for selected drug candidates
Thesis submitted to
The Tamilnadu Dr.M.G.R. Medical University, Chennai, India in partial fulfillment of the requirements
for the degree of
Doctor of Philosophy
Submitted by
D.NAGASAMY VENKATESH, M.Pharm.,
JANUARY 2010
J.S.S. COLLEGE OF PHARMACY
OOTACAMUND - 643001
DECLARATION
I hereby declare that the thesis entitled “Formulation and development of
oral sustained release systems for selected drug candidates”
submitted by me for the award of degree of Doctor of Philosophy of the
Tamilnadu Dr.M.G.R. Medical University, Chennai is a record of research work
done by me at J.S.S. College of Pharmacy, Ootacamund - 643 001, Tamilnadu,
India during the years 2006-2010 under the supervision of Dr.S.Sankar and that
the thesis had not previously formed the basis for the award of any degree,
diploma, associateship, fellowship or other similar title previously.
This is to certify that the thesis entitled “Formulation and development of
oral sustained release systems for selected drug candidates” is a
record of research work done by Mr.D. Nagasamy Venkatesh at J.S.S. College of
Pharmacy, Ootacamund - 643 001, Tamilnadu, India during the years 2006-2010
under my supervision and that the thesis had not previously formed the basis for
the award of any degree, diploma, associateship, fellowship or other similar title
previously. I also certify that the thesis represents independent work done by
the candidate.
Dr.S.Sankar
This is to certify that the thesis entitled “Formulation and development of
oral sustained release systems for selected drug candidates” is a
record of research work done by Mr.D. Nagasamy Venkatesh at J.S.S. College of
Pharmacy, Ootacamund - 643 001, Tamilnadu, India during the years 2006-2010
under my supervision as co-guide.
Dr.K.Santhi
J.S.S. MAHAVIDYAPEETHA
J.S.S. COLLEGE OF PHARMACY, OOTACAMUND
Off campus college of J.S.S. University, Mysore
CERTIFICATE
This is to certify that the thesis entitled “Formulation and development of
oral sustained release systems for selected drug candidates” is a
record of research work done by Mr.D. Nagasamy Venkatesh at J.S.S. College of
Pharmacy, Ootacamund - 643 001, Tamilnadu, India during the years 2006-2010
under the supervision of Dr.S.Sankar.
Dr.K.Elango
Principal i/c
ACKNOWLEDGEMENT
I express my gratitude to Dr.S.Sankar,
Supervisor for his support and
guidance throughout the course of this work. His trust on me to explore and
pursue my own interests gave me confidence that will remain with me for the
rest of my career. Most importantly, his patience and understanding allowed me
to complete this work even when faced with some very serious challenges, both
experimental and emotional.
I thank Dr. B. Suresh, Vice Chancellor, J.S.S. University, Mysore, for his
encouragement and the facilities extended to me for carrying out this project.
I thank Dr.K.Santhi for consenting to assume the role of my co-guide in
the area of pharmaceutics.
Words are not enough to express my sincere thanks and gratitude to Dr.
S.N.Meyyanathan, Head, Department of Pharmaceutical Analysis, for his
encouragement, valuable help, innovative ideas, constructive approach,
motivation and timely help in carrying out analytical method development and
studies during my project.
I thank Dr.M.J.Nanjan, Director, Post graduate studies and Research,
J.S.S.College of Pharmacy, Ootacamund for his support and advice.
I thank Dr.K.Elango, Principal i/c, J.S.S. College of Pharmacy,
Ootacamund for his valuable support and encouragement.
I thank Dr. Sanjay Vijayaraj, MD, Vijaya Hospital, Ootacamund for his
direct help in completing the bioavailability study.
I thank Dr. P.R.Anand Vijay Kumar, Head, Department of Pharmacy
Practice and Dr. Sabin Thomas, for their valuable help in completing the
bioavailability study.
Laboratories, Hyderabad and Mr. Phani Kumar, Nicholas Piramol Ltd, Mumbai
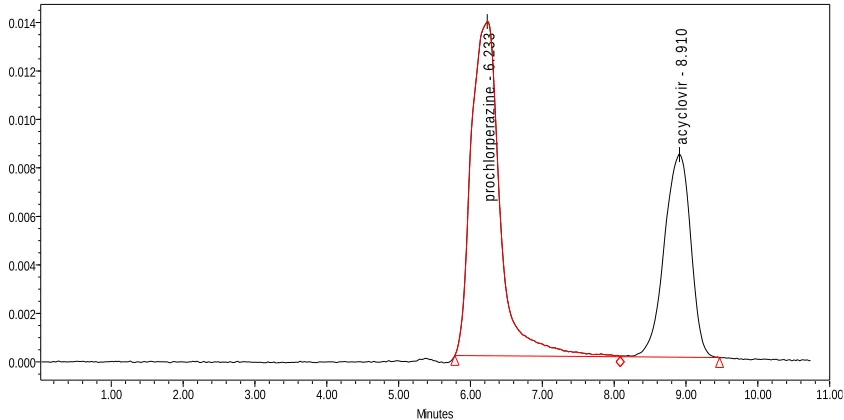
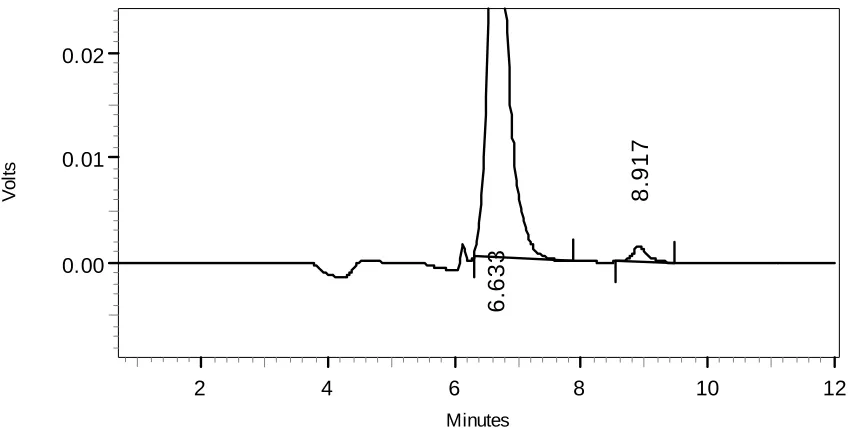
for providing me generous gift samples of Acyclovir and Prochlorperazine
maleate respectively for my project.
I thank Mr.S.Muralidharan, Research Scholar, Department of
Pharmaceutical Analysis, Mr. R. Shanmugam, Mr.K.S.Sumanth and
Mr.Narendra, II M Pharm Department of Pharmaceutical Analysis and
volunteers for their timely help in carrying out my bio analytical work.
I thank all the faculty, staff members, my fellow graduate students and
researchers of the J.S.S.College of Pharmacy, Ootacamund.
I thank Mr.S.Puttarajappa, Superintendent, J.S.S. College of Pharmacy,
Ootacamund for his encouragement.
My most sincere pranams to Lotus feet of His Holiness Jagadguru Sri
Shivarathri Deshikendra Mahaswamigalavaru, Suttur Mutt, Mysore for his
blessings to my endeavor.
I thank my parents, my wife and my daughter for always being there for
me, without their constant support and encouragement this work would not
have been possible. I thank my family members for their constant support.
Page No
1.0 Introduction
01
2.0 Aim and objectives
19
3.0 Review of literature
21
4.0
Place
of
research
work
57
5.0 Scope and plan of work
58
6.0
Materials
and
methods
61
7.0 Results and discussion
85
8.0 Summary and conclusion
101
Bibliography
Annexure I
Drug and Polymer profiles
Annexure II
Certificate of Analysis of drugs and polymers
Annexure III
Certificate copies of Institutional review board
Annexure IV
1
1. INTRODUCTION
The pharmaceutical industry today is caught between the downward pressure on prices and the increasing cost of successful drug discovery and development. The average cost and time for the development of a new chemical entity is much higher (approximately $500 million and 10–12 years) than those required to develop a Novel Drug Delivery System (NDDS) ($20–50 million and 3–4 years). An existing drug molecule in the form of an NDDS can get a new life, thereby increasing its market value, competitiveness and extending its patent life. Limited formularies, patent expiry with subsequent entry of generic competition and vertical integration have made the entire pharmaceutical industry focus today on designing and developing new and better methods of drug delivery. There has been a significant increase in approvals of NDDS in the past couple of years and this is expected to continue at an impressive rate in the near future1.
In the past few decades, significant medical advances have been made in the area of drug delivery with the development of novel dosage forms. The delivery of several classes of drugs, however, continues to be a challenge mainly due to their short half-life, poor membrane permeability and associated toxicity in the administered doses. Today we have a better understanding about the relationship between chemical properties of drugs and their movement in the body. Drug discovery scientists are, therefore, considering the pharmacokinetic properties of agents much earlier in the drug development process.
The rational development of a delivery system is sensible and expensive. Formulation development and optimization involves varying excipient levels, processing methods, identifying, discriminating dissolution methods and subsequent scale up of the final product.
2
ointments, liquids, aerosols, and injectables. Even today these conventional dosage forms are the primary pharmaceutical vehicles commonly seen in the prescription and over the counter drug market. The oral conventional types of drug delivery systems are known to provide a prompt release of drug. Therefore, to achieve as well as to maintain the drug concentration within the therapeutically effective range needed for treatment, it is often necessary to take this type of drug delivery system several times a day. This results in a significant fluctuation in drug levels often with sub-therapeutic and/or toxic levels and wastage of drug. Recently, several technical advancements have resulted in the development of new systems of drug delivery capable of controlling the rate of drug delivery, sustaining the duration of therapeutic activity, and/or targeting the delivery of drug to a tissue2-7.
Conventional Drug Therapy
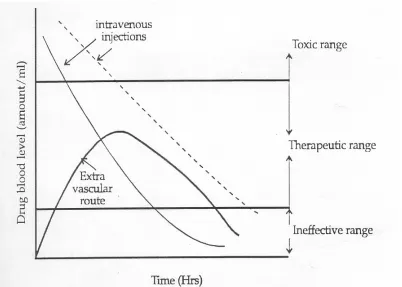
The goal of any drug delivery system is to promptly provide the required amount of a drug to the proper site in the body and to maintain the desired drug concentration. This idealized objective points to the two aspects most important to drug delivery systems, namely spatial placement and temporal delivery of a drug8. The term drug blood level refers to the concentration of a drug in the
blood or plasma.
It can be seen from the figure 1, that administration of a drug by either intravenous injection or an extra vascular route e.g. orally, intramuscularly or rectally does not maintain drug blood levels within the therapeutic range for extended periods of time. The short duration of action is due to the inability of a conventional dosage form to control delivery. If an attempt is made to maintain drug blood levels in the therapeutic range for longer periods by, for example, increasing the dose of an intravenous injection as shown by dotted line in the figure, toxic levels may be produced at early times. This is obviously undesirable.
3
There are several potentials inherent in multiple dose therapy:
• If the dosing interval is not appropriate for the biological half life of the drug, large “peaks” and “valleys” in the drug blood level may result. For example, a drug with a short half life requires frequent dosing to maintain constant therapeutic levels,
• The drug blood level may not be within the therapeutic range at sufficiently early times, an important consideration for certain disease states and
• Patient non-compliance with the multiple dosing regimen can result in failure in this approach.
Concept of Sustained Release Drug Delivery Systems
Sustained release systems include any drug delivery system that achieves slow release of a drug over an extended period of time. If the system is successful at maintaining constant drug levels in the blood or target tissues it is considered as a controlled release system. If it is successful at this stage but nevertheless extends the duration of action over that achieved by conventional delivery, it is considered as a prolonged release system which is illustrated in Figure 3.
There are several advantages associated with the development of sustained release drug delivery system:
• It improves patient compliance,
• It employs lesser quantity of the drug such as
o Minimizes or eliminates local side effects o Minimizes or eliminates systemic side effects
o Obtains less potentiation or reduction in drug activity with the
chronic use
o Minimizes drug accumulation with chronic dosing, • It improves the efficiency in treatment such as
o Cures or controls the condition more promptly
o Improves the control of condition like reduces fluctuations in the
4
o Improves bioavailability of some drugs and
• Economy: In comparison with conventional dosage forms the average cost of treatment over an extended period time may be less. Economy may also result from a decrease in nursing time and hospitalization. These specialized drug delivery systems are highly applicable to many drugs and in many therapeutic conditions, such as therapy of hypertension, arthritis, diabetes, fertility control, glaucoma, etc. Research and development activities in Novel Drug Delivery Systems (NDDS) have been in progress since 1950’s. Initially, pharmaceutical companies were slow to realize the potential of this new technology but with the successful incorporation of delivery systems in many therapeutics, pharmaceutical companies are now aggressively involved in increasing and improving their product pipeline9.
At present the drug delivery sector of global pharmaceutical market accounts for only 10 percent. But there is optimism among the industry executives that the world market may grow more than the current annual growth of 5 to 10 percent.
Oral Drug Delivery Systems
The oral route is the most ancient, convenient and commonly used route of administration. A few drugs are swallowed for their local action within the confines of the gastro intestinal tract, made possible by their insolubility and/or poor absorbability from this route. Compared with other routes of drug administration, the oral route has several advantages:
• It is safe,
• It is convenient,
• It is economical,
• Self medication is possible and
• Absorption is selective as per need (Iron in anemia).
5
The Anatomy and Physiological Consideration of the GIT
The human gastro intestinal tract (GIT) is a highly specialized region of the body, the primary functions of which involve the processes of secretion, digestion and absorption. Since all the nutrients needed by the body, with the exception of oxygen, must first be ingested orally processed by the GIT, and made available for absorption into the blood stream, the GIT represents an important barrier and interface with the environment. The human GIT can be divided into three distinct sections, namely, the stomach, the small intestine and the colon. After oral ingestion, materials are presented to the stomach, the primary functions of which is storage, mixing and reducing all components to slurry with the aid of gastric secretions and emptying these contents in a controlled manner into the upper small intestine of duodenum10.
The small intestine, with its enormous absorptive area of between 200 and 500 m2 is invariably the principal site of drug absorption. In contrast, the
stomach being a secretary rather than an absorptive organ and the colon having small absorptive area usually play very little role in the absorption of drugs. Nevertheless, particularly in the case of sustained release preparations, the colon may play an important absorptive role. Although some drugs like theophylline and metoprolol have been shown to be well absorbed in the colon, in general absorption from this part of this intestine is incomplete and erratic. Since transit times through the colon are highly variable ranging from less than 1 hour to more than 60 hour. Absorption from the distal part can be considered negligible since any drug will be embedded in semi solid faecal matter.
In view of the differences in the local environment and absorptive capacity of the three sections of the GIT, the duration of residence and transit times of a sustained release product in each section, can greatly affects its performance. Even if a product can be formulated to release its medicament independently of the local environment, its performance can still be influenced by the oracaecal transit time of the drug.
6
residence, the overall transit time of a dosage form can be extended. If the drug dissolves in the stomach contents, drug solution will pass in an unimpeded matter to the small intestine for subsequent absorption at the optimal site.
Once a drug molecule is in solution, it has the potential to be absorbed. Whether or not it is in a form available for absorption depends on the physicochemical characteristics of the drug and the characteristics of its immediate environment. For a drug molecule to be absorbed from the GIT and gain access to the body (systemic circulation) it must penetrate all the regions of the intestine.
All those variables in terms of pH, nature of luminal contents, length and surface area of the GIT may singly or in combination influence the drug release/absorption from a sustained release preparation. The other factors that influence the drug absorption from oral route are:
• Particle size of the drug: Smaller the particle size, greater is the absorption. A tablet containing larger aggregates cannot be disintegrated easily,
• Solubility and ionization: Nonionized drugs are more lipid soluble and therefore, absorbed more rapidly than the ionized drugs which are less lipid soluble,
• Concentration: Higher concentration favors absorption,
• Area of absorbing surface: Larger the absorption area, greater is the rate of absorption,
• Physical state of drug: Liquids are absorbed better than the solids and
• Functional state of the GIT: Diseased conditions may impair the absorption of drugs.
Oral Controlled Release Drug Delivery Systems (CRDDS) 11,12
7
steady-state blood concentration level within the therapeutic effective and non-toxic range for an extended period of time.
Oral route has been the commercially adopted and most convenient route for the drug delivery. Oral route of administration has been received more attention in the pharmaceutical field because of the more flexibility in the designing of dosage form than drug delivery design for other routes. The oral drug delivery design depends on various factors such as type of delivery system, the disease being treated, nature of patient, length of the therapy and the properties of the drug. Most of the CRDDS rely on diffusion, dissolution or combination of both mechanisms, to release the drug in a controlled manner to the gastrointestinal tract. By considering the conventional dosage form of a drug and the drug profile data, such as dose, absorption and elimination rate constants, metabolic properties, drug properties and the quantity of drug needed, one can determine the desired release rate of the drug from controlled release dosage form.
Novel oral drug delivery systems are broadly classified into two categories, as they may be controlled release dosage forms as well as targeting dosage forms. In general controlled drug delivery preparations release the drug in a controlled manner in the GIT for systemic uptake and no particular area of GIT specified. In contrast, targeted preparations are releasing the drug in a specified area or tissue of the GIT (e.g. colon specific drug delivery systems).
Oral controlled release dosage forms Vs conventional systems
Over the years there has been an enormous amount of work put into designing drug delivery systems that can eliminate or reduce the cyclical plasma concentrations seen after conventional drug delivery systems are administered to a patient according to a specified dosage regimen.
8
Delayed release indicates that the drug is not being released immediately following administration but at a later time, e.g. enteric coated tablets, pulsatile release capsules.
Repeat action indicates that an individual dose is released fairly soon after administration, and second or third doses are subsequently released at intermittent intervals.
Prolonged release indicates that the drug is provided for absorption over a longer period of time than from a conventional dosage form. However, there is an implication that onset is delayed because of an overall slower release rate from the dosage form.
Sustained release(SR) indicates an initial release of drug sufficient to provide a therapeutic dose soon after administration, and then a gradual release over an extended period.
Extended release (ER) dosage forms release drug slowly, so that plasma concentrations are maintained at a therapeutic level for a prolonged period of time (usually between 8 and 24 hours).
Controlled release (CR) dosage forms release drug at a constant rate and provide plasma concentrations that remain invariant with time.
Modified release (MR) dosage forms are defined by USP as those whose drug release characteristics of time course and/or location are chosen to accomplish therapeutic or convenience objectives not offered by conventional forms, whereas an extended-release (ER) dosage form allows a twofold reduction in dosing frequency or increase in patient compliance or therapeutic performance. It is interesting to note that the USP considers that the terms controlled release, prolonged release and sustained release are interchangeable with extended release.
Reasons for Developing Oral CRDDS
There is a clinical need to develop the oral CRDDS formulations to improve the drug therapy over that achieved with their conventional counterparts, especially in the cases of short elimination half-life (t½) and
9
half-life of a drug, larger will be the fluctuations between the maximum steady state concentration (Cmax) and the minimum steady state concentration (Cmin)
upon multiple dosing. If MEC is therapeutically required, either frequent dosing of a conventional drug product or development of a CR product is necessary.
Advantages of Oral CRDDS
They have many advantages over traditional, immediate release products.
Improved therapeutic efficacy: Reduction in drug plasma level fluctuations; maintenance of a steady plasma level of the drug over a prolonged time period, ideally simulating an intravenous infusion of a drug.
Reduced side effects: Drug plasma levels are maintained and improvement in tolerability within a narrow window with no sharp peaks and with AUC of plasma concentration versus time curve comparable with total AUC from multiple dosing with immediate release dosage forms. This greatly reduces the possibility of side effects.
Patient comfort and compliance: Oral drug delivery is the most common and convenient for patients, and a reduction in dosing frequency enhances compliance.
Reduced health care cost: The total cost of therapy of the controlled release product could be comparable or lower than the immediate-release product. With reduction in side effects, the overall expense in disease management also would be reduced.
Limitations of Oral CRDDS
On the other hand oral CRDDS suffer from a number of potential disadvantages:
• Relatively poor in vitro-in vivo correlation,
• Possible dose dumping,
• Reduced potential for dose change or withdrawal in the event of toxicity,
• Loss of effect due to diarrhoea (too fast transit time),
10
• Extensive first pass metabolism (except prodrugs),
• Extremely short elimination half life (low therapeutic index),
• Extremely long elimination half life (narrow therapeutic range),
• Bioavailability problems and
• Instability in GI environment.
Ideal drug candidates for CRDDS
The ideal characteristics of drugs to be used in the development of peroral CR dosage forms are given in Table 1.
To evaluate whether or not a drug is viable candidate for the design of peroral CR formulations, one must consider the pharmacokinetic parameters of the drug which are given in Table 2.
Types of Oral Sustained / Controlled Release Products:
I Dissolution controlled systems: A drug with a slow dissolution will demonstrate sustaining properties, since the release of the drug is limited by the rate of dissolution. Dexedine (Dextroamphetamine sulfate) and Nicobid (Nicotinic acid) are examples for this system.
II Diffusion controlled systems13-15.
They are further classified into,
a) Reservoir devices characterized by a core of drug reservoir surrounded by a polymeric membrane. Nico-100 (Nicotinic acid) and Nitrobid (Nitoglycerine) are examples for this system.
b) Matrix devices are those that consist of a drug dispersed homogenously throughout a polymer matrix. PBZ-SR (Tripelennamide Hcl) and Procan-SR (Procainamide Hcl) are examples for this system.
c) Bioerodable, combination diffusion and dissolution system: In this system the drug is homogenously dispersed in an erodable matrix polymer or the drug is linked directly to the polymer by a chemical bond. Generally the drug is released from the polymer by hydrolysis or enzymatic degradation.
11
to water or any body fluid, water will flow into the tablet to produce osmotic pressure and drug is released in a controlled manner.
e) Ion exchange system: In this system resins composed of water insoluble cross linked polymers are used. They contain salt forming functional groups in repeating positions of the polymer chain. The drug is bound to the resin and released by exchanging with appropriately charged ions in contact with ion exchange groups.
f) Prodrugs: A prodrug is a compound formed by the chemical modification of the biologically active compound that will liberate the active compound in vivo, by enzymatic or hydrolytic cleavage. The primary purpose of employing prodrugs for oral administration is to increase intestinal absorption and reduce local side effects16. The relative ease of production, cost of dissolution and
diffusion controlled systems compared with other methods of sustained or controlled delivery systems make them suitable delivery approaches17.
Monolithic Matrix System as Oral CRDDS18
In pharmaceutical oral CRDDS, matrix based systems are the most commonly used type of release controlling methodology owing to their simple manufacturing process. The preparation of a tablet with the matrix involves the direct compression of the blends of drug, release retardant and other additives, in which the drug is uniformly distribute throughout the matrix core of the release retardant. Alternatively, drug-release retardant blends may be granulated to make the mixture suitable for the preparation of tablets by wet granulation or beads.
To characterize and define the matrix systems the following properties of the matrix are considered:
• Chemical nature of the support,
• The physical state of the drug,
• The matrix and alteration in volume as the function of the time,
12
• The release kinetics model (in accordance with Higuchi equation, these system considered to release the drug as a function of square root of time).
The classification of the matrix-based systems is based on the following criteria:
• Matrix structure,
• Release kinetics,
• CR properties (diffusion, erosion and swelling),
• Chemical nature and
• Properties of the applied release retardant(s).
Based on the chemical nature of the release retardant(s), the matrix systems are classified as given in Table 3.
Mechanism of Drug Release from hydroxypropylmethylcellulose (HPMC) Matrix System19-20
Two important parameters for the release of drug from tablet matrices are the infiltration rate of medium into the matrix for those drugs with reasonable aqueous solubility, and the erosion rate of the matrix system for those drugs with poor aqueous solubility. In addition, the amount of drug loaded into the tablet also influences the release rate of the drug. The infiltration rate of medium into the matrix can be controlled by changes in the interspace volume of the matrix by the use of higher levels of materials such as lactose, which quickly rinse out of matrix system. The larger interspace volume produced by the higher ratio results in more rapid release of the drug. The viscosity of HPMC polymers is related to the molecular weight and has a large influence on the erosion rate of matrix tablet. The release rate of poorly soluble drug can be controlled by the rate of tablet erosion. The tablet erosion rate can also be adjusted by the choice of HPMC polymer viscosity or by mixing HPMC of different viscosity grades.
13
matrix followed by a relaxation process (hydration, gelation or swelling) and the other is the rate of erosion of the matrix. As a result of these simultaneous processes, two fronts are evident, a swelling front (glassy polymer/gel interface) and an eroding front (gel/medium interface). The distance between the two fronts (diffusion layer thickness) depends on the relative rates at which the swelling and eroding fronts move in relation to each other. The mathematical approaches most frequently used to describe the mechanism of drug release are those of Higuchi and Peppas.
Estimation of the Drugs in Biological Medium
Methods of measuring drugs in biological media are increasingly important problems related to the following studies and are highly dependent on biopharmaceutical analytical methodology;
• Bioavailability and bioequivalence studies,
• New drug development,
• Clinical pharmacokinetics and
• Research in basic biomedical and pharmaceutical sciences.
The most common samples obtained for biopharmaceutical analysis are blood and urine. Faeces are also utilized, especially if the drug or metabolite is poorly absorbed or extensively excreted in the bile. Other media that can be utilized include saliva, breath and tissue.
14
Detection of a drug or its metabolite in biological media is usually complicated by the matrix. Because of this, various types of clean up procedures involving techniques such as solvent extraction and chromatography are employed to effectively separate drug components from endogenous biological material. The ultimate sensitivity and selectivity of the assay method may be limited by the efficiency of the clean up methodology.
Separation or isolation of drugs and metabolites from biological samples is performed in order to partially purify the sample. In this manner, an analyst can obtain the selectivity and sensitivity needed to detect a particular compound and can do so with minimum interference from components of the more complex biological matrix. The number of steps in a separation procedure should be kept to a minimum to prevent loss of drug or metabolite. Sometimes, the separation steps are preceded by a sample pretreatment.
In order to avoid decomposition or other potential chemical changes in the drugs to be analyzed, biological samples should be frozen immediately upon collection and thawed before analysis. When drugs are susceptible to plasma esterase, the addition of esterase inhibitors such as sodium fluoride to blood samples immediately after collection helps to prevent drug decomposition.
In most cases, preliminary treatment of a sample is needed before proceeding to the measurement step. Drug analyses are required in samples as diverse as plasma, urine, faeces, saliva, bile, sweat and seminal fluid. Each of these samples has its own set of factors that must be considered before an appropriate pretreatment method can be selected. Such factors as texture and chemical composition of the sample, degree of drug-protein binding, chemical stability of the drug and types of interferences can affect the final measurement step.
15
samples can cause rapid deterioration of HPLC columns and also interfere the assay. Protein denaturation procedures include the use of tungstic acid, ammonium sulfate, heat, alcohol, trichloro acetic acid and perchloric acid.
Extraction of Drugs and Metabolites from Biological Samples
After pre treating biological material, the next step usually is the extraction of the drugs from the biological matrix. All separation procedures use one or more treatments of matrix-containing solute with some fluid. If the components are a liquid (extracting solvents) and a solid (e.g., lyophilized faeces), it is an example of liquid-solid extraction. If the extraction involves two liquid phases, it is an example of liquid-liquid extraction.
Liquid-solid extractions occur between a solid phase and a liquid phase. Either phase may initially contain the drug substance. Among the solids that have been used successfully in the extraction (usually via adsorption) of drugs from liquid samples are XAD-2 resin, charcoal, alumina, silica gel and aluminum silicate. Liquid-solid extraction is often particularly suitable for polar compounds that would otherwise tend to remain in the aqueous phase. The method could also be useful for amphoteric compounds that cannot be extracted easily from water.
The liquid solid extraction method provides a convenient isolation procedure for blood samples, thus avoiding solvent extraction, protein precipitation, drug losses and emulsion formulation. It is possible, however, that strong drug-protein binding could prevent sufficient adsorption of the drug to resin.
Liquid-liquid extraction is probably the most widely used technique because it can remove a drug or metabolite from larger concentrations of endogenous materials that might interfere with the final analytical determination and also this technique is simple, rapid and has a relatively small cost factor per sample.
16
solvents used. By knowing the partition coefficient for the extracted drug and the absolute volumes of the two phases to be utilized, the quantity of the drug extracted after a single extraction can be obtained. In multiple extraction methodology, the original biological sample is extracted several times with fresh volumes of organic solvent until maximum possible drug is obtained. As the combined extracts now contain the total extracted drug, it is desirable to calculate the number of extractions necessary to achieve maximum extraction. Factors that influence partition coefficient and hence recovery of drugs in liquid-liquid extraction are the choice of the solvent, pH and ionic strength of the aqueous phase. It is generally accepted that diethyl ether and chloroform are the solvents of choice for acidic and basic drugs, respectively, especially when the identity of the drugs in the samples is unknown. Chemically neutral drugs are extracted into either solvent depending on their relative partition tendencies.
The presence of metabolites or more than one drug in a biological sample usually demands a more sophisticated separation for their measurement especially, when two or more drugs are of similar physical and chemical nature. Chromatography is a separation technique that is based on differing affinities of a mixture of solutes between at least two phases. The result is a physical separation of the mixture into its various components. The affinities or interactions can be classified in terms of a solute adhering to the surface of a polar solid (adsorption), a solute dissolving in a liquid (partition) and a solute passing through or impeded by a porous substance based on its molecular size (exclusion).
Most of the drugs in biological samples can be analyzed by HPLC method because of several advantages like rapidity, specificity, accuracy, precision, ease of automation and eliminates tedious extraction and isolation procedures.
17
exchange chromatography, affinity chromatography and size exclusion chromatography (gel permeation and gel filtration chromatography).
Methods for analyzing drugs in biological samples can be developed, provided one has knowledge about the nature of the sample, namely, its molecular weight, polarity, ionic character and the solubility parameter. An exact recipe for HPLC, however, cannot be provided because method development involves considerable trial and error procedures. The most difficult problem usually is where to start, what type of column is worth trying with what kind of mobile phase. In general, one begins with reverse phase chromatography, when the compounds are hydrophilic in nature with many polar groups and are water soluble.
The organic phase concentration required for the mobile phase can be estimated by gradient elution method. For aqueous sample mixtures, the best way to start is with gradient reverse phase chromatography. Gradient can be started with 5 - 10 % organic phase in the mobile phase and the organic phase concentration (acetonitrile or methanol) can be increased up to 100 % within 20-30 min. Separation can be optimized by changing the initial mobile phase composition and the slope of gradient according to the chromatogram obtained from preliminary run. The initial mobile phase composition can be estimated on the basis of where the compounds of interest were eluted, namely, at what mobile phase composition.
Elution of drug molecules can be altered by changing the polarity of the mobile phase. The elution strength of a mobile phase depends upon its polarity, the stronger the polarity, higher is the elution. Ionic samples (acidic or basic) can be separated, if they are present in undissociated form. Dissociation of ionic samples may be suppressed by proper selection of pH.
18
Whenever acidic or basic samples are to be separated it is strongly advisable to control mobile phase pH by adding a buffer and the pH of the buffer should be adjusted before adding organic phase. The buffer selected for a particular separation should be used to control pH over the range of pKa ± 1.0. The buffer should transmit light at or below 220 nm so as to allow low UV detection.
19
2. AIM AND OBJECTIVES
Oral ingestion is the traditionally preferred route of drug administration, providing a convenient method of effectively achieving both local and systemic effects. In conventional oral drug delivery systems, there is very little control over the release of the drug and the effective concentration at the target site is achieved by intermittent administration of grossly excessive doses, which, in most situations, often results constantly changing, unpredictable and often sub or supra therapeutic plasma concentrations leading to marked side effects. However, peroral administration of drugs has disadvantages, such as hepatic first pass metabolism and enzymatic degradation within the gastrointestinal (GI) tract that prohibit oral administration of certain classes of drugs.
With a view to overcoming these problems, the current trend in pharmaceutical research is to design and develop new drug delivery systems so as to enhance the therapeutic efficacy of existing drugs. Moreover, the impetus for research in this area can be attributed to the exorbitant cost and large development period involved for “new drug development” when compared to the therapeutic advantages of newer drug delivery technologies. Sustained release (SR) technology of drugs has rapidly emerged over the past decades as a new interdisciplinary science that offers novel approaches to the delivery of bioactive agents into the systemic circulation. For the choice of the drug to be delivered, clinical needs and drug pharmacokinetics are some of the important considerations in the development of such formulations.
Several dosage forms have been developed and reported in literature for the sustained release of various bioactive materials of which matrix based tablets are one among them. Advantages associated with such dosage forms include high drug loading, simple and cost effective manufacturing process, the availability of a wider range of polymers and excipients for sustaining drug release and the possibility of using different mechanisms for sustaining the drug release.
20
varies with the nature of the matrix and its complex interaction of swelling, diffusion and erosion process. Release of drugs from such matrices can be sustained through their physical properties, the correct choice of gelling agent and setting up the conditions for fabrication. Among the hydrophilic polymers, hydroxypropylmethylcellulose (HPMC) and carbopol are widely used in controlled/sustained release dosage forms, because of their nontoxic in nature, its capacity to accommodate high levels of drug loading and their non pH dependence32. Acyclovir, a well known anti-viral drug used in the treatment of
herpes simplex virus mainly HSV-1, HSV-2 and varicella zoster19. The usual
dosing regimen of the drug in conventional dosage form is usually four to five times a day due to its shorter half-life of 2.5 hours and the oral bioavailability is 15–30%. Prochlorperazine maleate, a well known antiemetic and antivertiginoic used in treatment of nausea, vomiting (particularly in radiation induced cancer therapy) and in vertigo20. The usual dosing regimen of the drug is three to four
times a day due to its shorter half-life of 4 - 8 hours and the oral bioavailability is 5.7%.
However, a few literatures describe only in vitro experiments on acyclovir and prochlorperazine maleate21-22. There are no commercially
available sustained release formulations for the drugs of Acyclovir and Prochlorperazine maleate in India.
21
3. REVIEW OF LITERATURE
Several investigations have been carried out in the past on sustained /controlled release systems. A survey of literature was carried out in such investigations. In what follows, some of the important investigations are discussed.
Inmaculada Fuertes and coworkers21 have reported estimation of the
percolation thresholds in acyclovir hydrophilic matrix tablets. These statistical theory studies disordered or chaotic systems where the components are randomly distributed in a lattice. The application of this theory to study the release and hydration rate of hydrophilic matrices allows to explain the changes in release and hydration kinetics of swellable matrix type controlled delivery systems. The objective is to estimate the percolation threshold of HPMC K4M in
matrices of acyclovir and to apply the obtained result to the design of a hydrophilic matrices for the controlled delivery of this drug. Matrix tablets have been prepared using acyclovir as drug and HPMC K4M as matrix forming
material, employing five different excipient/drug percentages. Dissolution studies were carried out using the paddle method. Water uptake measurements were performed using a modified Enslin apparatus. In order to estimate the percolation threshold, the behavior of the kinetic parameters with respect to the excipient volumetric fraction at time zero plus initial porosity was studied. According to percolation theory, the critical points observed in dissolution and water uptake studies can be attributed to the excipient percolation threshold. This threshold was situated between 20.76% and 26.41% v/v of excipient plus initial porocity. The knowledge of percolation threshold of the components of the matrix formulations contributes to improve their design. First, reducing time to market and second, increasing their robustness when they are prepared at industrial scale, avoiding the formulations in the nearby of the percolation threshold.
S.Y.Vanarase and coworker22 have reported in vitro release studies of
22
Parojcic and coworkers23 have investigated the factors influencing drug
release from hydrophilic matrix tablets based on novel carbomer polymers. Drug release from hydrophilic matrix tablets can be strongly influenced by the proportion of matrix forming polymer and the dimensions and geometry of the tablets. A complete two-factor, three-level factorial design, followed by multiple regression analysis and response surface methodology, was applied to investigate the influence of polymer level and tablet size on drug release kinetics from hydrophilic matrix tablets prepared with Carbopol 971P and Carbopol 71G. Tablet diameter, radius-to-height ratio, tablet surface area, and surface-area-to-volume ratio were evaluated as independent variables in terms of their applicability to characterize tablet size and geometry. The results indicate that it may be possible to control the rate of drug release by modifying the proportion of carbomer in tablets and tablet dimensions. The practical benefit of these simulations is to optimize the geometry and dimensions of a controlled release device and reduce the number of experiments involved in the development of new controlled release dosage forms.
S.P.Hiremath and coworker24 have developed rifampicin oral controlled
release formulations of rifampicin using hydroxypropylmethylcellulose polymer at different ratios. They found from in vitro release data, that the release was extended with an increase of polymer proportion from 20% to 40%. However, increase in polymer beyond 40% resulted in no significant change in the release rate. But, there was a distinct difference in the release rate and release character due to variation in the compression force. The release kinetics was analyzed using Ritger and Peppas exponential equation. Stability studies at ambient storage conditions for 1 year showed that formulations were stable.
M.S. Bordaweka and coworkers25 have reported evaluation of polyvinyl
acetate dispersion as a sustained release polymer for tablets. Aqueous dispersion of poly vinyl acetate for the preparation of sustained release products for a water insoluble (ibuprofen) and water soluble drug (ascorbic acid). A 23 factorial design was employed to evaluate the optimum process
23
observed that a significant improvement on the flow properties of the granules. The dissolution rate of ibuprofen was decreased with the increase in the coating levels of poly vinyl acetate. Whereas in case of ascorbic acid, preparation of sustained release coated granules was difficult due to highly water soluble nature of drug. There was also a zero order type of release was observed on increasing the polymer concentration.
N.Vatsaraj and coworkers26 have optimized a sustained release tablet of
ketorolac tromethamine employing 23 full factorial design. They have studied
different variables such as amount of drug, ratios of hydroxylpropyl methylcellulose/sodium carboxy methylcellulose and ethyl cellulose. The swelling controlled matrix tablets were prepared by direct compression method. Analysis of variance (ANOVA) indicated that the release rate was affected by the HPMC/NaCMC ratio, amount of drug, two way and three way interactions, while the amount of drug, HPMC/NaCMC ratio, ethyl cellulose and interaction between the drug and polymer significantly affecting the diffusional exponent. The release mechanism of the drug from the matrix was super case II transport.
Z. Lu and coworkers27 have reported the in vitro dissolution, swelling
and erosion behavior of monolithic systems containing hydrophilic polymer hydroxypropylmethylcellulose, a combination of chitosan and poly carbophil in the form of inter poly electrolyte complex. They have reported that the in vitro
release profile of water soluble drug containing chitosan and poly carbophil complex exhibited higher mean dissolution time and slower drug release compared with other systems. The analysis also indicated that zero-order release kinetics was approached for some of the formulations containing the chitosan-polycarbophil complex, while this could not be achieved for those containing hydroxypropylmethylcellulose.
P.S. Hiremath and coworker28 have developed hydrophilic controlled
24
of viscosity, drug particle size, compression force and drug release. They have reported that the release rate of drug and mechanism of release from the polymer controlled by the viscosity grade of HPMC. A decrease in the drug particle size influenced the drug release.
J. Varshosaz and coworkers29 have developed sustained release
metoprolol tablets using natural gums and cellulose polymers by direct compression method. They have compared the physical characteristics of the tablets and water uptake studies with the standard formulation. Dissolution studies showed that formulations containing 100 and 80% of HPMC, 100% of guar, and 20% of xanthan followed the Higuchi model, while those containing 60 and 40% HPMC and 100 and 80% xanthan followed a zero order model. The tablets with 40% xanthan followed a Hixon-Crowell model. In cellulose derivatives, the highest MDT and dissolution efficiency until 8 hr (DE8%)
belonged to tablets with 40% HPMC, increasing the amount of CMC decreased the drug release rate, and formulations containing 60 and 40% of HPMC had the USP dissolution standards. While, in the gum formulations, the highest mean dissolution time and the lowest DE8% belonged to tablets with 100%
xanthan, increasing the xanthan decreased the release rate of metoprolol, and formulations containing 80 and 100% xanthan had the USP dissolution standards. Results showed that natural gums are suitable for production of sustained release tablets of metoprolol.
J. Varshosaz and coworkers30 have studied the sustained release systems
of metoprolol Tartrate prepared using different solid dispersion techniques such as melting and solvent methods, containing 7%, 15%, or 25% of the drug and different ratios of Eudragit RLPO and RSPO in ratios of 0:10, 3:7, 5:5, 7:3, and 10:0. The results showed that the drug release from dispersions was at a slower rate than pure drug and physical mixtures. Moreover, the formulations containing greater ratios of Eudragit RSPO showed slower release rates and smaller DE8% but larger mean dissolution time than those containing greater
25
those with the ratio of 3:7 (melting method) had similar release pattern to standard formulation (LopressorR) sustained release tablets by zero order and
Higuchi kinetics, respectively.
S. Mahesh Kumar and coworkers31 have developed a controlled release
(CR) matrix tablets of naproxen sodium were prepared by wet granulation using hydroxypropylmethylcellulose (HPMC-K-100 CR) as the hydrophilic rate controlling polymer. They have studied the effect of the concentration of the polymer and different fillers on the in vitro drug release rate. The studies indicated that the drug release can be modulated by varying the concentration of the polymer and the fillers. An optimized formulation subjected to accelerated stability studies for 3 months revealed that the developed CR tablets are stable. A complete cross-over bioavailability study of the optimized formulation of the developed CR tablets and marketed immediate release tablets was performed in healthy male volunteers. It was observed that the extent of absorption of drug from the CR tablets was significantly higher than that for the marketed naproxen sodium tablet due to lower elimination rate and longer half-life.
S.N. Meyyanathan and coworkers32 have developed sustained release
dextromethorphan hydrobromide tablets using hydroxylpropyl methylcellulose as rate controlling polymer. They have studied the effect of the concentration of the polymer and different fillers on the in vitro drug release rate. The studies indicated that the drug release can be modulated by varying the concentration of the polymer and the fillers. A complete cross-over bioavailability study of the optimized formulation of the developed CR tablets and marketed immediate release tablets was performed in healthy male volunteers. It was observed that the extent of absorption of drug from the CR tablets was significantly higher than that for the marketed naproxen sodium tablet due to lower elimination rate and longer half-life.
E.R. Rogelio and coworker33 have studied the influence of lactose on
26
pelanserin. Powder mixtures were wet-granulated with water and compressed in a hydraulic press at 55 MPa. Dissolution media were 900 ml HCl 0.1 N, the first 3 hours and phosphate buffer pH 7.4, 3 to 8 hours. Dissolution curves were described by peppas model, applied separately for each dissolution medium. Dissolution mechanism involved diffusion/relaxation with a linear trend favoring the diffusion mechanism with increasing lactose concentrations. Increasing lactose concentrations produced higher kinetic constants, in a cubic relationship. A double HPMC proportion produced slower dissolution rates, with a more gradual lactose release but including certain degree of erosion. High lactose concentrations compensated for a decreased solubility of pelanserin at pH 7.4.
N. Traconis and coworkers34 have reported the different technological
27
enclosed and bound to HPMC. Addition of EC produced no change in the Higuchi type release pattern from the matrix. Increasing proportions of EC corresponded to release rates that first decreased (1, 3, 5, 7, 9 and 10% EC; 34-27%/h½) and then increased (10, 30 and 50% EC; 27-42%/h½). These results are supposed to be the consequence of metronidazole diffusion through inter-particle pores and channels created by EC inter-particles loosely bound to HPMC, after percolation of void spaces at a critical EC concentration of approximately 13.8%.
L. Genc and coworkers35 have developed controlled release tablets of
dimenhydrinate with different polymers as MC, HEC, Carbopol 934, Eudragit RLPM and Eudragit NE 30 D at different concentrations (2.5–10%) by direct compression (DC) and wet granulation (WG) techniques. Magnesium stearate was the lubricant while starch gel was the binder. Pharmacotechnical parameters such as, weight deviation, hardness, friability, diameter–height ratio, content uniformity of the active substance and in vitro dissolution techniques were performed for different formulations.
Hsiu-O Ho and coworkers36 have reported the granulated excipients of
lactose or dicalcium phosphate with ethyl cellulose for their ability to be used as matrix materials to produce a controlled release dosage form by directs compression for captopril. The physical characteristics of both granulated excipients and their resulting tablets were evaluated. It was found necessary to add hydroxypropylmethylcellulose (HPMC) to improve the cohesion of granulated excipients. The hydrophobic nature of ethyl cellulose possibly hindered the release of captopril from all these matrix formulations. However, controllability of the in vitro release of captopril in lactose formulations appeared to be much better than that in dicalcium phosphate formulations. Three lactose tablet formulations with distinguishable in vitro dissolution rates were selected and compared with immediate release product for their in vivo
28
formulations tested indicating the sustainability of captopril release from matrix tablets using granulated excipients as a directly compressible matrix material.
Ifat Katzhendler and coworkers37 have investigated the influence of
hydroxypropylmethylcellulose (HPMC) on the crystal habit properties of carbamazepine in sustained release matrix tablets and in aqueous solutions using differential scanning calorimetry (DSC), X-ray powder diffraction and scanning electron microscopy (SEM). The results suggest that HPMC inhibits the transformation of carbamazepine to carbamazepine dihydrate in the gel layer of hydrated tablets and in aqueous solutions (depending on HPMC concentration), participates in its crystallization process and induces amorphism of carbamazepine crystals. The mechanism which explains these effects envisages the polymer serving as a template or microsubstrate for nucleation in the crystallization process. They assume that the interaction between the drug and polymer occurs by hydrogen bonding. The hydroxyl groups of the polymer may attach to the drug at the site of water binding, and thus its transformation to the dihydrate form, is inhibited. A more specific interaction involves structural matching (similar bond spacing distances) between inter atomic distances in the crystal lattice of carbamazepine dimer and intra-atomic distances along the polymer chain.
Amnon Hoffman and coworkers38 developed an oral sustained-release
29
The pharmacokinetics of the new formulation was evaluated in 12 healthy volunteers and compared to a conventional gelatin capsule with both formulations containing 500 mg amoxicillin. The plasma concentrations of active amoxicillin and penicilloic acid were determined by an HPLC method with a fluorometric detector. It was found that the area under the concentration–time curve and maximal serum amoxicillin concentrations following the sustained release preparation were lower than the immediate release formulation. However, the time over the required threshold concentrations, i.e. the minimal inhibitory concentration (MIC) as well as the more clinically relevant parameter — four times MIC of the drug against susceptible pathogens was found to be maintained for significantly longer periods. The results suggest that in order to achieve a twice daily dosing regimen that will provide therapeutic concentrations for the whole 12 hours dosing intervals, a larger dose of the new formulation should be given. (e.g. 750 mg or even 1 g twice daily). This dosing regimen will lead to increased patient compliance and improved therapeutic outcome.
Nobuyuki Tanaka and coworkers39 have designed a novel
sustained-release (SR) system, disintegration-controlled matrix tablet (DCMT), was developed for poorly water-soluble drugs. DCMT, consisting of wax and solid dispersion (SD) granules containing a disintegrant, could control the release of nilvadipine (NiD), a model compound by its disintegration. In the study, two DCMTs (DCMT-1 and DCMT-2) with different release rates of NiD were orally administered to beagle dogs, and in vivo absorption of NiD from DCMTs was compared with that from immediate-release (IR) tablets. DCMTs successfully sustained the absorption of NiD longer than IR tablets, while they did not decrease the bioavailability of NiD. DCMT-2, providing the slower release of NiD than DCMT-1, prolonged the absorption longer than DCMT-1. In vivo
30
of NiD after oral administration of DCMT-2. The results strongly indicate that the DCMT system would be a promising SR system, which could improve the solubility and sustain the absorption of poorly water-soluble drugs.
Y.Qiu and coworkers40 have developed hydrophilic matrix tablets for
oral controlled delivery of zileuton. The prototype formulations with drug loading of 50-60% were prepared and tested for in vitro using USP apparatus I, II and III. In vivo absorption of three formulations with differing release rates was evaluated using cross over designs in beagle dogs. The results indicated that the release of zileuton from the matrix tablets followed apparent zero order kinetics. Prolonged absorption of zileuton was achieved using the hydrophilic matrix system. In vivo drug release was estimated by deconvolution based on the data of both S-and-R-enantiomers was correlated with in vitro release. Linear relationships between in vitro and in vivo release were obtained with more rapid in vivo release than in vitro.
U.S.Toti and coworker41 have developed a polyacrylamide-grafted-guar
31
Ribeiro Laura and coworkers42 investigated the effect of multicomponent
complexation of vinpocetine, a poorly soluble base-type drug, with h-cyclodextrin, sulfobutylether h-h-cyclodextrin, tartaric acid, polyvinylpyrrolidone and hydroxypropylmethylcellulose (HPMC) on the design of controlled release hydrophilic HPMC tablets and to evaluate their in vitro release profiles by a pH gradient method. Multicomponent complexation led to enhanced dissolution properties of vinpocetine both in simulated gastric and intestinal fluids, and became possible the development of HPMC tablet formulations with more independent pH dissolution profiles. Drug release process was investigated experimentally using USP apparatus 3 and by means of model-independent parameters. Responses studied included similarity of dissolution profiles, time for 60% of the drug to dissolve (T60%), percent of vinpocetine released after 7
hours (PD7h) and the dissolution efficiency parameter at (DE12 h). Influence of
multicomponent complexation was proved to increase the release of vinpocetine from HPMC tablets and superior PD7h and DE12 h values were
obtained in formulations containing VP–CD–TA complexes. Results supported the use of HPMC matrices to provide a useful tool in retarding the release of vinpocetine and that dissolution characteristics of the drug may be modulated by multicomponent complexation in these delivery systems, suggesting an improvement on vinpocetine bioavailability.
Qing-Ri Cao and coworkers43 have studied the effect of incorporating
pharmaceutical excipients on the in vitro release profiles and the release mechanism of monolithic hydroxypropylmethylcellulose (4000 cps) matrix tablets (m-HPMC tablets) in terms of mimicking the dual drug release character of bi-layered TylenolR ER tablets was studied. They also compared the in vitro
release profiles of optimized m-HPMC matrix tablet and TylenolR ER tablet in
32
of pharmaceutical excipients incorporated. Starch 1500 (PrejelR) and sodium
lauryl sulfate (SLS) played a key role in determining the dissolution rate of m-HPMC tablets. Additional excipients, such as microcrystalline cellulose (Avicel PH101) and NaH2PO4 were used to tune the release profiles of m-HPMC tablets.
The effect of pharmaceutical excipients on drug release from HPMC-based matrix tablets was found to be mainly due to a change in hydrophilic gel expansion and on physical interactions between the drug and HPMC. The optimized m-HPMC tablet with a balanced ratio of PrejelR, SLS, AvicelPH101,
and NaH2PO4 in the formulation showed dual release profiles in water, pH 1.2
gastric fluids and pH 6.8 intestinal fluids in vitro. Dual release was defined as immediate drug release within few minutes followed by extended release over 8 h. The similarity factors of m-HPMC tablets and bi-layered TylenolR ER
tablets were 79.8, 66.1, and 82.7 in water, gastric fluid and intestinal fluid, respectively, indicating the equivalence of the two release profiles. No significant in vivo bioavailability differences were observed in healthy human volunteers. The developed m-HPMC tablet with dual release characteristics can be easily prepared using a conventional high-speed tablet machine and could provide an alternative to commercially available bilayered TylenolR ER tablets.
A.B. Silvina and coworkers44 developed uncoated HPMC matrix tablets
for evaluating the relationship and influence of different content levels of microcrystalline cellulose (MCC), starch, and lactose, in order to achieve a zero-order release of diclofenac sodium by wet granulation process. The in vitro
33
Higuchi and first order equations. The data obtained proved that the formulations are useful for a sustained release of diclofenac, due to the percentage released after 8 hours is nearly to 70%. The release of diclofenac sodium was influenced by the presence of MCC, and by the different concentrations of starch and lactose. Drug release kinetics from these formulations corresponded best to the zero-order kinetics. Compared to conventional tablets, release of the model drug from these HPMC matrix tablets was prolonged; as a result, an oral release dosage form to avoid the gastrointestinal adverse effects shall be achieved.
S. Ayhan and coworkers45 have prepared and evaluated the effect of
formulation variables on the release profile of diclofenac sodium from hydroxypropylmethylcellulose (HPMC) and chitosan. The matrix tablets were prepared by wet granulation and direct compression methods; employing different ratios of HPMC and chitosan. Physical properties of the prepared tablets were compared with the targeted commercial sustained release tablets. The drug release was studied in0.1 M HCl for 1 hour and phosphate buffer solution was added to reach pH value of 7.5. In vitro studies showed that 20% HPMC contained SR formulation by direct compression method was the optimum formulation due to its better drug release and best fitted formulation in to the zero order kinetics.
M. Selim Reza and coworkers46 investigated the effect of plastic,
34
among the drug release profile from different classes of polymeric matrices. The release kinetics was found to be governed by the type and content of polymer in the matrix system. Higher polymeric content (75%) in the matrix decreased the release rate of drug because of increased tortuosity and decreased porosity. At lower polymeric level (25%), the rate and extent of drug release was elevated. Carnauba wax was found to cause the strongest retardation of drug. The highest drug release was observed from HPMC matrices while Kollidon SR gave an intermediate release profile between these two polymers. Release rate was also found to be the function of physico-chemical nature of drug molecule. Theophylline and diltiazem HCl, being soluble in nature, released faster than diclofenac sodium from all matrix systems. The release mechanism was explored and explained with biexponential equation. Release profile showed a tendency to follow zero-order kinetics from HPMC matrix systems whereas Fickian (Case I) transport was predominant mechanism of drug release from Kollidon SR matrix system. The mean dissolution time (MDT) was calculated for all the formulations and the highest MDT value was obtained with Carnauba wax for all the drugs under investigation. The results generated in this study showed that the profile and kinetics of drug release were functions of polymer type, polymer level and physicochemical nature of drug.
L. Ochoa and coworkers47 have developed theophylline sustained release
matrix tablets based on the combination of hydroxylpropyl methylcellulose (HPMC K4M and K100M) and different meltable binders by melt granulation in a high-shear mixer. The dissolution profiles of each formulation were compared to those of TheoDur® 200 mg tablets and the mean dissolution time (MDT) and similarity factor (f2 factor) were calculated. The matrices swelling behavior was
35
mechanism. The investigation of matrices swelling behavior showed that the gel layer thickness increased continuously over the time period studied. Moreover, a correlation between gel layer thickness and strength with the percentage released was found. These results suggest that melt granulation could be an easy and fast method to formulate sustained release tablets.
S. Sumathi and coworker48 have examined the sustained release
behaviour of both water-soluble (acetaminophen, caffeine, theophylline and salicylic acid) and water insoluble (indomethacin) drugs from tamarind seed polysaccharide isolated from tamarind kernel powder. They have further investigated the effect of incorporation of diluents like microcrystalline cellulose and lactose on release of caffeine and partial cross-linking of the polysaccharide on release of acetaminophen. Applying exponential equation, the mechanism of release of soluble drugs was found to be anomalous. The insoluble drug showed near case II or zero order release mechanism. The rate of release was found to be in the decreasing order of caffeine, acetaminophen, theophylline, salicylic acid and indomethacin. An increase in release kinetics of drug was also observed on blending with diluents. However, the rate of release varied with type and amount of blends in the matrix. The mechanism of release due to effect of diluents was found to be anomalous. The rate of release of drug decreased on partial cross-linking and the mechanism of release was found to be super case II.
H. Abdelkader and coworkers49 have investigated on different types and
36
characteristics of the prepared granules significantly improved by virtue of granulation process. Also, the prepared matrix tablets showed good mechanical properties (hardness and friability). MC- and Alg-based tablet formulations showed high release-retarding efficiency, and good reproducibility and stability of the drug release profiles when stored for 6 months in ambient room conditions, suggesting that MC and Alg are good candidates for preparing modified release baclofen tablet formulations.
A.Hamid and coworkers50 have developed once-daily sustained release
tablet formulation of cefpodoxime using hydroxylpropylmethylcellulose. The formulation showed acceptable pharmacotechnical properties and assay requirements. In vitro dissolution studies indicated a sustained release pattern over a period of 24 hours. Drug release kinetics indicated that drug release was found to follow Higuchi’s equation with highest linearity and a close relationship with zero order kinetics. Koresmeyer’s plots indicated that the drug release was controlled by diffusion coupled with erosion.
P.R.Ravi and coworkers51 have designed oral controlled release matrix
tablets of lamivudine using hydroxylpropyl methylcellulose (HPMC) as the retardant polymer and studied the effect of various formulation factors such as polymer proportion, polymer viscosity, and compression force on the in vitro
37
but extended the release up to 20 hours. Mathematical analysis of the release kinetics indicated that the nature of drug release from the matrix tablets was dependent on drug diffusion and polymer relaxation and therefore followed non-fickian or anomalous release. No incompatibility was observed between the drug and excipients used in the formulation of matrix tablets. The developed controlled release matrix tablets of lamivudine, with good initial release (20%-25% in first hour) and extension of release up to 16 to 20 hours, can overcome the disadvantages of conventional tablets of lamivudine.
B.T. Sandip and coworkers52 have studied the effect of concentration of
hydrophilic (hydroxylpropylmethylcellulose [HPMC]) and hydrophobic polymers (hydrogenated castor oil [HCO], ethyl cellulose) on the release rate of tramadol. Hydrophilic matrix tablets were prepared by wet granulation technique, while hydrophobic (wax) matrix tablets were prepared by melt granulation technique and in vitro dissolution studies were performed using United States Pharmacopoeia (USP) apparatus type II. Hydrophobic matrix tablets resulted in sustained in vitro drug release (>20 hours) as compared with hydrophilic matrix tablets (<14 hours). The presence of ethyl cellulose in either of the matrix systems prolonged the release rate of the drug. Tablets prepared by combination of hydrophilic and hydrophobic polymers failed to prolong the drug release beyond 12 hours. The effect of ethyl cellulose and the presence of lactose and HPMC in the coating composition on the drug release was also investigated. Hydrophobic matrix tablets prepared using HCO were found to be best suited for modulating the delivery of tramadol hydrochloride.
Atul Kuksal and coworkers53 have developed extended release matrix