0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.23.14498–14506.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Analysis of Human Immunodeficiency Virus Type 1 Gag
Dimerization-Induced Assembly
Ayna Alfadhli, Tenzin Choesang Dhenub, Amelia Still, and Eric Barklis*
Vollum Institute and Department of Microbiology, Oregon Health & Science University, 3181 S.W. Sam Jackson Park Road, Portland, Oregon 97201-3098
Received 3 March 2005/Accepted 5 September 2005
The nucleocapsid (NC) domains of retrovirus precursor Gag (PrGag) proteins play an essential role in virus assembly. Evidence suggests that NC binding to viral RNA promotes dimerization of PrGag capsid (CA) domains, which triggers assembly of CA N-terminal domains (NTDs) into hexamer rings that are intercon-nected by CA C-terminal domains. To examine the influence of dimerization on human immunodeficiency virus type 1 (HIV-1) Gag protein assembly in vitro, we analyzed the assembly properties of Gag proteins in which NC domains were replaced with cysteine residues that could be linked via chemical treatment. In accordance with the model that Gag protein pairing triggers assembly, we found that cysteine cross-linking or oxidation reagents induced the assembly of virus-like particles. However, efficient assembly also was observed to be temperature dependent or required the tethering of NTDs. Our results suggest a multistep pathway for HIV-1 Gag protein assembly. In the first step, Gag protein pairing through NC-RNA interactions or C-terminal cysteine linkage fosters dimerization. Next, a conformational change converts assembly-restricted dimers or small oligomers into assembly-competent ones. At the final stage, final particle assembly occurs, possibly through a set of larger intermediates.
When expressed in cells, the precursor Gag (PrGag) pro-teins of retroviruses such as human immunodeficiency virus (HIV), Rous sarcoma virus (RSV), and murine leukemia vi-ruses are sufficient for the production of virus-like particles (VLP) (36). These proteins encode N-terminal matrix (MA) domains involved in membrane-binding (M) functions; vari-ably located late (L) domains, which are important for VLP budding; and protein-protein interaction (I) regions, com-posed of the capsid (CA) and nucleocapsid (NC) domains (36). The NC domains bind viral RNA, facilitating its encapsidation, and also have an assembly function (3, 5, 6, 7, 10, 11, 25, 26, 32, 33, 35, 37, 41, 42). Results from investigations on NC mutants are consistent with the notion that NC-RNA binding is essen-tial for efficient VLP assembly (3, 5, 6, 10, 11, 32, 33, 35, 41, 42). Previously, we showed that the assembly function of HIV type 1 (HIV-1) NC could be replaced by heterologous dimerization domains (42), an observation that was confirmed by others (1, 19), and suggested that the assembly role of the NC-RNA interaction is to foster dimerization of PrGag proteins. In vitro investigations on RSV Gag proteins carrying the CA N-terminal domains (NTDs), C-terminal domains (CTDs), and NC domains have supported this view (7, 25, 26) and led to a dimerization model for VLP assembly. As shown in Fig. 1A, Gag proteins composed of the CA NTD, CA CTD, and NC domains concentrate on RNA by virtue of the NC-RNA interaction. Close juxtaposition of the proteins leads to dimerization, which then triggers the assembly of higher-order oligomers.
Assuming that the assembly role of the NC-RNA interaction for all retroviruses is to usher the formation of
assembly-com-petent Gag dimers, then other mechanisms for pairing proteins should fulfill this function. To examine this assumption, we replaced the NC domain in HIV-1 Gag proteins with a glycine residue, followed by a readily accessible C-terminal cysteine residue. With these proteins, we were able to test the model in Fig. 1B to determine whether pairing of proteins by cysteine cross-linking or oxidation would lead to assembly. Interest-ingly, we found that cross-linking or oxidation of our C-termi-nal cysteine proteins did promote assembly of VLPs but that efficient assembly was temperature dependent. Alternatively, the temperature dependence could be obviated in C-terminal cysteine proteins that also have an N-terminal patch of basic residues, as long as polyanions were present in assembly reac-tions. Our results support the notion that Gag protein assem-bly is induced by dimerization promoted by NC-RNA interac-tions but that an additional environmental cue is needed for dimers to oligomerize efficiently into higher-order structures.
MATERIALS AND METHODS
Protein purification. The his-CASP1, CASP1, his-CASP1Cys, his2-CASP1Cys, and his2CASP1Cys mutant derivative proteins were expressed in Esch-erichia colistrain BL21(DE3)/pLysS (Novagen) from the bacterial expression plas-mids pET15-HIVCASP1, pET15-his2-HIVCASP1, pET15-HIVCASP1Cys, and pET15-his2-HIVCASP1Cys. The recombinant proteins are composed of the cap-sid domain and SP1 spacer, plus an N-terminal histidine tag of either MGSSH HHHHH SSGLV PRGSH MLED (his) or MGSSH HHHHH SSGLV PRGSH MLEDQ YGRKK RRQRR RD (his2) residues. The recombinant proteins ter-minate either precisely after the SP1 spacer (his-CASP1 and his2-CASP1) or after a glycine and a cysteine residue (his-CASP1Cys and his2-CASP1Cys). Bacterial expression and purification of proteins by nickel-chelate chromatogra-phy followed methods described previously (3, 27, 28). Purified fractions were desalted by buffer exchange in Sephadex G25 spin columns with 10 mM sodium phosphate (pH 7.8) and stored under nitrogen at⫺80°C. Purified (⬎95%) proteins at 0.8 to 3 mg/ml were evaluated after sodium dodecyl sulfate-polyac-rylamide gel electrophoresis (SDS-PAGE) (17, 29, 30, 41, 42), staining with 0.4% Coomassie blue R250 (Sigma) in 50% methanol–10% acetic acid, destaining in 50% methanol–10% acetic acid, and gel storage in 10% acetic acid. Proteins also
* Corresponding author. Mailing address: Vollum Institute and De-partment of Microbiology, Mail Code L220, Oregon Health & Science University, 3181 S.W. Sam Jackson Park Road, Portland, OR 97201-3098. Phone: (503) 494-8098. Fax: (503) 494-6862. E-mail: [email protected].
14498
on November 8, 2019 by guest
http://jvi.asm.org/
were evaluated by immunoblotting. As described previously (29, 41, 42), immu-noblot detection employed an anti-HIV capsid monoclonal antibody (Hy183) (29, 41, 42) as the primary antibody, an alkaline phosphatase-conjugated anti-mouse secondary antibody (Promega), and visualization of bands by using a color reaction of nitroblue tetrazolium plus 5-bromo-4-chloro-3-indolyl phosphate in 100 mM Tris hydrochloride (pH 9.5)–100 mM NaCl–5 mM MgCl2. To verify that
his-CASP1Cys and his2-CASP1Cys proteins retained their C-terminal cysteine tags, they were subjected to sulfhydryl quantitation with Ellman’s reagent (Pierce) and mass spectrometry. Mass spectrometry was performed by the Ore-gon Health & Science University Proteomics Core Facility on an Applied Bio-systems Qstar XL equipped with an ion spray source for acquisition of time-of-flight data. As observed previously (39), the results showed that⬎80% of the his-CASP1Cys and his2-CASP1Cys proteins were full length, except for the removal of the N-terminal formyl-methionine residue. The remaining species predominantly corresponded to full-length protein; no evidence for removal of C-terminal cysteines was observed.
Assembly incubations.For cross-linking, incubations of 10M of protein in 50 mM sodium phosphate (pH 7.5) were performed in 20-l reaction mixtures at 23°C, 30°C, or 37°C for 60 min in the presence or absence of 30M of the cysteine-specific cross-linking agent bis-maleimidohexane (BMH) (Pierce). For assembly induction by cysteine oxidation, incubations of protein (10M or 50
M) were performed in the presence or absence of 50M or 250MN,N,N⬘,N⬘ -tetramethylazo-dicarboxamide (diamide) (Sigma D3648) with or without 10% glycerol. To examine the effects of different treatments on protein assembly, proteins (10M) were incubated at 23°C for 60 min in the absence or presence of the following concentrations of reagents: 30 M diamide; 16.5g/ml of a single-stranded, 54-nucleotide (nt) DNA (5⬘ AGCTT GACTA CAAGG ACGAT GACGA TAAGA GAGGA TCTCA TCATC ATCAT TAAT 3⬘; In-vitrogen); 16.5g/ml heparin (Sigma); and 100M trypan blue (Sigma). After all incubations, reaction mixtures were centrifuged at 4°C for 10 min at 14,000⫻g, after which supernatant and pellet fractions were collected. Samples were frac-tionated on SDS–10% polyacrylamide gels, and proteins were visualized by Coomassie blue staining or by immunoblotting. For quantitation of assembly efficiencies, stained gels were scanned on an Epson Perfection 1240U scanner, and band intensities were quantitated using NIH image 1.61 software. Assembly efficiencies were defined as the percentages of total supernatant-plus-pellet pro-tein signals obtained in the pellet fractions.
Gradient centrifugation.Proteins at 50M were incubated for 60 min in the presence of 250M diamide at 23°C, 30°C, or 37°C. Samples then were applied onto 20 to 70% (wt/vol) sucrose gradients in 100 mM NaCl–50 mm HEPES (pH 8.0) and centrifuged at 105,000⫻gfor 2 h at 4°C. After centrifugation, fractions were collected from gradient tops to bottoms, separated by SDS-PAGE, and visualized by immunoblotting.E. coli70S ribosomes, purified following methods described previously (40), were used as markers for sedimentation analysis; spectrophotometric quantitation at 260 nm of rRNA in gradient fractions was used to detect ribosome fractions.
Microscopy.For fluorescence analysis, either buffer (50 mM sodium phos-phate [pH 7.5]) or 10M protein in buffer was treated for 1 h at 23°C with 30
M diamide prior to the addition of 1M fluorescein-conjugated single-stranded, 26-nt DNA oligonucleotide (5⬘TTGAC TCTCC CCCAG GAGGA GGTCTT) (Oligos Etc., Inc.). After DNA addition, samples were monitored for fluorescein fluorescence on a Zeiss fluorescence microscope equipped with a charge-coupled-device camera, at an original magnifications of⫻630.
For electron microscopy (EM) analysis, his-CASP1Cys assembly products were produced by treatment of 10M protein for 60 min at 23°C with 30M diamide, followed by a 5-min incubation at 37°C. For his2-CASP1Cys proteins, 10M protein was incubated for 1 h at 23°C in the presence of 30M diamide plus 16.5g/ml of a single-stranded, 54-nt DNA oligonucleotide. After incuba-tions, assembly products were lifted onto thin carbon-coated EM grids, nega-tively stained with 1.33% uranyl acetate, and imaged at 100 kV on a Philips CM120/Biotwin transmission EM equipped with a Gatan 794 multiscan charge-coupled-device camera at original magnifications of⫻2,850 to⫻37,000.
RESULTS
Assembly of C-terminal cysteine Gag proteins. To test
whether Gag proteins with NC domains replaced by cysteines could be triggered to assemble by cross-linking or cysteine oxidation, a set of recombinant Gag proteins was purified from bacteria. The proteins (Fig. 2) carried short N-terminal histi-dine (his) tags or longer, more basic histihisti-dine (his2) tags (27) for purification purposes. All proteins also carried the entire HIV-1 CA domain, as well as the natural spacer peptide (SP1) between CA and NC. However, the his-CASP1 and his2-CASP1 proteins terminated neatly at the end of SP1, while his-CASP1Cys and his2-CASP1Cys each had an additional gly-cine residue, followed by a C-terminal cysteine. All proteins were purified to⬎95% purity by using previously established protocols (3, 27, 28), and sulfhydryl quantitation and mass spectrometry analysis indicated that C-terminal cysteines were retained on the CASP1Cys proteins (see Materials and Meth-ods).
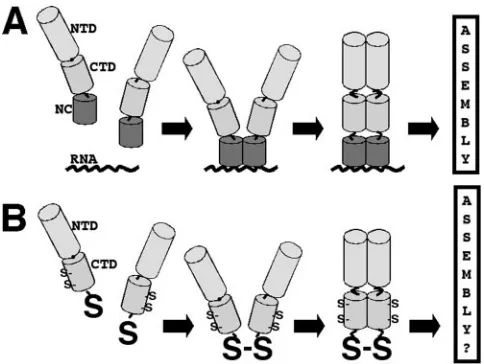
[image:2.585.42.284.68.250.2]Initially, we monitored whether BMH, a cysteine-specific cross-linker with a span of about 16 Å (17, 29, 30), could trigger FIG. 1. Nucleocapsid assembly function model. (A) Model for the
NC assembly function, based on observations that NC-RNA binding is needed for the assembly of wild-type Gag proteins and that protein dimerization domains can replace the NC-RNA interaction for virus particle assembly purposes. As illustrated, Gag proteins, composed minimally of the capsid NTD and CTD, the SP1 spacer peptide, and NC, concentrate on RNA targets permitting the appropriate dimer-ization of CTDs, which in turn induces NTD oligomerdimer-ization and assembly. (B) We hypothesize that replacement of NC with a readily accessible cysteine (S) residue will permit dimerization by cross-linking or oxidation, triggering the subsequent steps of the assembly process. Note that HIV-1 CA also has less accessible cysteines at CA residues 198 and 218.
FIG. 2. Recombinant HIV proteins. The primary product of the HIV-1gag gene is PrGag, which is N-terminally myristoylated and carries the MA, CA, NC, and p6 domains, along with spacer peptides between CA and NC (SP1) and between NC and p6 (SP2). The re-combinant HIV Gag proteins are composed of the capsid domain and SP1 spacer and an N-terminal histidine tag of either 24 (his) or 37 (his2) residues, and they terminate either precisely after the SP1 spacer (his-CASP1 and his2-CASP1) or following additional glycine plus cys-teine (S) residues (his-CASP1Cys and his2-CASP1Cys).
VOL. 79, 2005 HIV-1 Gag ASSEMBLY 14499
on November 8, 2019 by guest
http://jvi.asm.org/
the assembly of his-CASP1 and his-CASP1Cys proteins. Al-though his-CASP1 carries the two natural HIV-1 CA CTD Cys residues, little of this protein was cross-linked under our incu-bation conditions, and hardly any of the protein assembled into VLPs, as assessed by centrifugal separation into unassembled soluble fractions and assembled pellet fractions (Fig. 3B). In contrast to the CASP1 protein, BMH treatment of his-CASP1Cys proteins yielded a strong dimer band, as well as a secondary dimer band, which could represent cross-linking be-tween a C-terminal Cys and one of the CA CTD cysteines on a neighbor (Fig. 3A, lanes 3, 7, 11, and 12). BMH treatment of his-CASP1Cys proteins also generated higher-order cross-link products, suggesting that once proteins were cross-linked via C-terminal cysteines, CA cysteines became available for cross-linking to other neighbors, yielding small oligomers.
Despite the fact that small his-CASP1Cys oligomers were produced by BMH treatment at 23°C, higher-order complexes were not assembled, as indicated by the fractionation of pro-teins to the soluble rather than the pellet fraction (Fig. 3A, FIG. 3. Assembly induction by cross-linking. His-CASP1Cys (A) or his-CASP1 (B) proteins at 10M were incubated for 60 min at 23°C (lanes 1 to 4), 30°C (lanes 5 to 8), or 37°C (lanes 9 to 12) in the absence (lanes 1, 2, 5, 6, 9, and 10) or presence (lanes 3, 4, 7, 8, 11, and 12) of 30M BMH. After incubations, proteins in supernatant (S) and pellet (P) fractions were collected by centrifugation, separated by SDS-PAGE, and visualized by Coomassie blue staining. Marker sizes of 119, 96, 53, 37, and 28 kDa, determined from proteins run in a separate lane, are indicated by dashes to the right of the gels. Monomer, dimer, and oligomer bands, respectively, are indicated by single, double, and triple arrows. Note that apparent monomer and dimer doublet bands in panel A appear to correspond to alternatively BMH-conjugated or cross-linked forms.
FIG. 4. Assembly induction by cysteine oxidation. His-CASP1Cys (A to C) or his-CASP1 (D) proteins (50M) at 23°C (lanes 1 to 6), 30°C (lanes 7 to 10), or 37°C (lanes 11 to 14) either were untreated (lanes 3, 4, 7, 8, 11, and 12), salt treated (lanes 1 and 2), or oxidized with 250M diamide (lanes 5, 6, 9, 10, 13, and 14). After treatments, proteins in supernatant (S) and pellet (P) fractions were collected by centrifugation, separated by SDS-PAGE on either nonreducing (A, B, and D) or reducing (C) gels, and Coomassie blue stained (B to D) or immunoblotted to detect HIV CA proteins (A). Monomer, dimer, and oligomer bands, respectively, are indicated by single, double, and triple arrows. Note that doublet monomer and dimer bands presumably correspond to alternatively intra- and intermolecularly linked proteins. The molecular size markers (lanes 15) indicated by the dashes corre-spond to sizes of 119, 96, 53, 37, and 28 kDa.
on November 8, 2019 by guest
http://jvi.asm.org/
lanes 3 and 4). Because under some circumstances HIV-1 Gag proteins have been shown to assemble at 37°C and not 23°C (31) and because we have observed that elevated temperatures are necessary for optimal assembly of NC-containing HIV Gag proteins even in the presence of RNA (18), we examined the products of 30°C and 37°C his-CASP1Cys cross-linking reac-tions. As shown in Fig. 3A, while the results of 30°C incuba-tions (lanes 7 and 8) looked similar to those at 23°C (lanes 3 and 4), a substantial proportion of 37°C dimers and small oligomers was collected in the pellet fraction (lanes 11 and 12), suggesting that the cross-linked his-CASP1Cys proteins assem-bled into higher-order complexes at the higher temperature. Indeed, when we calculated the percentage of total protein detected in pellet fractions as a putative indicator of assembly efficiency, we found that BMH treatment at 37°C moved 52% of his-CASP1Cys to the pellet, versus 11% for the control his-CASP1 protein.
To verify and extend our observations, we examined his-CASP1Cys assembly on treatment withN,N,N⬘,N⬘ -tetrameth-ylazo-dicarboxamide (diamide), which covalently connects pro-teins by oxidation of free cysteines to cystine bonds (4, 20, 38). Because the bonds are sensitive to reduction, oligomer detec-tion required gel electrophoresis under nonreducing condi-tions. As shown in Fig. 4B, a small amount of our starting material was already oxidized (lane 3) but was not pelletable (lane 4). As a positive control for assembly, samples (lanes 1 and 2) at 23°C were treated with 2.5 M NaCl, which has been shown to induce assembly of HIV-1 CA monomers (21, 23). As predicted, salt treatment converted most of the his-CASP1Cys protein into an assembled, pelletable form (lane 2; cf. lane 1). As shown with BMH, diamide treatment at 23°C yielded dimers and oligomers, but less than 10% of the protein assem-bled into pelletable VLPs (Fig. 4B, lanes 5 and 6). However, raising the temperature to 30°C (lanes 9 and 10) and 37°C (lanes 13 and 14) resulted in a pronounced shift of dimer and oligomer products to the pellet fractions (43% and 77%, re-spectively), consistent with the BMH results. These results are in sharp contrast with the control his-CASP1 protein, where even 37°C diamide treatment yielded less than 15% pelletable material (Fig. 4D).
Although our his-CASP1Cys proteins were judged to be ⬎95% pure, it was of interest to verify that the observed dimer and oligomer bands in Fig. 4B were derived from HIV-1 CA. Thus, these protein fractions were electrophoresed on a sec-ond gel and immunoblotted with an anti-HIV CA antibody. As illustrated (Fig. 4A), monomer, dimer, and oligomer bands all were detected with the antibody. We also wished to demon-strate that diamide-induced dimers and oligomers were actu-ally linked via cystines. To do so, treated, centrifuged samples were subjected to electrophoresis on a reducing gel. Not sur-prisingly, the his-CASP1Cys bands coalesced as monomer-sized bands (Fig. 4C), indicating that dimers and oligomers induced by diamide indeed were linked by cystines. Taken together, the results in Fig. 3 and 4 suggest that covalent linkage of HIV-1 Gag proteins near their C termini can sub-stitute for the NC-RNA interaction and that efficient assembly requires an elevated temperature.
Analysis of assembly products.Although assembly reactions
routinely were performed at pH 7.5 in the absence of NaCl, similar results were obtained at pH 6.5, 7.0, and 8.0 and at salt
FIG. 5. Gradient fractionation of assembly products. His-CASP1Cys (A to C) and his-CASP1 (D) proteins at 50M were incubated for 60 min in the presence of 250M diamide at 23°C (A), 30°C (B), or 37°C (C and D) and then subjected to rate centrifugation at 105,000⫻gfor 2 h at 4°C. Samples were collected from gradient tops (lane 1) to bottoms (lane 12), fractionated by SDS-PAGE, and visualized by immunoblotting. For panel E,E. coli70 S ribosomes were centrifuged and detected by spectropho-tometric quantitation of rRNA in gradient fractions at 260 nm. Protein monomer and dimer bands are indicated by single and double arrows, respectively. Dashes indicate the migration positions for 53- and 28-kDa marker proteins run in parallel lanes.
VOL. 79, 2005 HIV-1 Gag ASSEMBLY 14501
on November 8, 2019 by guest
http://jvi.asm.org/
concentrations of up to 125 mM. However, assembly reactions were somewhat restrictive with regard to optimal cross-linker concentrations. In particular, assembly efficiency was greatest when the molar ratio of protein to BMH or the alternative cysteine-specific cross-linker bis-maleimidoethane (linker span, 8Å) ranged from 1:1 to 1:3. When protein-to-cross-linker ra-tios were either higher (3:1) or lower (1:10), assembly efficien-cies were markedly reduced, suggesting that assembly products were not simply highly cross-linked protein aggregates. This interpretation was supported by the inability of a primary amine cross-linker [bis(sulfosuccinimidyl)suberate; linker span 11Å (2)] to promote assembly in our reactions at protein-to-cross-linker concentrations of from 1:1 to 1:10.
To characterize assembly products, samples from diamide assembly reactions performed at 23°C, 30°C, and 37°C were subjected to rate centrifugation, after which fractions were separated on nonreducing SDS-polyacrylamide gels and pro-teins were detected by immunoblotting. As shown in Fig. 5A, products from his-CASP1Cys reactions at 23°C concentrated in the upper part of the centrifugation gradient, indicating that most of the monomer and dimer units were not assembled into higher-order structures. Similarly, reactions at 37°C with the control his-CASP1 protein showed high concentrations of
ma-terial in the top five gradient fractions, reduced levels in the middle fractions, and very little protein in the bottom fraction (Fig. 5D). In contrast, fractionation patterns of reactions at 30°C and 37°C showed shifts towards the bottoms of gradient tubes, as well as peaks in fractions 6 and 7 (Fig. 5B and C). Presumably, proteins at the bottoms of gradients (fractions 12) have assembled into VLPs and high-order assembly products. We speculate that the material in fractions 6 and 7, sediment-ing at a somewhat higher rate than bacterial 70S ribosomes (Fig. 5E), may correspond to previously observed assembly intermediates (22, 24, 31).
As a second means for characterizing assembly products, we resorted to transmission EM. For this, diamide assembly reac-tions were performed at 23°C, and mixtures were either kept at 23°C or shifted to 37°C prior to lifting of the products onto carbon coated EM grids, negative staining, and imaging. As expected, control reactions with his-CASP1 showed no evi-dence of higher-order assembly complexes (data not shown). Similarly, his-CASP1Cys reactions which were kept at 23°C also showed no complex formation (data not shown). In con-trast, reactions that were shifted to the higher temperature yielded a mixture of VLPs and small assembly aggregates (Fig. 6). The assembly aggregates (Fig. 6B, bottom right, and C, top half) frequently featured strings, crescents, and spirals of protein (Fig. 6C). The VLPs were roughly spherical with diameters of 136⫾23 nm, which is in the size range of HIV particles produced in vivo (14, 15). Thus, our observations support the view that chemically induced dimerization of his-CASP1Cys proteins can substitute for the NC-RNA interac-FIG. 6. Morphology of his-CASP1Cys assembly products.
His-CASP1Cys assembly products produced by treatment of 10M pro-tein for 60 min at 23°C with 30M diamide followed by a 5-min incubation at 37°C were lifted onto carbon-coated EM grids, negatively stained, and imaged. Assembly products were either particles or small aggregates (bottom right of panel B and top half of panel C). Particle diameters were 136⫾23 nm (n⫽20).
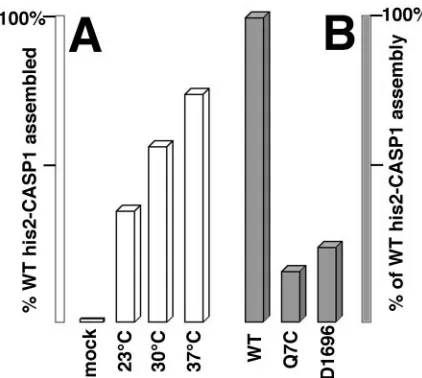
FIG. 7. Efficiency of his2-CASP1Cys assembly. His2-CASP1Cys pro-teins were induced to assemble by diamide treatment, fractionated into assembled pellet and unassembled supernatant fractions, separated by SDS-PAGE, and stained as described for Fig. 4. Band intensities from all protein species were quantitated densitometrically and used to calculate assembly efficiencies, defined as the percentage of total protein in the assembled (pellet) fraction. (A) Wild-type (WT) his2-CASP1Cys proteins were either mock treated or incubated in the presence of diamide at the indicated temperatures. (B) his2-CASP1Cys proteins either with the wild-type CA sequence or carrying NTD (Q7C) or CTD (D1696) mutations were assembled in the presence of diamide at 30°C. Assembly efficiencies were normalized to that of the WT protein.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.585.314.525.68.257.2]tion in HIV-1 Gag protein assembly but that efficient assembly also requires elevated temperature.
Alternative assembly reactions.In addition to his-CASP1Cys
proteins, we also tested the assembly properties of his2-CASP1Cys proteins, which carry an alternative, more basic N-terminal histidine tag (Fig. 2). Under our standard incuba-tion condiincuba-tions, his2-CASP1Cys proteins behaved similarly to their his-CASP1Cys counterparts, with regard both to pre-ferred molar protein-to-cross-linker ratios and to the improved assembly at elevated incubation temperatures (Fig. 7A). We also examined the assembly properties of two his2-CASP1Cys variants, his2-CASP1CysD1696 and his2-CASP1CysQ7C. When expressed in cell culture from proviral constructs, D1696 (with a five-codon insertion in the CA CTD) and Q7C (with a sub-stitution in the NTD)produce proteins which fail to assemble virus particles (reference 34 and data not shown). Significantly, when incorporated into the his2-CASP1Cys backbone and sub-jected to diamide-induced assembly conditions, these proteins assembled only 16 to 24% as efficiently as the wild-type his2-CASP1Cys protein (Fig. 7B). These results strongly support the notion that dimerization-induced assembly mimics the nat-ural assembly pathway.
Interestingly, we found that the elevated temperature re-quirement for efficient wild-type his2-CASP1Cys protein as-sembly was abrogated in the presence of polyanions. Figure 8A shows that compared to 23°C incubations with diamide alone (lanes 1 and 2) or DNA oligonucleotide alone (lanes 3 and 4), the combination of these two reagents markedly increased the levels of pelletable products in assembly reactions (lanes 5 and 6). Control incubations with his-CASP1Cys proteins failed to generate much pelletable assembly product (Fig. 8B), indicat-ing that the his2 tag was an essential component of these assembly reactions. Control incubations with his2-CASP1 pro-teins also failed to yield higher-order assembly products, dem-onstrating the requirement for the reactive C-terminal cys-teines.
The results from Fig. 8A to C suggested that as opposed to the normal function of nucleic acid in assembly reactions of NC-containing Gag proteins, oligonucleotides here were serv-ing to link or align CA NTDs to foster assembly. Given this
assumption, we tested alternative polyanions for their abilities to substitute for the temperature effect in his2-CASP1Cys as-sembly. As shown in Fig. 8D, both heparin (lanes 5 and 6) and trypan blue (lanes 3 and 4; four negative charges per molecule) worked successfully in conjunction with diamide to facilitate his2-CASP1Cys assembly, whereas neither set of treatments FIG. 8. Effects of different treatments on protein assembly. His2-CASP1Cys (A and D), his-CASP1Cys (B), and his2-CASP1 (C and E) proteins at 10M were incubated at 23°C for 1 h in the absence (⫺) or presence (⫹) of the following concentrations of reagents: 30M diamide (XL); 16.5g/ml of a single-stranded, 56-nt DNA oligonucleotide (DNA); 16.5g/ml heparin (hep); and 100M trypan blue (tb). After incubations, proteins in supernatant (S) and pellet (P) fractions were collected by centrifugation, subjected to SDS-PAGE on nonreducing gels, and visualized by Coomassie blue staining. Dashes on the right side of each panel indicate the migration positions of 96-, 53-, and 28-kDa marker proteins run on the same gels.
FIG. 9. Time course of his2-CASP1Cys particle assembly. Either buffer (A) or 10M his2-CASP1Cys protein (B to D) samples were treated for 1 h at 23°C with 30M diamide prior to the addition of 1/10 volume of 10M fluorescein-conjugated single-stranded, 26-nt DNA oligonucleotide. As quickly as possible after DNA addition, samples were imaged by fluorescence microscopy. Note that the mock image was collected within 1.5 min of DNA addition, which appears as the quickly quenching faint haze in the upper left of panel A. Immediately after DNA addition, protein samples appeared similar to that in panel A, but within 2.5 min (B), fluorescent foci were observed, with larger fluorescent aggregates appearing on prolonged incubation (C and D).
VOL. 79, 2005 HIV-1 Gag ASSEMBLY 14503
on November 8, 2019 by guest
http://jvi.asm.org/
worked for his2-CASP1 assembly. In contrast, similar concen-trations of several alternative anions, including citrate (three negative charges per molecule) and EDTA (four negative charges per molecule), failed to promote assembly, suggesting both charge and spacing requirements for the polyanion.
The ability of polyanions to promote his2-CASP1Cys assem-bly in combination with cysteine linkage allowed us to visualize the assembly process by fluorescence microscopy. To do so, proteins pretreated at 23°C with diamide were supplemented with a fluorescein-conjugated 26-nt DNA oligonucleotide and monitored microscopically. In control incubations, either with buffer (Fig. 9A) or with the his2-CASP1 protein (data not shown), oligonucleotide addition produced only a rapidly quenched haze of fluorescence. However, with his2-CASP1Cys incubations, as early as we could image (2.5 min), bright fluo-rescent spots appeared (Fig. 9B), indicating the concentration of fluorescent oligonucleotides into small assemblies. At later time points (Fig. 9C and D), a number of the smallest fluores-cent spots remained, along with some larger fluoresfluores-cent dots, suggesting a clustering of assembly products.
What is the morphology of these cysteine-linked and polyan-ion-treated his2-CASP1Cys assemblies? To address this question, assembly reaction products were lifted onto EM grids, stained,
and imaged by transmission EM. In contrast with control his2-CASP1 incubations, which showed no apparent assembly prod-ucts (data not shown), the his2-CASP1Cys assembly prodprod-ucts were remarkably uniform (Fig. 10). Although an occasional con-nected pair of spheres was observed (Fig. 10B), nearly all of the assembly products appeared as spherical VLPs. Often the VLPs showed a central spot (Fig. 10C to H), and frequently spiral tracks of protein could be seen (Fig. 10C, F, and H). While the polya-nion-induced his2-CASP1Cys VLPs were slightly smaller than their temperature-induced his-CASP1Cys counterparts, they still varied little in diameter (106⫾20 nm). Thus, induction of HIV-1 Gag protein assembly by our alternative means appears to have yielded a consistent product.
DISCUSSION
Efficient assembly of retroviruses such as HIV-1 generally re-quires an interaction between PrGag protein NC domains and the viral RNA (3, 5, 6, 7, 10, 11, 25, 26, 32, 33, 35, 37, 41, 42). In the absence of viral RNA, cellular RNAs appear to be used to fulfill this assembly function (33). In the absence of NC, assembly is impaired, unless the NCs are replaced by protein domains capa-ble of dimerization (1, 19, 42). These observations have led to a dimerization model for retrovirus Gag protein assembly, in which cobinding to RNA of NC domain-containing Gag proteins leads to dimerization, which then leads to assembly of higher-order products (Fig. 1A) (25, 26).
Our results indicate a flexibility with regard to the avenue by which dimerization is initiated. As depicted in Fig. 1B, replace-ment of NC domains by reactive thiols allowed us to initiate assembly via a cross-link mechanism. Indeed, assembly initia-tion of CASP1Cys proteins but not CASP1 proteins could be accomplished with the cysteine-specific cross-linkers BMH and bis-maleimidoethane or with the cysteine oxidation reagent diamide. In using any cross-linking agent, a natural concern is that putative assembly products might simply be highly cross-linked random protein aggregates. Several lines of evidence suggest that this is not the case here. Efficient assembly re-quired molar protein-to-cross-linker ratios in the range of 1:1 to 1:3, and both lower and higher ratios were less efficient. The amine-specific cross-linker bis(sulfosuccinimidyl)suberate did not substitute for cysteine-specific reagents. Capsid protein mutants which fail to assemble virus particles in vivo also were impaired for assembly in vitro (Fig. 7). Both dimers and larger covalent oligomers were found in assembled fractions, but they were not assembled in the absence of additional environmental cues (either polyanion addition or increased temperatures) (Fig. 3 to 5 and 8). Moreover, morphologies of assembly prod-ucts appeared to be regular, and VLPs were in the size range of virus particles (Fig. 6 and 10). While we speculate that the CASP1Cys protein packing in VLPs may reflect an immature hexameric tight-packing arrangement rather than a mature-form loosely packed organization (18, 27), proof of this as-sumption will require a detailed analysis of VLP Fourier spac-ings (27).
Although his-CASP1Cys and his2-CASP1Cys assembly was induced by protein linkage via C-terminal cysteines, the link-age was not sufficient for efficient assembly. However, raising incubation temperatures from 23°C to 30°C and 37°C yielded increasing amounts of pelletable assembly product (Fig. 3 to FIG. 10. Morphology of oligonucleotide-induced assembly
prod-ucts. his2-CASP1Cys protein (10M) was incubated for 1 h at 23°C in the presence of 30M diamide plus 16.5g/ml of a single-stranded, 56-nt DNA oligonucleotide. After incubation, assembly products were lifted onto carbon-coated EM grids, negatively stained, and imaged. The size bar for panels A and B is shown in panel A, while the size bar for panels C to H is in panel G. Particle diameters were 106⫾20 nm (n⫽50).
on November 8, 2019 by guest
http://jvi.asm.org/
5). For his2-tagged proteins, the addition of polyanions to reaction mixes could substitute for the increased temperature (Fig. 8). The 37°C preference for HIV-1 Gag protein in vitro assembly has been observed previously. Morikawa et al. dem-onstrated that bacterially expressed HIV-1 Gag proteins lack-ing the p6 domain preferentially assembled at 37°C (31). With similar NC-containing HIV-1 Gag proteins, we also observed an assembly temperature dependence, and while assembly lev-els increased with the addition of RNA at all temperatures, the temperature dependence was not removed (18). Consequently, our results imply a multistep mechanism for the initiation of HIV-1 Gag protein assembly. In the first step, Gag proteins must be paired by virtue of the NC-RNA interaction (7, 8, 9, 13, 25, 26), heterologous dimerization domains (1, 19, 42), or, in the current study, C-terminal cysteine linkage. Second, Gag dimerization occurs. We envision the third step, facilitated by increasing the temperature or by polyanion addition in our systems, to be a conformation change from assembly-restricted dimers or small oligomers to assembly-competent ones. At that stage, final particle assembly can occur, possibly via a set of larger assembly intermediates (22, 24, 31) (Fig. 5).
The assembly pathway outlined above is consistent with a number of observations. It is consistent with the temperature requirement for efficient HIV-1 Gag assembly (18, 31) (Fig. 3 and 4) and accounts for the observation of two different types of HIV-1 CA dimer (12). Our results implying a conforma-tional switch also are consistent with the observation that RSV Gag protein dimers do not self-assemble at pH 8.0 but can be induced to assemble at pH 6.5 (25, 26). Nevertheless, several reports have not noted a temperature influence on HIV-1 Gag assembly in vitro (8, 9, 13, 16). Variations in incubation con-ditions could be responsible for these differences; indeed, very high salt concentrations (2 to 2.5 M NaCl) induce the assembly of HIV-1 CA proteins, completely circumventing the need for an NC-RNA interaction (21, 23). The histidine tags also could have an impact, although we note that temperature effects were observed with our N-terminally His-tagged proteins, as well as the C-terminally His-tagged proteins described by Morikawa et al. (31). A third factor might be protein concen-tration, which would be expected to have an exponential effect on assembly rates. However, in the context of our own exper-iments, it is clear that protein concentrations are high enough to observe dimers and small oligomers but that these are not sufficient for the formation of higher-order products (Fig. 3 to 5 and 8).
Assuming that temperature increases and polyanion addi-tion (for his2-CASP1Cys) have similar effects on assembly, how do they exert their effects? Given that protein folding is temperature dependent (36), our observed temperature effects suggest that a conformational change must occur after dimer-ization. In this scenario, raised temperatures would increase the configurational diffusion (36) of Gag dimers, allowing them to find an assembly-competent conformation more readily. With regard to polyanion effects on his2-CASP1Cys protein as-sembly, two potential roles seem possible. One role might be the simple neutralization of basic charges in the histidine tag. Alter-natively, cobinding of one polyanion to two his2-CASP1Cys pro-teins might help align NTDs to facilitate the formation of assem-bly-competent dimers. We prefer the latter model, since it more readily explains why some polyanions (oligonucleotides, heparin,
and trypan blue) promoted assembly, while smaller polyanions (citrate and EDTA) did not. If this is the case, our results suggest that natural HIV-1 PrGag assembly in cells may be initiated by NC-RNA-induced dimerization and that subsequent conforma-tional alignment of capsid NTDs then leads to assembly. It is possible that membrane binding contributes to this NTD align-ment, thereby leading to a preference of lentiviruses for assembly on membranes (37).
ACKNOWLEDGMENTS
We are grateful for grant support from the National Institute of General Medical Sciences (NIGMS GM060170) to E.B. and for an American Foundation for AIDS Research Postdoctoral Fellowship Grant (amfAR 106523-35-RFNT) to A.A.
We thank Doug Huseby and Dalbinder Colman for electron micros-copy assistance and Isabel Scholz for help and advice.
REFERENCES
1.Accola, M., B. Strack, and H. Gottlinger.2000. Efficient particle production by minimal Gag constructs which retain the carboxy-terminal domain of human immunodeficiency virus type 1-capsid-p2 and a late assembly domain. J. Virol.74:5395–5402.
2.Alfadhli, A., E. Steel, L. Finlay, H. Bachinger, and E. Barklis.2002. Hanta-virus nucleocapsid protein coiled-coil domains. J. Biol. Chem.277:27103– 27108.
3.Barklis, E., J. McDermott, S. Wilkens, E. Schabtach, M. D. Schmid, S. Fuller, S. Karanjia, Z. Love, Y. Rui, X. Zhao, and D. Thompson.1997. Structural analysis of membrane-bound retrovirus capsid proteins. EMBO J.
16:1199–1213.
4.Boise, L., and C. Thompson.1999. Bcl-XL can inhibit apoptosis in cells that have undergone Fas-induced protease activation. J. Biol. Chem.274:27891– 27897.
5.Bowzard, J., R. Bennett, N. Krishna, S. Ernst, A. Rein, and J. Wills.1998. Importance of basic residues in the nucleocapsid sequence for retrovirus Gag assembly and complementation rescue. J. Virol.72:9034–9044.
6.Burniston, M., A. Cimarelli, J. Colgan, S. Curtis, and J. Luban.1999. Human immunodeficiency virus type 1 Gag polyprotein multimerization requires the nucleocapsid domain and RNA and is promoted by the capsid-dimer interface and the basic region of matrix protein. J. Virol.73:8527– 8540.
7.Campbell, S., and V. Vogt.1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol.69:6487–6497.
8.Campbell, S., and A. Rein.1999. In vitro assembly properties of human immunodeficiency virus type 1 Gag protein lacking the p6 domain. J. Virol.
73:2270–2279.
9.Campbell, S., R. Fisher, E. Towler, S. Fox, H. Issaq, T. Wolfe, L. Phillips, and A. Rein.2001. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc. Natl. Acad. Sci. USA98:10875–10879. 10.Cimarelli, A., S. Sandin, S. Hoglund, and J. Luban.2000. Basic residues in
human immunodeficiency virus type 1 nucleocapsid promote virion assembly via interaction with RNA. J. Virol.74:3046–3057.
11.Dawson, L., and X.-F. Yu.1998. The role of nucleocapsid of HIV-1 in virus assembly. Virology251:141–157.
12.Ehrlich, L., T. Liu, S. Scarlata, B. Chu, and C. Carter.2001. HIV-1 capsid protein forms spherical (immature-like) and tubular (mature-like) particles in vitro: structure switching by pH-induced conformational changes. Bio-phys. J.81:586–594.
13.Feng, Y.-X., T. Li, S. Campbell, and A. Rein.2002. Reversible binding of recombinant human immunodeficiency virus type 1 Gag protein to nucleic acids in virus-like particle assembly in vitro. J. Virol.76:11757–11762. 14.Fuller, S., T. Wilk, B. Gowen, H. Krausslich, and V. Vogt. 1997.
Cryo-electron microscopy reveals ordered domains in the immature HIV-1 parti-cle. Curr. Biol.7:729–738.
15.Gelderblom, H.1991. Assembly and morphology of HIV: potential effect of structure on viral function. AIDS5:617–638.
16.Gross, I., H. Hohenberg, T. Wilk, K. Wiegers, M. Grattinger, B. Muller, S. Fuller, and H.-G. Krausslich. 2000. A conformational switch controlling HIV-1 morphogenesis. EMBO J.19:103–113.
17.Hansen, M., and E. Barklis.1995. Structural interactions between retroviral Gag proteins examined by cysteine crosslinking. J. Virol.69:1150–1159. 18.Huseby, D., R. L. Barklis, A. Alfadhli, and E. Barklis.2005. Assembly of human
immunodeficiency virus precursor proteins. J. Biol. Chem.280:17664–17670. 19.Johnson, M., H. Scobie, Y. Ma, and V. Vogt.2002. Nucleic acid-independent
retrovirus assembly can be driven by dimerization. J. Virol.76:11177–11185.
VOL. 79, 2005 HIV-1 Gag ASSEMBLY 14505
on November 8, 2019 by guest
http://jvi.asm.org/
20.Kosower, N., E. Kosower, B. Wertheim, and W. Correa.1969. Diamide, a new reagent for the intracellular oxidation of glutathione to the disulfide. Biochem. Biophys. Res. Commun.37:593–596.
21.Lanman, J., J. Sexton, M. Sakalian, and P. Prevelige.2002. Kinetic analysis of the role of intersubunit interactions in human immunodeficiency virus type 1 capsid protein assembly in vitro. J. Virol.76:6900–6908.
22.Lee, Y., B. Liu, and X.-F. Yu.1999. Formation of virus assembly intermediate complexes in the cytoplasm by wild-type and assembly-defective mutant human immunodeficiency virus type I and their association with membranes. J. Virol.73:5654–5662.
23.Li, S., C. Hill, W. Sundquist, and J. Finch.2000. Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature407:409–413. 24.Lingappa, J., R. Hill, M. Wong, and R. Hegde.1997. A multistep
ATP-dependent pathway for assembly of human immunodeficiency virus capsids in a cell-free system. J. Cell Biol.136:567–581.
25.Ma, Y., and V. Vogt.2002. Rous sarcoma virus Gag protein-oligonucleotide interaction suggests a critical role for protein dimer formation in assembly. J. Virol.76:5452–5462.
26.Ma, Y., and V. Vogt.2004. Nucleic acid binding-induced Gag dimerization in the assembly of Rous sarcoma virus particles in vitro. J. Virol.78:52–60. 27.Mayo, K., D. Huseby, J. McDermott, B. Arvidson, L. Finlay, and E. Barklis.
2003. Retrovirus capsid protein assembly arrangements. J. Mol. Biol.325:
225–237.
28.Mayo, K., M. Vana, J. McDermott, D. Huseby, J. Leis, and E. Barklis.2002. Analysis of Rous sarcoma virus capsid protein variants assembled on lipid monolayers. J. Mol. Biol.316:667–678.
29.McDermott, J., L. Farrell, R. Ross, and E. Barklis.1996. Structural analysis of human immunodeficiency virus type 1 Gag protein interactions, using cysteine-specific reagents. J. Virol.70:5106–5114.
30.McDermott, J., S. Karanjia, Z. Love, and E. Barklis.2000. Crosslink analysis of N-terminal, C-terminal, and N/B determining regions of the Moloney murine leukemia virus capsid protein. Virology269:190–200.
31.Morikawa, Y., T. Goto, and F. Momose.2004. Human immunodeficiency virus type 1 Gag assembly through assembly intermediates. J. Biol. Chem.
279:31964–31972.
32.Muriaux, D., S. Costes, K. Nagashima, J. Mirro, E. Cho, S. Lockett, and A. Rein.2004. Role of murine leukemia virus nucleocapsid protein in virus assembly. J. Virol.78:12378–12385.
33.Muriaux, D., J. Mirro, D. Harvin, and A. Rein.2001. RNA is a structural element in retrovirus particles. Proc. Natl. Acad. Sci. USA98:5246–5251. 34.Reicin, A., S. Paik, R. Berkowitz, J. Luban, I. Lowy, and S. Goff.1995. Linker
insertion mutations in the human immunodeficiency virus type 1gaggene: effects on virion particle assembly, release, and infectivity. J. Virol.69:642– 650.
35.Sandefur, S., R. Smith, V. Varthakavi, and P. Spearman.2000. Mapping and characterization of the N-terminal I domain of human immunodeficiency virus type 1 Pr55(Gag). J. Virol.74:7238–7249.
36.Scalley, M., and D. Baker.1997. Protein folding kinetics exhibit an Arrhenius temperature dependence when corrected for the temperature dependence of protein stability. Proc. Natl. Acad. Sci. USA94:10636–10640.
37.Swanstrom, R., and J. Wills.1997. Synthesis, assembly and processing of viral proteins, p. 263–334.InJ. Coffin, S. Hughes, and H. Varmus (ed.), Retroviruses. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
38.Woo, M., C. Losasso, H. Guo, L. Pattarello, P. Benedetti, and M.-A. Bjornsti.
1997. Locking the DNA topoisomerase I protein clamp inhibits DNA rota-tion and induces cell lethality. Proc. Natl. Acad. Sci. USA94:3759–3764. 39.Yoo, S., D. Myszka, C.-Y. Yeh, M. McMurray, C. Hill, and W. Sundquist.
1997. Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol.269:780–795.
40.Yusupov, M., and A. Spirin.1988. Hot tritium bombardment technique for ribosome surface topography. Methods Enzymol.164:426–439.
41.Zhang, Y., and E. Barklis.1997. Effects of nucleocapsid mutations on human immunodeficiency virus assembly and RNA encapsidation. J. Virol.71:6765– 6776.
42.Zhang, Y., H. Qian, Z. Love, and E. Barklis.1998. Analysis of the assembly function of the human immunodeficiency virus type 1 Gag protein nucleo-capsid domain. J. Virol.72:1782–1789.