune clearance (rather than direct repression
of HBV genes by HDV gene products). Alternatively, it is
pos-sible that suppression of HBV in HDV-infected cells is
medi-ated by host factors induced by LHDAg or that the inductive
effect of LHDAg observed here is countered by a repressive
effect of another viral gene product, e.g., SHDAg (39).
Finally, the transactivation function described here might
have no function in present-day HDV replication but may
simply represent a vestige of the evolutionary history of
LHDAg. We recently identified a host gene, termed dipA,
whose product interacts with HDAg (5). Characterization of
this gene showed that it is distantly related to that for LHDAg,
suggesting that present-day HDV may have been derived from
the capture of a cellular gene by a primitive, viroid-like
repli-con. Of note is the fact that dipA also shares limited homology
with members of the fra-encoded family of transcription
fac-tors (10), and the dipA gene is directly adjacent to fra-1 in the
mouse genome (5a), again suggesting a relationship. If these
tantalizing evolutionary connections to cellular transcription
factors are correct, then the activation function of LHDAg may
simply be the vestigial remnant of an activity that was once its
central mission but which now is only a secondary or accessory
function.
ACKNOWLEDGMENTS
We are grateful to Rob Brazas for many insightful discussions and
for helpful comments on the manuscript. We thank David Lazinski for
plasmid pDL452; David Lukac, James Alwine, and Robert Kingston
for the promoter constructs based on hsp70 and SV40 early promoters;
and T.-S. Benedict Yen for pPreS1CAT, pSCAT, and pUCAT9. We
thank F. Bergametti, D. Sitterlin, and C. Transy for help with the
luciferase assay.
REFERENCES
1. Bichko, V., S. Barik, and J. Taylor. 1997. Phosphorylation of the hepatitis delta virus antigens. J. Virol. 71:512–518.
2. Bichko, V. V., and J. M. Taylor. 1996. Redistribution of the delta antigens in cells replicating the genome of hepatitis delta virus. J. Virol. 70:8064–8070. 3. Bonino, F., K. H. Heermann, M. Rizzetto, and W. H. Gerlich. 1986. Hepatitis delta virus: protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 58:945–950.
4. Bonino, F., F. Negro, M. Baldi, M. R. Brunetto, E. Chiaberge, M. Capalbo,
E. Maran, C. Lavarini, N. Rocca, and G. Rocca.1987. The natural history of chronic delta hepatitis, vol. 234. Alan R. Liss, New York, N.Y.
5. Brazas, R., and D. Ganem. 1996. A cellular homolog of hepatitis delta antigen: implications for viral replication and evolution. Science 274:90–94. 5a.Brazas, R., and D. Ganem. Unpublished data.
6. Chang, F. L., S. J. Chen, S. J. Tu, M. N. Chiu, C. J. Wang, and D. S. Chen. 1991. The large form of hepatitis delta antigen is crucial for the assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 88:8490–8494.
7. Chang, M. F., S. C. Baker, L. H. Soe, T. Kamahora, J. G. Keck, S. Makino,
S. Govindajajan, and M. M. C. Lai.1988. Human hepatitis delta antigen is a nuclear phosphoprotein with RNA-binding activity. J. Virol. 62:2403–2410. 8. Chao, M., S.-Y. Hsieh, and J. Taylor. 1990. Role of two forms of the hepatitis delta virus antigen: evidence for a mechanism of self-limiting genome rep-lication. J. Virol. 64:5066–5069.
9. Cicchetti, P., B. J. Mayer, G. Thiel, and B. Baltimore. 1992. Identification of a protein that binds to the SH3 region of abl and is similar to Bcr and GAP-rho. Science 257:803–806.
10. Cohen, D. R., and T. Curran. 1988. fra-1, a serum-inducible, cellular imme-diate-early gene that encodes a Fos-related antigen. Mol. Cell. Biol. 8:2063– 2069.
11. Doria, M., N. Klein, R. Lucito, and R. J. Schneider. 1995. The hepatitis B virus HBx protein is a dual specificity cytoplasmic activator of Ras and nuclear activator of transcription factors. EMBO J. 14:4747–4757. 12. Engler-Blum, G., M. Meier, J. Frank, and G. A. Mu¨ller. 1993. Reduction of
background problems in nonradioactive Northern and Southern blot analy-ses enables higher sensitivity than 32P-based hybridizations. Anal. Biochem.
210:235–244.
13. Fourel, G., C. Transy, B. C. Tennant, and M. A. Buendia. 1992. Expression of the woodchuck N-myc2 retroposon in brain and in liver tumors is driven by a cryptic N-myc promoter. Mol. Cell. Biol. 12:5336–5344.
14. Fourel, G., C. Tre´po, L. Bougueleret, B. Henglein, A. Ponzetto, P. Tiollais,
and M. A. Buendia.1990. Frequent activation of N-myc genes by hepadna-virus insertion in woodchuck liver tumours. Nature 347:294–298. 15. Fu, T.-B., and J. Taylor. 1993. The RNAs of hepatitis delta virus are copied
by RNA polymerase II in nuclear homogenates. J. Virol. 67:6965–6972. 16. Glenn, J. S., J. A. Watson, C. M. Havel, and J. M. White. 1992. Identification
of a prenylation site in delta virus large antigen. Science 256:1331–1333. 17. Guo, W., M. Chen, T. S. B. Yen, and J. Ou. 1993. Hepatocyte-specific
expression of the hepatitis B virus core promoter depends on both positive and negative regulation. Mol. Cell. Biol. 13:443–448.
18. Hwang, S. B., and M. M. C. Lai. 1994. Isoprenylation masks a conforma-tional epitope and enhances trans-dominant inhibitory function of the large hepatitis delta antigen. J. Virol. 68:2958–2964.
19. Kuo, M. Y.-P., M. Chao, and J. Taylor. 1989. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: role of delta antigen. J. Virol. 63:1945–1950.
20. Lai, M. M. C. 1995. The molecular biology of hepatitis delta virus. Annu. Rev. Biochem. 64:259–286.
21. Lazinski, D. W., and J. M. Taylor. 1994. Recent developments in hepatitis delta virus research. Adv. Virus Res. 43:187–231.
22. Lazinski, D. W., and J. M. Taylor. 1993. Relating structure to function in the hepatitis delta virus antigen. J. Virol. 67:2672–2680.
23. Lin, J.-H., M.-F. Chang, S. C. Baker, S. Govindarajan, and M. M. C. Lai. 1990. Characterization of hepatitis delta antigen: specific binding to hepatitis delta virus RNA. J. Virol. 64:4051–4058.
24. Lukac, D. M., J. R. Manuppello, and J. C. Alwine. 1994. Transcriptional activation by the human cytomegalovirus immediate-early proteins: require-ments for simple promoter structures and interactions with multiple com-ponents of the transcription complex. J. Virol. 68:5184–5193.
[image:7.612.73.266.68.269.2]25. Luo, G., M. Chao, S.-Y. Hsieh, C. Sureau, K. Nishikura, and J. Taylor. 1990. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 64:1021–1027.
FIG. 6. Activation of HBV promoters by LHDAg. One microgram of the indicated HBV-CAT reporter construct was cotransfected into HepG2 cells with
4mg of pcDNA3 (vector) or derivatives encoding SHDAg or LHDAg. CAT
activity was measured 48 h after transfection, and the results for each reporter were normalized to the level produced in the presence of the pcDNA3 vector alone, which was set at 1.0.
on November 9, 2019 by guest
http://jvi.asm.org/
26. MacNaughton, T. B., E. J. Gowans, S. P. McNamara, and C. J. Burrell. 1991. Hepatitis delta antigen is necessary for access of hepatitis delta virus RNA to the cell transcriptional machinery but is not part of the transcriptional complex. Virology 184:387–390.
27. Otto, J. C., and P. J. Casey. 1996. The hepatitis delta virus large antigen is farnesylated both in vitro and in animal cells. J. Biol. Chem. 27:4569–4572. 28. Ponzetto, A., P. J. Cote, H. Popper, B. H. Boyer, W. T. London, E. C. Ford,
F. Bonino, R. H. Purcell, and J. L. Gerin.1984. Transmission of the hepatitis B-associated delta agent to the eastern woodchuck. Proc. Natl. Acad. Sci. USA 81:2208–2212.
29. Rizzetto, M., M. G. Canese, J. Arico, O. Crivelli, F. Bonino, C. G. Trepo, and
G. Verme.1977. Immunofluorescence detection of a new antigen-antibody system associated to the hepatitis B virus in the liver and in the serum of HDsAg carriers. Gut 18:997–1003.
30. Rizzetto, M., M. G. Canese, J. L. Gerin, W. T. London, D. L. Sly, and R. H.
Purcell.1980. Transmission of the hepatitis B virus-associated delta antigen to chimpanzees. J. Infect. Dis. 141:590–602.
31. Rizzetto, M., B. Hoyer, M. G. Canese, J. W.-K. Shih, R. H. Purcell, and J. L.
Gerin.1980. Delta agent: association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc. Natl. Acad. Sci. USA 77:6124–6128.
32. Saracco, G., S. Macagno, F. Rosina, and M. Rizzetto. 1988. Serologic markers with fulminant hepatitis in persons positive for hepatitis B surface antigen. A worldwide epidemiologic and clinical survey. Ann. Intern. Med. 108:380.
33. Taylor, I. C. A., and R. E. Kingston. 1990. Factor substitution in a human HSP70 gene promoter: TATA-dependent and TATA-independent interac-tions. Mol. Cell. Biol. 10:165–175.
34. Taylor, J., W. Mason, J. Summers, J. Goldberg, C. Aldrich, L. Coates, J.
Gerin, and E. Gowans.1987. Replication of human hepatitis delta virus in primary cultures of woodchuck hepatocytes. J. Virol. 61:2891–2895. 35. Ueda, K., Y. Wei, and D. Ganem. 1996. Activation of N-myc2 gene expression
by cis-acting elements of oncogenic hepadnaviral genomes: key role of en-hancer II. Virology 217:413–417.
36. Ueda, K., Y. Wei, and D. Ganem. 1996. Cellular factors controlling the activity of woodchuck hepatitis virus enhancer II. J. Virol. 70:4714–4723. 37. Wang, J.-G., and S. M. Lemon. 1993. Hepatitis delta virus antigen forms
dimers and multimeric complexes in vivo. J. Virol. 67:446–454.
38. Weiner, A. J., Q.-L. Choo, K.-S. Wang, S. Govindarajan, A. G. Redeker, J. L.
Gerin, and M. Houghton.1988. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta
anti-gen polypeptides p24dand p27d. J. Virol. 62:594–599.
39. Wu, J.-C., P.-J. Chen, M. Y. P. Kuo, S.-D. Lee, D.-S. Chen, and L.-P. Ting. 1991. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. J. Virol. 65:1099–1104.
40. Zhou, D., and T. S. B. Yen. 1990. Differential regulation of the hepatitis B virus surface gene promoters by a second viral enhancer. J. Biol. Chem.
265:20731–20734.