Copyright © 1988, American Society for Microbiology
Functional
Organization
of the Murine Leukemia Virus
Reverse
Transcriptase:
Characterization
of
a
Bacterially
Expressed
AKR DNA
Polymerase
Deficient in RNase H Activity
JUDITHG.
LEVIN,'*
ROBERT J. CROUCH,' KLARA POST,' STELLA C. HU,1t DOROTHY McKELVIN,1 MARTINZWEIG,2 DONALD L. COURT,3 ANDBRENDA I. GERWIN4Laboratory ofMolecularGenetics, NationalInstitute of ChildHealth andHuman Development, Bethesda, Maryland208921;Program Resources,Inc.,2 andLaboratory ofMolecular Oncology, NationalCancer Institute,3
Frederick Cancer Research Facility, Frederick, Maryland 21701; and Laboratory ofHuman Carcinogenesis, National CancerInstitute, Bethesda, Maryland 208924
Received11 April 1988/Accepted 15 July 1988
The functional organization of the murine leukemia virus reverse transcriptase was investigated by
expressing a molecular clone containing AKR MuLV reverse transcriptase-coding sequences inEscherichia
coli. Apurified preparation oftheexpressed enzyme(pRT250reversetranscriptase) consisted primarily ofa
69-kilodaltonproteinthat has normal levels ofmurineleukemiaviruspolymerase activitybut10-fold-reduced levelsofRNase H compared withthe viralenzyme. Thedeficit inRNaseH activity wascorrelatedwith the
absence of 60 to 65 amino acids normally present at the carboxyl end of murine leukemia virus reverse
transcriptase. The results provide additional experimental evidence for the localization of polymerase and RNase Hdomainsto theN- andC-terminal regions ofreversetranscriptase, respectively.
The reverse transcriptase enzyme is encoded by the pol gene and is synthesized as part ofa 200-kilodalton (kDa) gag-pol precursor that is cleaved during virus assembly to
give the mature virion protein (4). It functions early in the
replication cycle by catalyzing transcription of the viral RNA genome into a double-stranded DNA copy, which is
ultimately integrated into the host chromosome (33). The enzyme consistsofasingle polypeptide with several activi-ties(33).Theseactivities includethefollowing:
RNA-depen-dentDNApolymeraseactivity;DNA-dependent DNA poly-merase activity; and RNase H activity, which enables the enzyme todegrade RNAassociated with DNA in an RNA-DNA hybrid (18, 24, 29). Although the process of reverse transcription is complex (10) and requires the coordinated
interaction of each of theseactivities, in vitro studies with
inhibitors suggestedtheexistence ofindependentactive sites
forthepolymerase and RNase Hfunctions (3, 33).
The present study investigates the relationship between the genetic structure and functional activities of murine
leukemia virus (MuLV) reverse transcriptase. We report results showing that a purified bacterially expressed AKR enzyme which is missing 60 to 65 amino acids from the extremeC terminus ofthe viralproteinhas normal
polymer-ase activity but only 10% of the normal level of RNase H
activity. Our findings provide experimental support for the concept of separatefunctional domains within reverse
tran-scriptase.
Tocarry outthiswork, wemodified anearlierMuLVpol
clone,pRT24(15), and obtaineda newclone, pRT250,which should express only reversetranscriptase. Acomparisonof the structures of the pRT250 and AKR MuLV reverse
transcriptase proteins is illustrated in Fig. 1. At the N terminus of the pRT250protein, the XcIIpeptideconsisting of 13 aminoacids (15, 31) isjoinedtoamino acid 5ofreverse
*Correspondingauthor.
tPresentaddress: LaboratoryofBiochemistry,NationalCancer Institute, Bethesda, MD'20892.
transcriptase; at the C terminus, there is a substitution at amino acids 658 through 660 and residues 661 through 671 aredeleted. The molecular masspredicted for thisprotein is 74.6 kDa(75 kDa).
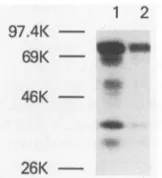
Clone pRT250was expressed inEscherichia coli DC520 (15, 31). Analysis of the bacterial proteins in Coomassie blue-stained sodium dodecyl sulfate-polyacrylamide gels showed that a unique 75-kDa protein is expressed and localized primarily in the insoluble fraction of a bacterial cell lysate (datanotshown).Westernblot(immunoblot)analysis of whole cell extracts with an anti-cII peptide monoclonal antibody (34) revealed the presence of the 75-kDa protein and indicated that it is the major cII peptide-containing product (Fig. 2). Some smallerproteins, including one which comigrates with the 69-kDa bovineserum albumin marker, were detected when ahigher concentration of extract was tested(Fig. 2, lane 1). Control experiments withextractsof the vector clonepWS50 (15, 31) did notreveal cII
peptide-containingproteins (data notshown) (34). The 75-kDa pro-tein and the lower-molecular-weight products could also be
immunoprecipitated from [35S]methionine-labeled extracts with anti-MuLV reversetranscriptase (data not shown).
Although much of the expressed protein is in theinsoluble cellfraction, standard biochemical procedures (see legendto Fig. 3) could be used to obtain enough enzyme from the soluble fraction for purification and characterization. The overall increase in the specific activity of the pRT250 en-zymewasabout 80-fold(datanotshown)andwas compara-ble to the specific activity achieved for a highly purified preparation of the viral enzyme (7,8). Atthe final phospho-cellulosechromatography step, the ratio ofRNA-dependent
polymerase activitytoDNA-dependent polymeraseactivity, as measured by comparing the relative responses to
poly(rA)-oligo(dT) and poly (dA)-oligo (dT), was 160 (data not shown).
The purity of individual fractions at each stage in the purification was assessed by electrophoresis in a sodium
dodecyl
sulfate-polyacrylamide
gel followedby silver stain-4376on November 10, 2019 by guest
http://jvi.asm.org/
GCTTAATTAATTAAGC
Icc GG
pRT250 pol Insert
22a5
,--I
--4682
SmaI end4825RT
5080
XmnI
AC1Ipeptide
pRT250Protein
1MVRANKRNEAL-RI
DQAARiNi5 658
AKR MuLV RT DQAAR| IKTPPDTSTLL
5 668 671
FIG. 1. Mapof clonepRT250polinsertand structuresofpRT250and AKR MuLVreversetranscriptaseproteins.ThepRT250clonewas
derived from the lacZ+ plasmid pRT24 (15), which containssequencesencoding the bacteriophage X cllpeptidejoinedtotheAKRMuLV reversetranscriptase coding region (minus the first12bases) plus 456 nucleotidesencoding the5'portion of theintegration protein(p46). The mapofthe pRT250 AKR MuLVpolfragment (nucleotidepositions numbered accordingto Herr[13]) shows the insertion ofauniversal termination linker(5'-GCTTAATTAATTAAGC)at auniqueSmalsitenearthe3'endofthe reversetranscriptase-coding region(nucleotide 4582). This insertioncreates aframeshift betweenreversetranscriptase andthep46andlacZsequences. The nucleotidesequenceatthe5' terminus ofthepolinsert was identical to thatofpRT24 (15). The sequence in the region of the termination linker wasverified with a
Sequenase kit(U.S. Biochemical Corp.). The reading frame is indicated by the brackets.Theamino acidsequencesof thepRT250protein andAKR MuLV reverse transcriptase (RT) wereinferred fromthenucleotidesequences and areidentical from amino acid residues5 to657. pRT250 and AKR MuLVaminoacid sequenceswhichdifferareboxed. Thefigure isnotdrawn toscale.
1 2 97.4K
-69K - s
46K
-26K
FIG. 2. Western blotanalysis of proteins expressed by plasmid clonepRT250inE.coli DC520 cells. ClonepRT250 in E. coli DC520 (15, 31)wasgrownat32°C in L broth containing 50 ,ug of ampicillin permluntil the opticaldensityat600nm was0.6. Toexpress genes
underthe control of the APLpromoter, the culturewas shiftedto 42°C for 15 min and then incubated at 37°C for 30 min. These induction conditions were chosento increase the yield of soluble pRT250 protein. The bacterial cell pelletwassuspended in loading
buffer (15), heated for 2minat100°C, sonicated for 15to30s,and
spunfor 1 min in anEppendorf microcentrifuge. The supernatant
fluid(approximately 1mgof proteinperml)wasdiluted 1:4 (lane 1)
or 1:16 (lane 2). Samples of 10 ,ul of each dilution were further diluted with10,ul of loading buffer, and the 20-,ul sampleswerethen
reheatedat 100°C for 2 min and subjected toelectrophoresis in a
12.5% sodium dodecyl sulfate-polyacrylamide gel. Protein bands reactingwithananti-cll peptide monoclonal antibodyweredetected
by incubation with the antibody as described previously (34) and
then with 200,000 cpm of 125I-labeled protein A per ml in Tris-bufferedsaline-0.5% Tween 20-0.5% bovineserumalbumin for 1 h at25°C. After the membraneswerewashed with thesamebuffered
solution, theywereair dried andexposedto Kodak XAR-5 filmat -70°C withaDupont Lightning-Plus intensifying screen. The
mo-lecular size markers shown onthe left are phosphorylase B (97.4 kDa), bovine serum albumin (69 kDa), ovalbumin (46 kDa), and
a-chymotrypsinogen (26 kDa).
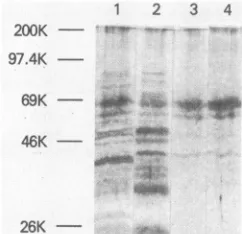
ing (Fig. 3). The DEAEflowthroughfraction andthe ammo-nium sulfate precipitate both contained many proteins,
al-though there appeared to be a somewhat prominent band comigratingwith the bovine serum albumin marker(69 kDa) (Fig. 3, lanes 1and 2). The
(dT)12-18-cellulose
fraction (lane 3), by contrast, contained predominantly the 69-kDa band and small amounts oflower-molecular-weight proteins. Thephosphocellulose-purified enzyme gave similar results, ex-cept that the concentration of the 69-kDa protein was increased relative to the smaller bands (lane 4). As the enzymewaspurified,theincrease in the 69-kDaproteinwas concomitant with theincrease inspecific polymeraseactivity
(data not shown), suggesting that the major polymerase
activity of the preparation is associated with the 69-kDa
protein.
Surprisingly, the purified fractions did not appear to contain the 75-kDaprimarytranslationproduct (Fig. 2).The 69-kDaprotein and the minorproteins present in the phos-phocellulose-purified material reactedwiththeanti-cII pep-tide monoclonal antibody (data not shown), indicating that C-terminal amino acids from the 75-kDaproductaremissing
in the 69-kDa protein. Similarly, extracts of two other
clones, pRT24andpRT235,alsocontainedlargeamountsof a69-kDaprotein (15) whichreactswith theanti-clIantibody (unpublished observation). In all likelihood, the 69-kDa
proteinisabacterialcleavage productformedbyC-terminal processing oftheprimary translation productasthe
overex-pressed viralprotein is solubilized within the bacterial cell.
Instability of the carboxyl-terminal structure ofbacterially expressed Moloney MuLV reverse transcriptase has also been observed (6).
The requirementsfor polymerase
activity
were examined with the phosphocellulose-purified pRT250 enzyme. Thebacterially expressed enzyme showed the same template preferencesandrequirementfor the divalentcationMn2+ as theAKRviral
protein
(7)and,asexpected, wasinhibitedbyon November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.140.499.72.252.2] [image:2.612.147.228.411.500.2]1 2 3 4 200K
97.4K
-69K
26K -
-FIG. 3. Gelanalysis of fractions obtainedduring the purification
ofthe pRT250 MuLV reverse transcriptase. The pRT250enzyme waspurifiedasfollows.Abacterial cell pellet (froma2-liter culture)
was suspended ina solution containing 10% sucrose, 10 mM Tris hydrochloride (pH 7.4), 1 mM EDTA, 0.3 M NaCl, and 1 mM phenylmethylsulfonyl fluoride (atacell/bufferratio of1:4) andwas incubated firstwithlysozyme(finalconcentration,1mg/ml) for 10to
20minoniceandthenwith'NonidetP-40ata'finalconcentrationof
0.2%at 37°Cfor30' min.Thelysatewascentrifuged inaBeckman
Ti6Orotor at30,000rpmfor30minat4°C.Thesupernatantfluidwas
removed and dialyzed overnight at 4°C against bufferA (50 mM Tris-HCI [pH 7.5],1mMEDTA,1mMdithiothreitol, 0.1%Nonidet
P-40, and 5% glycerol) containing 0.1 M NaCI. The dialysate (soluble extract) was applied to a DE-52 column (2.6 by 12 cm) equilibratedinbuffer Acontaining0.1MNaCI butwithoutglycerQl.
Theflowthroughfractionwascollected andbroughtto40%
satura-tionwithsolid ammoniumsulfate. Theammoniumsulfatepelletwas furtherpurifiedby sequential column chromatographyon (dT)1218-cellulose and phosphocellulose (7, 8). Peak fractions from the columns were concentrated 30-fold with the Cen'tricon-30 filter system (Amicon Corp.). Protein concentration was determined by
themethod of Bradford(1). Samples weresubjectedto
electropho-resis in a 10% sodium dodecyl sulfate-polyacrylamide gel, and
proteins weredetected bysilverstaining with the PierceGelCode
kit. Lanes: 1, DEAEflowthroughfraction(4 .gofprotein); 2,0to
40% ammonium sulfate precipitate (5.5' ,ug of protein); 3, pooled
(dT)
,-cellulose
fractions(0.45 ,ugofprotein); 4, pooledphospho-celluJsefractions (1.9 jigofprotein).Themolecularsize markers shown on the left are the same as in Fig. 2 with the addition of
myosin(200 kDa).
N-ethylmaleimide and the immunoglobulin G fraction of anti-MuLV reversetranscriptase (datanotshown). In addi-tion,thepRT250reversetranscriptase catalyzed synthesisof full-length cDNA in response to poly(A)+ mRNA from Xenopuslaevis embryos (datanotshown). Similar observa-tions have been made with other bacterially expressed MuLV reversetranscriptases (20, 30).
The studies on the DNA polymerase activity of the pRT250enzymedemonstrate thatminormodificationsatthe N'terminusof MuLVreversetranscriptase (Fig. 1)andmore
extensive alteration of the C terminus are compatible with expression of high levels of activity (Table 1; data not
shown). In thisconnection, it was alsoofinterest to assess thelevel ofRNase Hactivity andtodetermine whetheritis similar to that of the MuLV enzyme. Table 1 comparesthe polymeraseandRNase H activities of the purified viral and pRT250enzymes. The assays were performedunder condi-tions whereactivity was linear with enzyme concentration. The DNA polymerase specific activity values were very similarfor the twoenzymepreparations,whereasthere was a strikingdifference in the RNase Hspecific activities. The
[image:3.612.111.232.75.192.2]RNase Hactivityof thepRT250enzymewasalmost 10-fold lower than that of the AKR enzyme, resulting in a 15-fold higher ratio of polymerase activity to RNase H activity (3,500) for the bacterially expressed enzyme. Theseresults
TABLE 1. Comparisonofpolymeraseand RNase H activitiesof viral andbacteriallyexpressed MuLV reversetranscriptase
Reverse Polymerase RNase H Polymerase/
transcriptase activity activityb RNase Hratio
AKR MuLV' 1,236 5.5 225
pRT250 2,370 0.67 3,537
" Picomoles of[3H]dTMP incorporated permicrogram ofprotein in
re-sponsetopoly(rA)-oligo(dT). Polymerase activitywasassayedasdescribed
previously (25),exceptthatthe manganeseacetateconcentrationwas0.5 mM
andreactions wereincubatedfor 40minat37C.
bPicomolesof[3H]poly(rA)-poly(dT) degradedpermicrogramofprotein.
RNase H activity was assayed in 50-RI reactions containing 40 mM Tris
hydrochloride (pH 7.9), 1 mM manganese acetate, 40 mM KCI, 2 mM
dithiothreitol,and 20pmolof[3Hlpoly(rA)-poly(dT) (specificactivity,10,000 cpm/pmol).Reactionswereincubated for30 minat37C,andtrichloroacetic
acid-solubleradioactivitywasdetermined(11).The 3H-labeled substratewas
preparedby incubating10 mMTrishydrochloride (pH 8.0), 10mMMgCI2,14
mM2-mercaptoethanol,0.2A260unit ofpoly(dT),0.1 mMunlabeledATP,0.5
mCi of[3H]ATP(52.2 Ci/mmol;NewEnglandNuclearCorp.),and 18 UofE.
coli RNA polymeraseat 37°C for 30min in a final volume of 0.5 ml. The
labeledRNA-DNAhybridwaspurifiedasdescribedby HallandCrouch(11).
The viral enzyme was purified from 6 mg ofsucrose-densitygradient
purifiedAKRvirus (Electronucleonics;lot5029-6-55)asdescribedpreviously
(7,8).
indicate that the pRT250 reverse transcriptase has normal levels of polymerase activity but is deficient in RNase H activity. Recently it was reported that a bacterially ex-pressed mutant reverse transcriptase (pRT603), witha mo-lecular mass of approximately the same as (21) or smaller than(6) the 69-kDapRT250 protein, exhibits normal RNase Hactivity (21). Since the precise C termini of the proteins in question are not known, the apparent contradiction in the data cannot be resolved at present.
Our finding that a drastic reduction in RNase H activity
(Table 1) is correlated withdeletion of 60 to 65amino acids upstream of the C terminus of reversetranscriptase (Fig. 4)
provides strong experimental support for the existence ofa C-terminal RNase H domain. Other studies reporting evi-dence for an RNase H domain at the C termini of human immunodeficiency virus (12) and MuLV reverse
transcrip-tase (21, 32) have also appeared. Localization of the
poly-merase domain to the N-terminal two-thirds of MuLV re-verse transcriptase has been demonstrated by studies on a viral mutant reverse transcriptase (MuLV clone 23) (9, 26, 27) as well as by in vitro mutagenesis of MuLV molecular clones(21, 32). These results are in accord with a computer-based model previously proposed by Johnson et al. (16) suggesting that the reversetranscriptase protein consists of two separate domains, i.e., an N-terminal polymerase do-main and a C-terminal RNase Hdomain, joined togetherby
P DADHTUV TDG S S F LQEGQRX AGA AU T T ET E U I UARAL PA GTSAQ RAELI ALT Q L nAE G N R L U YTDSRYA FATA H I H
C E I Y RR RC L L T S H KS E I L ALL KAL F L P N R L S I I H C LGHQK G D S A E A RGN R L A D S A A R E AA Il T P P D T S T L L
FIG. 4. Carboxyl-terminal regionof AKR MuLV reverse tran-scriptase.The amino acid sequencewasobtainedbytranslationof the nucleotide sequence (13) and represents residues 515 though 671, where number 1 is the first amino acid ofreversetranscriptase. The boxed sequence indicates the approximate location of the C terminusof the 69-kDa pRT250protein. Aminoacids presentatthe
samerelative position in all sequences examined in the alignment (see text) of Itaya et al. (in preparation) are indicated with an
asterisk(*).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.309.549.96.143.2]a poorly conserved "tether" region. In an independent study, Itayaetal. (M. Itaya, D. McKelvin, S. F. Chatterjie, M.-L.Dirksen,and R. J.Crouch, manuscriptinpreparation) havealignedthe C-terminal amino acidsequencesof several viral reverse transcriptases (Rous sarcoma virus, human immunodeficiency virus, and Moloney and AKR MuLV) with the Saccharomyces cerevisiae (Itayaetal., in
prepara-tion) and E. coli(17) RNase H sequences. The data are in agreement with the RNase H domain boundaries predicted by Johnsonetal. (16)and showhomologieslocated through-out the sequences examined. Eleven amino acids are
con-served in all of the examples (see residues marked with asterisks in AKRsequence, Fig. 4). It should be noted that roughly half of the RNase H domain is eliminated in the 69-kDa pRT250 protein, including 3 of the 11 conserved residues (Fig. 4). One of these missing amino acids is a
highly conserved histidine which could be important for enzymatic activity (2).
Inconceptualizingthe organizationof the multifunctional
reverse transcriptase protein, it is important to consider whetherreversetranscriptase isatruly modularenzymeand toask, for example,whetherthe polymerasedomain is fully active in the absence of the RNase H domainor vice versa.
Experimentally itis foundthat smallfragmentsof avian(22) and MuLV (5) reverse transcriptase as well as defined C-terminalfragments of the human immunodeficiencyvirus (12) and MuLV (32) enzymes exhibit RNase H activity without polymerase activity. The present results and work
on theclone 23 mutant(9, 26, 27) and bacterially expressed Moloney MuLV reverse transcriptases lacking RNase H activity (21, 32) show that polymerase activity can be expressedinthe absence of C-terminalsequences.Although polymerase and RNase H activities can be separated in vitro, this situation may not represent normal biological activity. Thus, the truncated proteins having RNase H activitybutnopolymerase activityhave lost theabilityto act inaprocessivemanner (5, 12, 28). In thecase of the human immunodeficiency virus reverse transcriptase, removal of C-terminalsequences (12, 14, 23), even asfew as 23 amino acids (14), drastically reduces polymerase activity.
More-over, transfection studieswith amolecularly cloned MuLV
ecotropic endogenous virus have shown that a naturally
occurring singleamino acidchangefrom alaninetoprolinein
the "tether" region is correlated with lowered polymerase activityand loss ofinfectivityin progeny particles (19).
These observations suggest that the
polymerase
and RNase H domains interacttoanextentwhichmay varywith individual retroviralenzymes. This isnot surprisingin view of the fact that catalytic function is often enhanced by protein folding, which places distant residues in close con-tact with one another. The "tether" region, althoughnon-conserved, could also play a structural role in maintaining
the proper configuration of the protein. Furthermore, in
considering the implications for virus replication, it should berecognizedthatreversetranscriptaseand viral RNAform
a functional unit, possibly as part of a larger replication complex. Each representsasingle molecule with morethan
one function: reverse transcriptase has several enzymatic activities, and the RNA functions as both template and substrate for these activities. The interaction is dynamic, and there is evidence that polymerization of minus-strand DNA and RNase H digestion of the viral RNA template proceed simultaneously (33). Clearly, additional experimen-talapproachesareneededtoobtainadetailedunderstanding
of the structure-function relationships of the reverse
tran-scriptaseenzyme.
It is a pleasure to thank Michael Seddon and Steve Joe for outstanding technical assistance. WeareindebtedtoThomas Sar-gentforagiftofX.laevisembryonicmRNA and forgenerousadvice andhelpwithcDNAanalysis.WearealsogratefultoAnthonyFaras and AnnaMarie Skalkafor valuablediscussion andtoAlanRein for critical readingofthe manuscript. Kathleen Shoobridge
provided
expertassistance in thepreparationof themanuscript.LITERATURECITED
1. Bradford, M. M. 1976. Arapid and sensitive method for the quantitation of microgram
quantities
of proteinutilizing
the principleofprotein-dyebinding. Anal. Biochem. 72:248-254. 2. Crestfield,A.M.,W. H.Stein,and S.Moore. 1963.Alkylationand identification of the histidine residuesattheactive site of ribonuclease. J. Biol. Chem.238:2413-2420.
3. Crouch, R. J.,and M. L. Dirksen. 1982. Ribonucleases H, p. 211-241. In S. Linn and R. Roberts (ed.), Nucleases. Cold SpringHarborLaboratory,ColdSpring Harbor, N.Y. 4. Dickson, C., R. Eisenman, H. Fan, E. Hunter, and N. Teich.
1982. Protein biosynthesis and assembly, p. 513-648. In R. Weiss, N. Teich, H. Varmus, and J. Coffin (ed.), Molecular biology oftumor viruses, RNA tumor viruses, 2nd ed. Cold SpringHarborLaboratory, ColdSpring Harbor, N.Y. 5. Gerard,G. F. 1981. Mechanism of action ofMoloney murine
leukemia virusRNase H III. J. Virol.37:748-754.
6. Gerard,G.F.,J.M.D'Alessio,M.L.Kotewicz,andM.C. Noon.
1986. InfluenceonstabilityinEscherichiacoliof the
carboxy-terminal structure of cloned Moloney murine leukemia virus
reversetranscriptase. DNA 5:271-279.
7. Gerwin, B..I., and J. G. Levin. 1977. Interactions of murine leukemia virus core components: characterization of reverse
transcriptase packagedin theabsence of 70SgenomicRNA. J.
Virol. 24:478-488.
8. Gerwin, B. I., and J. B. Milstien. 1972. An oligonucleotide
affinity column for RNA-dependent DNA polymerase from
RNAtumorviruses. Proc. Natl.Acad.Sci. USA69:2599-2603.
9. Gerwin,B. I.,A.Rein,J.G.Levin,R.H.Bassin,B.M.Benjers,
S.V. S.
Kashmairi,
D.Hopkins,and B.J. O'Neill. 1979.Mutant ofB-tropicmurine leukemia virussynthesizinganaltered poly-merasemolecule. J.Virol. 31:741-751.10. Gilboa, E., S. W. Mitra, S. Goff, and D. Baltimore. 1979. A detailed model of reverse transcription and tests of crucial aspects.Cell 18:93-100.
11. Hall,S. H.,and R.J.Crouch.1977.Isolation and characteriza-tion of two enzymatic activities from chick embryos which degradedouble-stranded RNA. J. Biol. Chem. 252:4092-4097. 12. Hansen, J., T. Schulze, W. Mellert, and K. Moelling. 1988.
Identification and characterization ofHIV-specificRNaseHby
monoclonalantibody. EMBO J. 7:239-243.
13. Herr, W. 1984. NucleotidesequenceofAKVmurine leukemia
virus. J.Virol.49:471-478.
14. Hizi, A., C. McGill, and S. H. Hughes. 1988. Expression of soluble, enzymatically active, human immunodeficiency virus
reverse transcriptase in Escherichia coli and analysis of
mu-tants. Proc.Natl. Acad. Sci. USA 85:1218-1222.
15. Hu,S.C.,D.L.Court,M.Zweig,andJ.G. Levin.1986.Murine leukemia viruspol geneproducts: analysiswith antisera gener-ated against reverse transcriptase and endonuclease fusion
proteins expressedinEscherichiacoli. J;Virol.60:267-274. 16. Johnson,M.S.,M. A. McClure, D. F.Feng,J.Gray,and R. F.
Doolittle. 1986. Computer analysis of retroviral pol genes: assignment ofenzymatic functions to specific sequences and homologieswithnonviralenzymes. Proc. Natl.Acad. Sci.USA 83:7648-7652.
17. Kanaya,S.,andR.J.Crouch. 1983. DNAsequence
of
thegenecodingforEscherichiacoliribonuclease H. J. Biol. Chem.258: 1276-1281.
18. Keller, W., and R. Crouch. 1972. Degradation of DNA-RNA hybrids by ribonuclease H and DNA polymerases of cellular and viralorigin. Proc. Natl. Acad. Sci. USA 69:3360-3364. 19. King, S.R., B. J. Berson, andR. Risser. 1988. Mechanism of
interaction between endogenous ecotropic murine leukemia viruses
int
(Balb/c x C57BL/6)hybridcells. Virology162:1-11.on November 10, 2019 by guest
http://jvi.asm.org/
20. Kotewicz, M. L., J. M. D'Alessio, K. M. Driftmier, K. P. Blodgett,andG. F. Gerard. 1985. Cloning andoverexpressionof Moloney murine leukemiavirusreverse transcriptasein Esch-erichia coli. Gene35:249-258.
21. Kotewicz, M. L., C. M. Sampson, J. M. D'Alessio, and G. F. Gerard. 1988. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. NucleicAcids Res. 16:265-277.
22. Lai, M.-H. T., and I. M. Verma. 1978. Reversetranscriptaseof RNA tumorviruses.V. Invitro proteolysis ofreverse transcrip-tasefrom avian myeloblastosis virusandisolation ofa polypep-tide manifestingonly RNase Hactivity.J. Virol.25:652-663. 23. Lardcer, B., D. Purifoy, K. Powell, and G. Darby. 1987. AIDS
virusreverse transcriptasedefined by high level expressionin Escherichia coli. EMBOJ.6:3133-3137.
24. Leis, J. P.,I. Berkower, and J. Hurwitz. 1973. Mechanism of action of ribonuclease H isolated from avian myeloblastosis virus and Escherichiacoli.Proc.Natl. Acad. Sci. USA 70:466-470.
25. Levin, J. G., P. M. Grimley, J. M. Ramseur, and I. K. Berezesky. 1974. Deficiency of 60 to 70S RNA in murine leukemia virus particles assembledincellstreated with actino-mycinD. J.Virol. 14:152-161.
26. Levin, J. G., S. C. Hu, A. Rein, L. I. Messer, and B.I.Gerwin. 1984. Murine leukemia virus mutant with a frameshift in the reverse transcriptase codingregion: implications for polgene structure.J.Virol.51:470-478.
27. Messer, L. I., K. M. Currey, B. J. O'Neill, J. V. Maizel, Jr., J. G. Levin, and B. I. Gerwin. 1985. Functional analysis of reverse transcription by a frameshift pol mutant of murine
leukemiavirus.Virology146:146-152.
28. Moelling, K. 1976. Furthercharacterization of the Friend
mu-rineleukemia virusreversetranscriptase-RNaseHcomplex.J. Virol. 18:418-425.
29. Moelling, K., D. P. Bolognesi, H. Bauer, W. Busen, H. W. Plassmann, and P. Hausen. 1971. Association of viral reverse
transcriptase with an enzyme degrading the RNA moiety of RNA-DNAhybrids. Nature(London)New Biol.234:240-243. 30. Roth, M.J., N. Tanese, and S. P.Goff. 1985. Purification and
characterization of murine retroviral reverse transcriptase
ex-pressedinEscherichia coli. J. Biol. Chem.260:9326-9335. 31. Sisk,W. P.,J.G.Chirikjian, J. Lautenberger,C.Jorcyk,T. S.
Papas, M, L. Berman, R. Zagursky, and D. L. Court. 1986. A plasmid vector for cloningand expression of gene segments: expressionofanHTLV-1envelopegene segment.Gene 48:183-193.
32. Tanese, N., and S. P. Goff. 1988. Domain structure of the Moloney murine leukemia virus reverse transcriptase:
muta-tionalanalysisand separate expression ofthe DNApolymerase and RNase H activities. Proc. Natl. Acad. Sci. USA 85:1777-1781.
33. Varmus, H., and R. Swanstrom. 1982. Replication of retrovi-ruses, p. 369-512. In R. Weiss, N. Teich, H. Varmus, and J. Coffin (ed.). Molecular biology of tumorviruses, RNA tumor
viruses, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor,N.Y.
34. Zweig, M., S. D. Showalter, G. C. DuBois, W. P. Sisk, and D. L. Court. 1987. Detection ofheterologousfusionproteinsin Esch-erichia coliwithamonoclonalantibody. Gene 55:47-53.