World Alphaviruses Mediate Formation of Distinct, Virus-Specific
Protein Complexes
Niall J. Foy, Maryna Akhrymuk, Ivan Akhrymuk, Svetlana Atasheva, Alain Bopda-Waffo,* Ilya Frolov, Elena I. Frolova
Department of Microbiology, University of Alabama at Birmingham, Birmingham, Alabama, USA
Alphaviruses are a group of single-stranded RNA viruses with genomes of positive polarity. They are divided into two geographi-cally isolated groups: the Old World and the New World alphaviruses. Despite their similar genome organizations and virion structures, they differ in many aspects of pathogenesis and interaction with the host cell. Here we present new data highlighting previously unknown differences between these two groups. We found that nsP3 proteins of Sindbis virus (SINV) and Venezue-lan equine encephalitis virus (VEEV) form cytoplasmic complexes with different morphologies and protein compositions. Un-like the amorphous aggregates formed by SINV nsP3 and other Old World alphavirus-specific nsP3s, VEEV nsP3 forms unique, large spherical structures with striking symmetry. Moreover, VEEV nsP3 does not interact with proteins previously identified as major components of SINV nsP3 complexes, such as G3BP1 and G3BP2. Importantly, the morphology of the complexes and the specificity of the interaction with cellular proteins are largely determined by the hypervariable domain (HVD) of nsP3. Replace-ment of the VEEV nsP3 HVD with the corresponding domain of SINV nsP3 rendered this protein capable of interaction with G3BPs. Conversely, replacement of the SINV nsP3 HVD with that of VEEV abolished SINV nsP3’s interaction with G3BPs. The replacement of natural HVDs with those from heterologous viruses did not abrogate virus replication, despite these fragments demonstrating very low levels of sequence identity. Our data suggest that in spite of the differences in morphology and composi-tion of the SINV- and VEEV-specific nsP3 complexes, it is likely that they have similar funccomposi-tions in virus replicacomposi-tion and modifi-cation of the cellular environment.
T
heAlphavirusgenus of theTogaviridaefamily contains over 30 different members, many of which represent an unquestion-able but often underappreciated public health threat. These vi-ruses are widely distributed on all continents and circulate in mostly subtropical and tropical areas, between mosquitoes and amplifying vertebrate hosts. In mosquitoes, they cause a life-long infection without interfering with the vector’s biological func-tions. However, upon transmission to amplifying hosts during the mosquito blood meal, alphaviruses induce acute infection. This infection results in a high-titer viremia that is essential for trans-mission to new mosquito vectors (1). Alphaviruses are divided into two distinct groups, the Old World and New World alphavi-ruses, based on geographical origin. The Old World alphavialphavi-ruses, exemplified by Sindbis virus (SINV) and Semliki Forest virus (SFV), usually induce mild diseases in vertebrates, characterized by rash, fever, and arthritis. However, some of them, such as chi-kungunya virus (CHIKV), are capable of producing excruciating joint pain and severe, persistent polyarthritis (2–4). In general, New World alphaviruses are more virulent, causing outbreaks of highly debilitating disease. Symptoms often include severe, and frequently fatal, encephalitis and other neurological sequelae. Venezuelan (VEEV), eastern (EEEV), and western (WEEV) equine encephalitis viruses are members of the New World group of alphaviruses and circulate in the Central, South, and North Americas, causing periodic outbreaks of disease among equids and humans (5,6). Interestingly, while there are marked differ-ences in the severity of disease, virus-host interactions, and patho-genesis caused by Old World and New World alphaviruses, these viruses are identical in their genome replication strategy and ex-hibit obvious similarities in virion structure (7). Additionally,they demonstrate similar capacities for persistent, noncytopathic replication in insect cells and mosquito vectors.
The alphavirus genome is a single-stranded RNA of positive polarity which encodes only a few proteins (1). The nonstructural proteins, nsP1 to -4, are translated directly from the genomic RNA, and depending on the virus species, three or four structural proteins, ultimately composing viral particles, are translated from the subgenomic RNA. Alphavirus nonstructural proteins are of particular interest because their functions have been poorly stud-ied and thus are insufficiently understood. These proteins not only are directly involved in replication of the viral genome and tran-scription of the subgenomic RNA but also interact with numerous cellular proteins, target replication complexes to particular cellu-lar compartments, and appear to mediate a variety of other aspects of virus-host interactions (7–13). Among the structural proteins, so far only the capsid protein, with its ability to inhibit nucleocy-toplasmic trafficking (14) and to bind to ribosomes, has been shown to exhibit any other function beyond its role in virion for-mation (15). However, the biological effect on cellular translation of capsid binding to ribosomes has yet to be determined.
Received11 October 2012Accepted26 November 2012
Published ahead of print5 December 2012
Address correspondence to Elena I. Frolova, [email protected].
* Present address: Alain Bopda-Waffo, Department of Biological Science, Alabama State University, Montgomery, Alabama, USA.
Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.02853-12
on November 7, 2019 by guest
http://jvi.asm.org/
The research carried out to date has successfully ascribed functions to three of the four alphavirus nonstructural pro-teins. Capping of the viral genomic and subgenomic RNAs is carried out by nsP1 (16). nsP2 functions as an RNA helicase, protease (17–20), and cellular transcription inhibitor (8,21), while nsP4 is directly engaged in synthesis of positive and neg-ative strands of virus-specific RNAs (22). However, the func-tions of another nonstructural protein, nsP3, have yet to be elucidated fully. What is known about nsP3 is that it is colocal-ized with viral double-stranded RNA (dsRNA) intermediates (23) and is required for viral RNA replication (24). Mutations in nsP3 or the cleavage site between nsP2 and nsP3 both change the balance between synthesis of genomic and subgenomic RNAs and indirectly affect nsP2 nuclear functions (25,26). It has also been shown that some mutations in the RNA promoter elements can cause adaptive mutations in nsP3, suggesting its direct or indirect function in promoter recognition (27). Of further interest is the observation from SINV-infected cells that nsP3 is coprecipitated with other viral nsPs and distinct sets of cellular proteins, characterized by the presence of G3BP1 and G3BP2 (or their insect homolog, Rasputin) at high concentrations (9,23). Normally, G3BP1 and G3BP2 proteins are principal components of cellular stress granules. In addi-tion, nsP3 proteins from SINV and other Old World alphavi-ruses have been shown to form at least two types of complexes. One type is colocalized with plasma membranes and, at later times postinfection, with endosome/lysosome-like membra-nous organelles. Another type of complex appears not to be associated with lipid membranes but is likely bound to cellular filaments (23,28). One of the potential functions of the fila-ment-bound complex is to compete with formation of func-tional stress granules. This hypothesis has yet to be confirmed experimentally.
The study of alphavirus nsP3 function(s) is complicated by its unusual structure. This protein has two distinct domains. The amino-terminal domain is highly conserved among the alphavi-ruses and exhibits a high level of homology with protein domains present in the nonstructural proteins of many other positive-strand RNA viruses and some cellular proteins, such as macroH2A histone (29). In contrast, the carboxy-terminal domain has very low sequence homology, even between closely related alphavi-ruses, and is thus called the hypervariable domain (HVD). These two domains appear to be involved in very different functions in alphavirus replication.
This study presents new data aimed at further understanding the function of nsP3 in replication of VEEV. We show that VEEV and SINV nsP3 proteins form distinct cytoplasmic complexes with unique morphological appearances. Moreover, cellular pro-teins which were previously identified as components of SINV nsP3-specific complexes, such as G3BPs, do not interact with VEEV nsP3. This specificity of nsP3 interaction with cellular pro-teins is determined by the HVD. Interestingly, swapping of the HVDs between VEEV and SINV nsP3 proteins changed the spec-ificity of the interaction with the cellular proteins and complexes formed but did not profoundly affect virus replication, if at all. Taken together, the data presented reveal that the New World and Old World alphavirus nsP3 proteins demonstrate strong differ-ences in virus-host cell interactions on the molecular level, and these need to be investigated further.
MATERIALS AND METHODS
Cell cultures.BHK-21 cells were kindly provided by Paul Olivo (Wash-ington University, St. Louis, MO). NIH 3T3 cells were obtained from the American Type Culture Collection (Manassas, VA). These cell lines were maintained at 37°C in alpha minimum essential medium (␣MEM) sup-plemented with 10% fetal bovine serum (FBS) and vitamins. Mosquito C7/10 cells were obtained from Henry Huang (Washington University, St. Louis, MO) and were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated FBS and 10% tryptose phosphate broth (TPB).
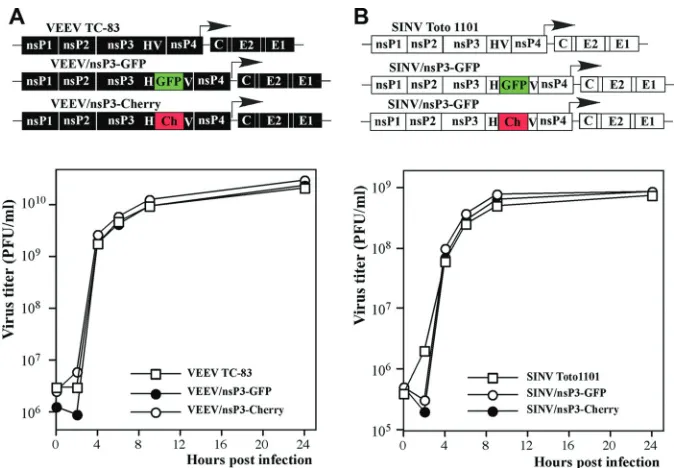
Plasmid constructs.Plasmids carrying VEEV TC-83 and SINV ge-nomes, i.e., pVEEV and pToto1101, respectively, have been described elsewhere. pSINV/GFP and pVEEV/GFP encode VEEV TC-83 and SINV genomes, respectively, in which the green fluorescent protein (GFP) cod-ing sequence is cloned under the control of a second subgenomic pro-moter (30, 31). pVEEV/nsP3-GFP and pVEEV/nsP3-Cherry contain VEEV TC-83 genome variants in which the GFP and Cherry coding se-quences, respectively, are cloned between codons corresponding to amino acids (aa) 391 and 392 of nsP3. GFP and pSINV/nsP3-Cherry have essentially the same design, but the GFP and pSINV/nsP3-Cherry genes are cloned in frame between the codons for aa 389 and 390 of SINV nsP3. pVEEV/nsP3sinvHVF/GFP contains a DNA fragment encoding aa 335 to 538 of VEEV nsP3 replaced by a corresponding fragment for SINV nsP3. In SINV/nsP3veevHVF/GFP, a DNA fragment encoding aa 335 to 544 of SINV nsP3 is replaced by a corresponding fragment for VEEV nsP3. pSINV/nsP3veevHVF-GFP and pVEEV/nsP3sinvHVF-GFP encode GFP insertions in heterologous HVDs of nsP3 as in the above-described pVEEV/nsP3-GFP and pSINV/nsP3-GFP constructs, respectively. The schemes of the viral genomes are presented in the relevant figures. The recombinant genomes were assembled using relatively standard, PCR-based approaches and were sequenced before virus rescue to eliminate any possible additional PCR-mediated mutations. All of the sequences and details of the cloning procedures can be provided upon request.
RNA transcription and virus rescue.Plasmids were purified by ctrifugation in CsCl gradients. They were linearized using restriction en-zymes recognizing unique restriction sites located immediately down-stream of the poly(A) sequences. RNAs were synthesized by SP6 RNA polymerase in the presence of a cap analog under previously described conditions (32). The yield and integrity of transcripts were analyzed by gel electrophoresis under nondenaturing conditions, and transcription reac-tion mixtures were used for electroporareac-tion, without addireac-tional purifica-tion (33). A fracpurifica-tion of the electroporated cells was used for an infectious-center assay to evaluate the infectivity of thein vitro-synthesized RNA. This assay was always applied to rule out the possibility that the designed constructs needed additional, adaptive mutations for efficient virus rep-lication. Released viruses were harvested at 24 h postelectroporation, and titers were determined by plaque assay on BHK-21 cells (34).
Viral replication analysis.Cells were seeded into 35-mm dishes. After a 4-h incubation at 37°C, monolayers were infected at the multiplicities of infection (MOIs) indicated in the figure legends, washed with phosphate-buffered saline (PBS), and overlaid with 1 ml of complete medium. At the times indicated in the figures, the medium was replaced with fresh me-dium, and virus titers were determined by a plaque assay on BHK-21 cells as previously described (34).
Antibody production.A fragment of the VEEV nsP3 protein (aa 1 to 328) was produced as a fusion with 6⫻His-SUMO inEscherichia coliand purified by immobilized-metal-ion affinity chromatography. The fusion protein 6⫻His-SUMO-nsP3⌬was used for mouse monoclonal antibody (MAb) production at the UAB Hybridoma Core Facility. Hybridomas were screened by enzyme-linked immunosorbent assay (ELISA) with the untagged protein, and two positive clones, MAB1-35 and MAB6-30, were selected. Both MAbs specifically recognized nsP3 on Western blots and in immunofluorescence assays.
Immunofluorescence assay. For confocal microscopy, cells were seeded onto 8-well-slides (Ibidi GmbH, Munich, Germany), infected at Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
an MOI of ca. 20 PFU/cell, and incubated at 37°C in a CO2incubator. At
the indicated times, they were fixed in 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature and permeabilized with 0.5% Triton X-100 in PBS. They were then blocked with 5% goat serum and stained with a mouse monoclonal antibody against dsRNA (MAb J2; Scicons, Hungary) or a rabbit polyclonal antibody against G3BP1 (a gift from Richard Lloyd) or G3BP2 (Epitomics), followed by appropriate secondary antibodies labeled with Alexa Fluor 555 (Invitrogen) or DyLight 649 (Jackson ImmunoResearch). Images were acquired on a Zeiss LSM700 confocal microscope with a 63⫻ 1.4-numerical-aperture (NA) Plan-Apochromat oil objective. The three-dimensional (3D) image stacks were further processed using Huygens Professional (Scientific Volume Imag-ing, Hilversum, Netherlands) for deconvolution, using experimental point spread function (PSF), and Imaris was used for 3D rendering (Bit-plane AG, St. Paul, MN). The colocalization was quantitated using decon-volved images in Huygens Professional.
RESULTS
Fluorescent tag insertions in nsP3 protein have no negative ef-fect on VEEV and SINV replication.Insertion of fluorescent tags into virus-specific proteins provides an opportunity to investigate their intracellular compartmentalization and temporal-spatial co-localization with other viral or cellular proteins during virus rep-lication. The distinguishing feature of the alphavirus nsP3 protein is the presence in its carboxy terminus of the HVD, which dem-onstrates low sequence homology between alphaviruses. To date, no defined secondary structures have been predicted for HVDs of any of the known alphaviruses. Previously, we and others demon-strated that the HVDs of the SINV and SFV nsP3 proteins could be used for the in-frame insertion of large heterologous sequences without causing deleterious effects on virus replication (9,10,35). Therefore, in this study, to further investigate structures of the nsP3-specific complexes formed by representative members of the Old World and New World alphaviruses, i.e., SINV and VEEV, respectively, we inserted different fluorescent proteins, GFP and Cherry, into their nsP3-specific HVDs (Fig. 1A). Insertion of these genes did not affect the replication rate of either virus (Fig. 1B). The engineered variants demonstrated essentially the same rates of replication as their wild-type (wt) parents. Thus, further appli-cation of these insertion mutants was expected to generate biolog-ically relevant data.
The VEEV nsP3 protein forms unique cytoplasmic com-plexes called spheroids.Data that we obtained from other lines of research strongly suggested that the Old World and New World alphaviruses have fundamental differences in numerous aspects of their replication and virus-host interactions (7). In this study, this was also found to be the case for nsP3-specific protein complex formation.
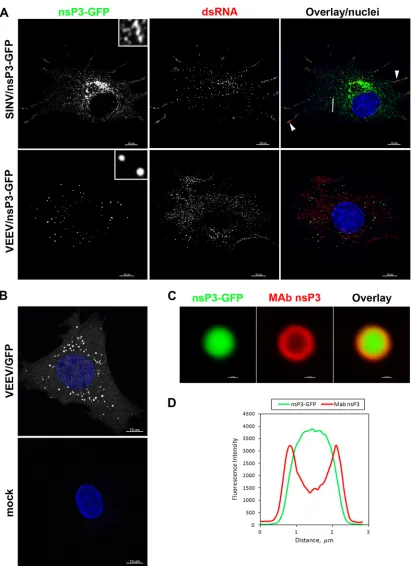
In the initial experiments, we analyzed the distribution of nsP3-GFP fusion proteins 4 h postinfection with VEEV/nsP3-GFP or SINV/nsP3-GFP. Cells were additionally stained with an anti-body against dsRNA to mark the localization of viral replicative complexes (vRCs). In good correlation with our previously pub-lished data (23), at 4 h postinfection (p.i.) a major fraction of SINV nsP3-GFP accumulated in large amorphous cytoplasmic complexes (type 2 complexes). This protein was also readily de-tectable at the plasma membrane and in endosomes (type 1 com-plexes), where its signal overlapped with vRCs identified by dsRNA staining (Fig. 2A). Surprisingly, at the same time p.i., VEEV nsP3-GFP formed very different complexes. At this time point, we were unable to find VEEV nsP3 localized with dsRNA
staining and thus associated with vRCs (type 1 complexes). The large, cytoplasmic complexes formed by VEEV nsP3 (type 2 com-plexes) had a strikingly different morphology from that of SINV nsP3-specific cytoplasmic complexes (Fig. 2A). These large com-plexes had various sizes, ranging from barely visible to⬃1m in diameter, and a regular spherical shape. Because of their shape, we called these complexes spheroids. The insets inFig. 2Aclearly demonstrate the difference in morphology of the VEEV and SINV nsP3-specific cytoplasmic complexes. The apparent difference in the amount of nsP3 in VEEV- and SINV-infected cells was a result of a denser accumulation of VEEV nsP3-GFP in the spheroids.
To exclude the possibility that the unusual structure of the type 2 VEEV nsP3-GFP complexes was an artifact of nsP3 fusion with the fluorescent protein, we developed two monoclonal antibodies to VEEV nsP3. Staining of cells infected with VEEV TC-83 encod-ing an unmodified nsP3 protein by use of two different MAbs demonstrated a distribution of nsP3 indistinguishable from that of nsP3-GFP (Fig. 2Band data not shown). Importantly, the un-modified nsP3 protein also formed spheroids of different sizes. However, their internal part, below the 0.5-m layer, was poorly accessible to MAbs, which is indicative of a high protein density. Similarly, nsP3-GFP-based spheroids demonstrated only surface staining with MAbs (Fig. 2CandD). These data suggest that VEEV nsP3 is strongly different from its SINV-specific homolog in terms of cytoplasmic complex formation.
Similar to SINV nsP3, VEEV nsP3 accumulates near viral replicative complexes. The experiments described above were performed at 4 h p.i. At this point in the infection, VEEV TC-83 variants have already released very high levels of infectious virions, and spheroids have already formed. These spheroids exhibit a very high fluorescence intensity, which strongly complicates detection of other, relatively weaker signals. Therefore, to determine whether VEEV nsP3-GFP accumulates in the vicinity of vRCs, as was demonstrated for SINV nsP3, we analyzed the distribution of VEEV nsP3-GFP at earlier times postinfection, when VEEV nsP3 is present at a relatively low concentration and is not assembled into large complexes. Indeed, at 1 h p.i., a readily detectable frac-tion of VEEV nsP3-GFP colocalized with dsRNA at the plasma membrane (Fig. 3), similar to the type 1 SINV nsP3 complexes. At 2 h p.i., this protein started to assemble into additional, larger and brighter spherical structures which were not associated with dsRNA. By 3 h p.i., nsP3-specific spheroids became too large and bright, and this made the detection of nsP3 colocalization with dsRNA by confocal microscopy an impossible task.
In conclusion, the presented data demonstrate that distribu-tion of the VEEV nsP3 protein in infected cells is different from that previously described for nsP3 proteins of representative members of the Old World alphaviruses, i.e., SINV and SFV (23,
35). As expected, a detectable fraction of VEEV nsP3 was associ-ated with the membrane-bound, dsRNA-containing vRCs. How-ever, the major fraction of nsP3 was assembled in large, high-density complexes which were different from those previously described for the Old World alphaviruses in terms of their size and morphology. This suggests the likelihood of different composi-tions, and potentially different funccomposi-tions, of the New World and Old World alphavirus nsP3-specific cytoplasmic complexes.
VEEV and SINV nsP3 proteins do not colocalize in coin-fected cells.To further demonstrate that nsP3-specific cytoplas-mic complexes formed during VEEV and SINV infections are dis-tinct, we carried out a series of experiments utilizing viruses
on November 7, 2019 by guest
http://jvi.asm.org/
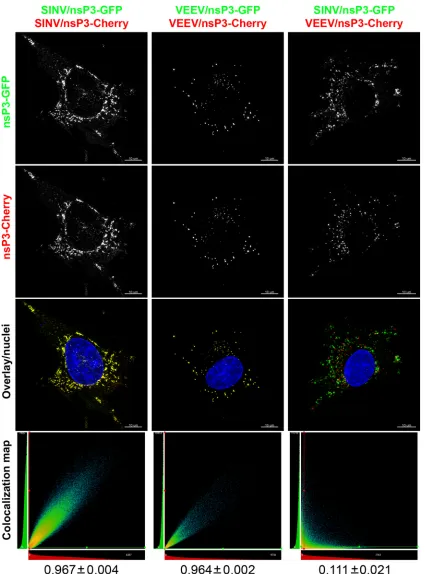
expressing nsP3 fused with different fluorescent markers, either GFP or Cherry. First, we analyzed the distribution of nsP3 pro-teins tagged with GFP or Cherry in cells coinfected with the ho-mologous virus pair SINV/nsP3-GFP plus SINV/nsP3-Cherry or VEEV/nsP3-GFP plus VEEV/nsP3-Cherry. For both pairs, ho-mologous nsP3-GFP and nsP3-Cherry were found colocalized in the same cytoplasmic complexes, with a Pearson’s coefficient above 0.96 (Fig. 4). Importantly, the ratios of GFP and nsP3-Cherry in the cytoplasmic complexes in the same cell were iden-tical (Fig. 4) and proportional to the overall expression levels of these proteins in cells as judged by analyzing the different levels of expression of nsP3-GFP and nsP3-Cherry (data not shown). These data suggested that the nsP3-containing complexes were formed from the total available cytoplasmic nsP3 pool, without a detectable selection bias for the original virus. In contrast, coin-fection with the heterologous viruses SINV/nsP3-GFP and VEEV/ Cherry led to development of two distinct types of nsP3-containing complexes, nsP3-containing exclusively the GFP or Cherry marker. Thus, each type of complex contained nsP3 derived from a single virus, SINV or VEEV, and the structures formed were morphologically indistinguishable from those formed in infec-tions carried out with a single virus. These results suggest a num-ber of potential explanations for the observed data. Formation of these virus-specific nsP3-containing cytoplasmic complexes may be driven by nsP3 oligomerization or by interaction with different cellular proteins. It may also be the case that both components determine the specificity of complex formation.
nsP3 HVDs of distantly related alphaviruses are inter-changeable.VEEV and SINV are representative members of very distant, geographically isolated clades, and in our previous stud-ies, they demonstrated fundamental differences in pathogenesis and interaction with host cells (7). Accumulated data about the
sequences and functions of nsP3 proteins of different alphaviruses suggest that these proteins contain at least three structural do-mains (Fig. 5). The first two domains, the macro domain and the alphavirus-specific domain, are very conserved between all alpha-viruses (Fig. 5). The carboxy-terminal domain of nsP3 is highly variable among the members of the alphavirus genus (hence the term hypervariable domain), and it also differs in size. Although it demonstrates some homology between members of the same geo-graphic group, it can vary in sequence and size even between closely related members (36,37), and partial deletions in this do-main have a negligible effect on virus replication (26, 38,39). VEEV and SINV nsP3 HVDs have similar lengths (220 aa in VEEV TC-83 and 222 aa in SINV) but demonstrate no significant ho-mology at the amino acid level (Fig. 5), which might be suggestive of different functions in the formation of virus-specific cytoplas-mic protein complexes, modification of cellular functions, and/or RNA replication.
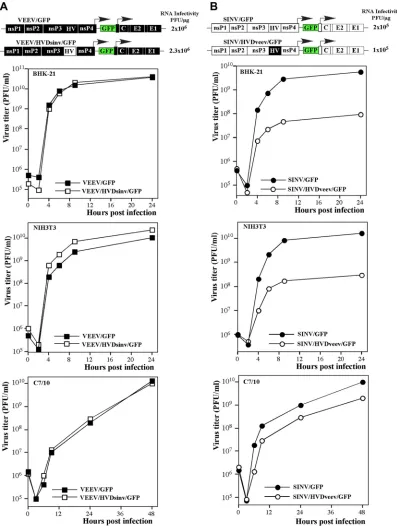
To further define the role of the HVD in virus replication, in the next constructs we replaced the HVD in the VEEV genome with that of SINV (VEEV/HVDsinv/GFP) and made a reciprocal replacement in the SINV genome to produce SINV/HVDveev/ GFP (Fig. 6). Both chimeric viruses were viable, their replication induced CPE, and in the infectious-center assay, thein vitro-syn-thesized RNAs demonstrated infectivity similar to those of the nonchimeric parental viruses, VEEV/GFP and SINV/GFP (Fig. 6). Thus, the viability of these viruses did not require additional adap-tive mutations. VEEV/HVDsinv/GFP replicated at the same rates and to the same final titers as did VEEV/GFP in BHK-21 and C7/10 cells, but it reproducibly demonstrated a noticeably higher replication rate in NIH 3T3 cells. Compared to that of SINV/GFP, replication of SINV/HVDveev/GFP was less efficient in all of the tested cell lines; however, in numerous experiments, the titers of FIG 1In-frame insertions of different fluorescent proteins into alphavirus nsP3-specific HVDs do not interfere with virus replication. Schematic representations of VEEV TC-83 (A) and SINV (B) genomes containing GFP and Cherry fluorescent protein insertions in their nsP3-specific HVDs, indicated as HV, are shown, as well as the rates of their replication in BHK-21 cells. The GFP and Cherry coding sequences were inserted into nsP3 after aa 391 and 389 in VEEV and SINV, respectively. BHK-21 cells (5⫻105) in 6-well Costar plates were infected with the indicated viruses at an MOI of 20 PFU/cell. At the indicated times postinfection,
the medium was replaced, and virus titers were determined by plaque assay on BHK-21 cells. Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.123.460.64.298.2]FIG 2VEEV- and SINV-specific nsP3 proteins demonstrate different distributions in infected cells. (A) BHK-21 cells in Ibidi 8-well-slides were infected with the indicated viruses at an MOI of 20 PFU/cell. At 4 h postinfection, cells were fixed with 4% paraformaldehyde, permeabilized, and stained with a dsRNA-specific MAb, and nuclei were stained with Hoechst dye. Images are presented as multiple-intensity projections (MIPs) of 3D stacks after deconvolution. Insets (6⫻ zoom) in the first panels demonstrate the morphologies of the complexes formed by VEEV and SINV nsP3-GFP. The arrowheads point to the colocalization of SINV nsP3-GFP with dsRNA (red). (B) BHK-21 cells were infected with VEEV TC-83 encoding GFP under the control of the subgenomic promoter. At 6 h postinfection, cells were fixed, permeabilized, and stained with a VEEV nsP3-specific MAb and an Alexa Fluor 555-labeled secondary antibody. The lower panel presents stained mock-infected cells, which were processed and imaged identically to infected cells. Images are presented as MIPs of 6 optical sections. (C) Enlarged, representative single optical section images presenting one of the spheroids demonstrated in the cell in panel B. Note that antibodies did not penetrate efficiently into the nsP3-GFP-containing spheroid. (D) Fluorescence intensity profile of the spheroid image presented in panel C.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.87.498.61.627.2]this virus exceeded 108PFU/ml in BHK-21 and NIH 3T3 cells and
109PFU/ml in C7/10 cells (Fig. 6). Thus, both chimeric viruses retained the ability to replicate in the tested cell lines, albeit with different efficiencies. Thus, the HVDs were able to function in
different ns polyprotein contexts, despite demonstrating no ho-mology at the aa level.
To further analyze the role of the HVD in the genesis of nsP3-specific cytoplasmic complexes, we designed chimeric viruses with FIG 3nsP3-containing spheroids are gradually formed at early times post-VEEV infection and contain no dsRNA. BHK-21 cells in Ibidi 8-well-slides were infected with VEEV/nsP3-GFP at an MOI of 20 PFU/cell, fixed at the indicated times postinfection, and, after permeabilization, stained with the dsRNA-specific MAb followed by the Alexa Fluor 555-labeled secondary antibody and Hoechst dye (nuclei). Images are presented as MIPs of 3D stacks after deconvolution. The overlap between nsP3-GFP and dsRNA structures appears as yellow staining at 1 h p.i. (see bottom panels). Very few overlapping spots could be detected at 2 and 3 h p.i.
Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.81.503.62.614.2]FIG 4SINV and VEEV nsP3-specific cytoplasmic complexes do not colocalize. BHK-21 cells in Ibidi 8-well-slides were coinfected at an MOI of 20 PFU/cell with pairs of recombinant VEEV and SINV encoding different fluorescent proteins within the nsP3 HVDs. At 6 h postinfection, cells were fixed and counter-stained with Hoechst dye (nuclei). Images are presented as MIPs of 3D stacks after deconvolution. nsP3-GFP and nsP3-Cherry expressed by homologous viruses were completely colocalized, as shown in yellow in the overlay images and evident from the presented colocalization maps and Pearson’s coefficient values (indicated below the columns). No nsP3-GFP and nsP3-Cherry colocalization was detected in the case of infection with the heterologous viruses SINV/nsP3-GFP and VEEV/nsP3-Cherry.
on November 7, 2019 by guest
http://jvi.asm.org/
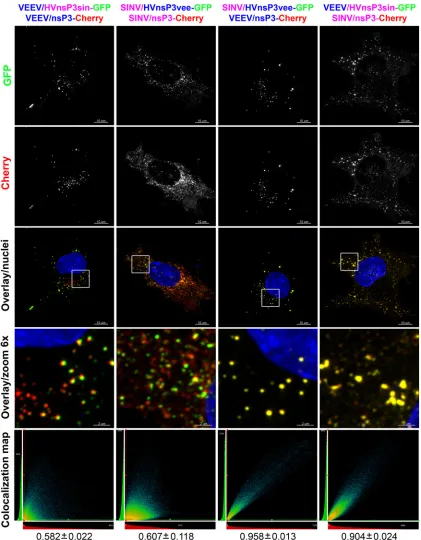
[image:7.585.81.506.64.638.2]a GFP insertion in the HVD, namely, VEEV/HVDsinv-GFP and SINV/HVDveev-GFP. Insertion of GFP into the HVDs of chime-ric viruses did not have any detectable negative effect on virus replication. Replication of SINV/HVDveev-GFP was even more efficient than that of SINV/HVDveev/GFP (data not shown). An analysis of the distribution of chimeric nsP3-GFP at 6 h p.i. re-vealed that in the infected cells, chimeric nsP3 proteins also formed large cytoplasmic complexes (data not shown). To further define the nature of these complexes, we analyzed their interaction with those formed by nonchimeric nsP3. BHK-21 cells were in-fected with the following pairs of viruses: VEEV/nsP3-Cherry plus GFP, SINV/nsP3-Cherry plus VEEV/HVDsinv-GFP, SINV/nsP3-Cherry plus SINV/HVDveev-VEEV/HVDsinv-GFP, and VEEV/ nsP3-Cherry plus SINV/HVDveev-GFP. A complete colocaliza-tion of heterologous nsP3 proteins (Pearson’s coefficient above 0.9) was found in cells infected with SINV/nsP3-Cherry plus VEEV/nsP3sinvHVD-GFP or VEEV/nsP3-Cherry plus SINV/ nsP3veevHVD-GFP (Fig. 7), suggesting that the HVD plays a crit-ical role in large complex formation. Importantly, the morphol-ogy of the nsP3 cytoplasmic complexes was also determined by the HVD source. The VEEV/nsP3-Cherry-plus-SINV/HVDvee-GFP
pair formed VEEV-specific spheroids, while coinfection with SINV/nsP3-Cherry and VEEV/HVDsinv-GFP resulted in SINV-specific amorphous aggregates. Infection with the homologous viruses, with one expressing a chimeric nsP3 protein containing a heterologous HVD, led to formation of distinct chimeric nsP3-GFP- or wt nsP3-Cherry-containing complexes. Interestingly, al-though the wt and chimeric proteins did not colocalize in the same complexes, some overlap in the complexes could be detected (the overlap appears as a yellow border between closely located GFP-and Cherry-specific complexes inFig. 7) that was indicative of homologous interaction of the N-terminal domain of nsP3.
Taken together, these data imply that formation of the large cytoplasmic nsP3-specific complexes is mediated either directly by the HVD or possibly indirectly, through its interaction with cellular proteins. At the same time, the overlap of the wt and chimeric nsP3 complexes of the same virus suggests that N-termi-nal domains mediate some additioN-termi-nal nsP3 interaction, perhaps dimerization or oligomerization.
The HVD of nsP3 determines the specificity of its interaction with G3BPs.SINV replication leads to accumulation of a distinct set of cellular proteins in the nsP3-containing protein complexes. FIG 5Domain structure and sequence alignment of nsP3 proteins of representative members of the New World and Old World alphaviruses. The schematic representation of the nsP3 protein shows the three predicted structural domains: the macro domain, the alpha domain, and the HVD. The sequence alignment of nsP3 proteins of different alphaviruses was performed with Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo/), and the figure was prepared with Jalview (http://www.jalview.org). The domain sequences are underlined with the same colors as those used in the schematic presentation. Sequences were derived from the following viruses: VEEV (GenBank accession no.P27282.2), SINV (GenBank accession no.P03317.1), SFV (GenBank accession no. NP_740667.1), CHIKV (GenBank accession no. NP_690588.1), and EEEV (GenBank accession no.Q4QXJ8.2).
Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.44.540.68.407.2]The hallmark of these complexes is accumulation of cellular G3BP1 and G3BP2 (or their homolog Rasputin in mosquito cells), which are the major components of cellular stress granules formed in response to a variety of abnormal conditions (9, 10, 23). In
mock-treated or mock-infected cells, these proteins are diffusely distributed in the cytoplasm. However, within a few hours after infection with SINV and other Old World alphaviruses, G3BP1 and G3BP2 demonstrate a profound redistribution and become FIG 6Alphaviruses with heterologous HVDs are capable of efficient replication in a variety of cell lines. (A) Schematic representation of VEEV genomes encoding either wt nsP3 or chimeric nsP3 with the SINV-specific HVD, infectivities of thein vitro-synthesized RNAs in the infectious-center assay, and replication rates of the rescued viruses in the indicated cell lines. (B) Schematic representation of SINV genomes encoding either wt nsP3 or chimeric nsP3 with the VEEV-specific HVD, infectivities of thein vitro-synthesized RNAs in the infectious-center assay, and replication rates of the rescued viruses in the indicated cell lines. In the experiments presented in both panels, subconfluent cells in 6-well Costar plates were infected with the indicated viruses at an MOI of 20 PFU/cell. At the indicated time points, the medium was replaced, and virus titers were measured by plaque assay on BHK-21 cells.
on November 7, 2019 by guest
http://jvi.asm.org/
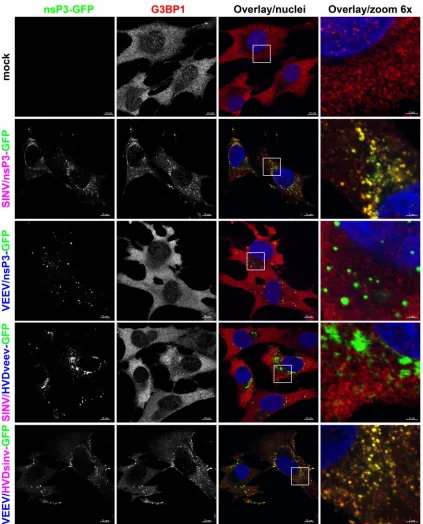
[image:9.585.92.489.67.593.2]colocalized with nsP3, which is present in both the membrane-associated and large cytoplasmic complexes (23,40,41). To test whether VEEV nsP3 is capable of interacting with G3BPs, BHK-21 cells were infected with the VEEV/nsP3-GFP or SINV/nsP3-GFP
recombinant virus and, at 6 h postinfection, stained with antibod-ies against G3BP1 or G3BP2. In agreement with previous data, G3BPs strongly accumulated in SINV nsP3-specific complexes
(Fig. 8and data not shown [for G3BP2]). However, no
colocaliza-FIG 7The HVD determines the formation of virus-specific nsP3 complexes. The viruses used in this study encoded either homologous nsP3 HVDs with Cherry insertions (VEEV/nsP3-Cherry and SINV/nsP3-Cherry) or heterologous nsP3 HVDs with GFP insertions (VEEV/HVDsinv-GFP and SINV/HVDveev-GFP). BHK-21 cells in Ibidi 8-well-slides were coinfected with the indicated pairs of viruses at an MOI of 20 PFU/cell. Cells were fixed at 6 h postinfection and counterstained with Hoechst dye (nuclei). Images are presented as MIPs of 3D stacks after deconvolution. Pearson’s coefficient values for each pair are indicated below the columns.
Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.82.503.65.605.2]tion of G3BPs with VEEV nsP3-GFP was detected. At 6 h p.i., G3BPs remained diffusely distributed in the cytoplasm and nuclei, as observed in mock-infected cells (Fig. 8). No relocalization of G3BPs into nsP3 complexes was detected at later times post-VEEV/nsP3-GFP infection (data not shown).
The experiments described above suggested that the nsP3
HVD plays a critical role in formation of nsP3-containing com-plexes. Thus, we next evaluated whether swapping the HVDs of VEEV and SINV would alter the nsP3 interaction with G3BPs. Indeed, the presence of the SINV HVD in the VEEV nsP3 protein rendered it capable of interacting with G3BP1 and induced relo-calization of this protein into nsP3-specific complexes (Fig. 8). On FIG 8Colocalization of G3BP1 with nsP3 is determined by the HVD and is specific for SINV, not VEEV. BHK-21 cells in Ibidi 8-well-slides were coinfected at an MOI of 20 PFU/cell with the indicated viruses, encoding either homologous or heterologous nsP3 HVDs with GFP insertions. At 6 h postinfection, cells were fixed, permeabilized, and stained with a G3BP1-specific antibody, a secondary Alexa Fluor 555-labeled antibody, and Hoechst dye (nuclei). Colocalization of nsP3-GFP and G3BP1 is indicated in yellow in the overlay images.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.81.504.64.588.2]the other hand, SINV nsP3 containing the VEEV HVD could no longer colocalize with G3BP1 and induce its aggregation. Once again, we observed the influence of the HVD on the formation of nsP3-containing complexes, as the VEEV nsP3-HVDsinv chime-ric protein no longer formed spheroids and was distributed more diffusely in the cytoplasm, resembling the distribution of SINV nsP3. The SINV nsP3-HVDveev chimera formed two types of dense aggregates: spheroid-like and, occasionally, rod-like. Thus, these results imply that the HVD mediates the interaction of nsP3 with cellular proteins and determines the morphology of the nsP3-specific cytoplasmic complexes. Further identification of VEEV nsP3-interacting proteins will be needed to confirm that this is true for the VEEV nsP3 protein.
DISCUSSION
Alphaviruses encode only 4 nonstructural and 3 main structural proteins (1). This small set of proteins is responsible for a diverse array of functions, including the control of viral RNA replication, packaging of viral genomes into infectious virions, altering of the intracellular environment to facilitate viral RNA synthesis, and downregulation of the innate immune response to prevent induc-tion of an antiviral state in as yet uninfected cells. The wide geo-graphical distribution of alphaviruses is also a testament to the adaptability of this small set of proteins, as they have successfully adjusted to replication in various species of vertebrate hosts and mosquito vectors. This adaptation is a multicomponent process which requires accumulation of mutations in both structural and nonstructural proteins to achieve better interaction with host fac-tors. Among the virus-specific proteins, nsP3, and its carboxy-terminal domain HVD in particular, is the most variable. More-over, the HVD demonstrates a high level of diversity even among members of the same serological group (36). As a result of these observations, within the last few years nsP3 has begun to emerge as a protein that might be strongly responsible for host-specific ad-aptation. However, its functions in virus replication are still poorly understood.
The intracellular compartmentalization of the nsP3 protein and its formation of complexes with other cellular and viral pro-teins were at least partially characterized through analysis of infec-tion with several alphaviruses belonging to the Old World group, primarily SINV (9,10,12,23,40–42). All of the Old World alpha-viruses demonstrated similar intracellular distributions of nsP3 and the formation of at least two different types of large protein complexes. The first type of complex was found in close proximity to the viral replicative complexes formed at the plasma or endo-somal membranes. Importantly, these large complexes are not the vRCs themselves and are unlikely to interact with dsRNA, but rather they accumulate at the cytoplasmic side of the neck con-necting membrane-bound spherules with the cytoplasm (43). We previously demonstrated that the vRC-specific nsP3 protein is represented by a very small number of molecules, which can be detected only by the fluorescence signal amplification technique (44). Another type of Old World alphavirus nsP3-containing complex is not associated with the membranes and is tightly bound to the cellular cytoskeleton. These complexes appear as large aggregates of smaller granule-like structures (10), which rap-idly increase in size and number during virus replication. It has been found that both types of SINV nsP3-specific complexes con-tain several cellular proteins, such as G3BP1, G3BP2, and YBX1, which are common components of stress granules. Interaction
with G3BPs was also demonstrated for CHIKV nsP3 and SFV nsP3 (40,41), suggesting that this might be a common feature of all Old World alphaviruses. The current working hypothesis in the field is that interaction of nsP3 with G3BPs might prevent formation of stress granules that would interfere with virus replication. Knock-down of either G3BP1 or G3BP2 leads to a detectable reduction of virus replication (9; I. Frolov and E. I. Frolova, unpublished data), suggesting that these proteins are important but are probably re-dundant or have a cell-specific role. Unfortunately, knockdown of both genes induces cell death (I. Frolov, unpublished data), and therefore its effect cannot be evaluated directly by experimenta-tion. Recently, another protein, amphiphysine-2 (AMPH2), was also found to accumulate in nsP3-containing complexes during SINV and SFV infections (42). AMPH2 interacts with the proline-rich motif located in the nsP3 HVD, and knockdown of AMPH2 has a small, but detectable effect on SINV release from HeLa cells but almost no effect on its replication in other cell types. These data support the hypothesis that nsP3 interacts with cellular pro-teins through its HVD, in a cell-specific mode, and appears to have distinctive importance for virus replication in different cell types. Until this report, there were no data on complex formation by nsP3 proteins of the New World alphaviruses. In this study, we found that a significant fraction of VEEV nsP3 protein was also associated with the membrane-bound vRCs and was particularly visible at early times postinfection. These complexes appeared to be equivalent to the type 1 complexes described above for the Old World alphaviruses. However, other VEEV nsP3-specific cyto-plasmic complexes differed greatly from those formed by SINV nsP3. VEEV nsP3 forms very dense, three-dimensionally symmet-rical structures which we termed spheroids. Until now, no protein structures of similar size and density have been described for any other virus. The distinct differences in appearance of VEEV and SINV nsP3-specific complexes suggest that their morphological differences may result from interaction with different cellular pro-teins. Indeed, we found that VEEV nsP3 did not induce relocal-ization of the main component of SINV nsP3-specific complexes, i.e., the G3BP1/2 proteins. Characterization of the cellular pro-teins interacting with VEEV nsP3 is an ongoing project.
The lack of association of the cytoplasmic nsP3 complexes with vRCs suggested that they are formed from the cytoplasmic, not membrane-associated, pool of nsP3, and coinfection-based ex-periments strongly supported this possibility. Homologous nsP3 proteins labeled with different fluorescent proteins and expressed from different replicating genomes were concentrated in the same complexes. However, in cells infected with heterologous viruses, VEEV and SINV, the corresponding nsP3 proteins, containing different tags, never mixed and were found in two distinct types of complexes morphologically identical to those formed during a single infection with each specific virus. Importantly, the HVDs were found to critically determine the specificity of complex for-mation. nsP3 proteins containing the same HVD but expressed from heterologous viruses readily formed mixed complexes (Fig. 7). Remarkably, a chimeric VEEV containing the SINV HVD gained the ability to interact with G3BP1/2 proteins, while SINV with the VEEV-specific HVD no longer induced G3BP relocaliza-tion and accumularelocaliza-tion into virus-specific complexes. Thus, these data suggested that the HVD controls interaction with cellular proteins and, ultimately, complex formation. Alas, it is not that simple, as HVDs alone or as a fusion with an unrelated protein, such as GFP, do not efficiently form aggregates and demonstrate a Foy et al.
on November 7, 2019 by guest
http://jvi.asm.org/
diffuse cytoplasmic and nuclear distribution (28; data not shown). Moreover, complexes formed by homologous nsP3 proteins with heterologous HVDs were found in close contact, suggesting that the N-terminal domain is also important in complex formation. Thus, based on the accumulated data, we propose that the forma-tion of large nsP3-specific complexes is determined by two types of interactions: (i) interaction of the carboxy-terminal domains with cellular proteins (G3BP1/2 in the case of the Old World al-phaviruses) and (ii) further oligomerization of complexes through the conserved amino-terminal domain. This certainly does not rule out the possibility that lipids, RNA, or other macro-molecules might also be involved in these interactions, but to date we have not detected the presence of lipids in spheroids by use of the available lipid-specific dyes (data not shown).
As we demonstrated above (Fig. 5), VEEV- and SINV-specific HVDs demonstrate a very low level of identity at the aa level. Therefore, it was surprising to find that replacement of the natu-rally occurring HVDs with those derived from heterologous vi-ruses did not abolish virus replication. An explanation for this phenomenon may be extrapolated from our data, which suggest that these domains are likely to be involved in different interac-tions. For example, the SINV-specific HVD appears to bind dif-ferent proteins from those bound by the VEEV HVD, with the cellular G3BP1/2 proteins being good examples. Thus, we hypoth-esize that the primary role of the HVD is modification of the cellular environment for productive virus replication. Impor-tantly, since viral variants containing heterologous HVDs are ca-pable of replication, whereas those containing a complete HVD deletion are not (Frolov, unpublished data), it is likely that the VEEV and SINV HVDs modify the same cellular processes by different means. Such a concept is not new for alphaviruses. We have previously shown that the Old World and New World alpha-viruses use different viral proteins and distinct mechanisms to achieve the same goal, i.e., global inhibition of cellular transcrip-tion, which is the most efficient way to block the antiviral re-sponse.
In conclusion, a comparative analysis of nsP3 proteins derived from different alphaviruses, determination of their interactions with cellular proteins and organelles, and identification of host-specific cellular partners should bring us closer to a complete un-derstanding of the cell-specific modification required for efficient virus replication. The information gleaned from this research will be vital to revealing a cellular target that can be exploited for an-tiviral drug development.
ACKNOWLEDGMENTS
We thank Mary Accavitti-Loper and the staff of the UAB Hybridoma Core Facility for assistance in production of nsP3 MAbs.
This work was supported by NIH NIAID grants R01AI073301 and R56AI091705 to E.I.F. and R01AI070207 and R01AI095449 to I.F.
REFERENCES
1.Strauss JH, Strauss EG.1994. The alphaviruses: gene expression, repli-cation, and evolution. Microbiol. Rev.58:491–562.
2.Rulli NE, Melton J, Wilmes A, Ewart G, Mahalingam S. 2007. The molecular and cellular aspects of arthritis due to alphavirus infections: lesson learned from Ross River virus. Ann. N. Y. Acad. Sci.1102:96 –108. 3.Staples JE, Breiman RF, Powers AM.2009. Chikungunya fever: an epi-demiological review of a re-emerging infectious disease. Clin. Infect. Dis. 49:942–948.
4.Toivanen A.2008. Alphaviruses: an emerging cause of arthritis? Curr. Opin. Rheumatol.20:486 – 490.
5.Weaver SC, Ferro C, Barrera R, Boshell J, Navarro JC.2004. Venezuelan equine encephalitis. Annu. Rev. Entomol.49:141–174.
6.Zacks MA, Paessler S.2010. Encephalitic alphaviruses. Vet. Microbiol. 140:281–286.
7.Garmashova N, Gorchakov R, Volkova E, Paessler S, Frolova E, Frolov I.2007. The Old World and New World alphaviruses use different virus-specific proteins for induction of transcriptional shutoff. J. Virol.81: 2472–2484.
8.Akhrymuk I, Kulemzin SV, Frolova EI. 2012. Evasion of the innate immune response: the Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 86:7180 –7191.
9.Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, MacDonald MR. 2006. Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem.281:30269 –30278.
10. Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. 2006. Formation of nsP3-specific protein complexes during Sindbis virus replication. J. Virol.80:4122– 4134.
11. Laakkonen P, Auvinen P, Kujala P, Kaariainen L. 1998. Alphavirus replicase protein NSP1 induces filopodia and rearrangement of actin fila-ments. J. Virol.72:10265–10269.
12. Salonen A, Vasiljeva L, Merits A, Magden J, Jokitalo E, Kaariainen L. 2003. Properly folded nonstructural polyprotein directs the Semliki Forest virus replication complex to the endosomal compartment. J. Virol.77: 1691–1702.
13. Tamm K, Merits A, Sarand I.2008. Mutations in the nuclear localization signal of nsP2 influencing RNA synthesis, protein expression and cytotox-icity of Semliki Forest virus. J. Gen. Virol.89:676 – 686.
14. Atasheva S, Fish A, Fornerod M, Frolova EI.2010. Venezuelan equine encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol.84:4158 – 4171.
15. Wengler G.1984. Identification of a transfer of viral core protein to cellular ribosomes during the early stages of alphavirus infection. Virology 134:435– 442.
16. Ahola T, Kaariainen L.1995. Reaction in alphavirus mRNA capping: formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. U. S. A.92:507–511.
17. Ding MX, Schlesinger MJ.1989. Evidence that Sindbis virus NSP2 is an autoprotease which processes the virus nonstructural polyprotein. Virol-ogy171:280 –284.
18. Gomez de Cedron M, Ehsani N, Mikkola ML, Garcia JA, Kaariainen L. 1999. RNA helicase activity of Semliki Forest virus replicase protein NSP2. FEBS Lett.448:19 –22.
19. Lulla A, Lulla V, Merits A.2012. Macromolecular assembly-driven pro-cessing of the 2/3 cleavage site in the alphavirus replicase polyprotein. J. Virol.86:553–565.
20. Vasiljeva L, Valmu L, Kaariainen L, Merits A.2001. Site-specific pro-tease activity of the carboxyl-terminal domain of Semliki Forest virus rep-licase protein nsP2. J. Biol. Chem.276:30786 –30793.
21. Gorchakov R, Frolova E, Frolov I.2005. Inhibition of transcription and translation in Sindbis virus-infected cells. J. Virol.79:9397–9409. 22. Strauss EG, Strauss JH.1991. RNA viruses: genome structure and
evo-lution. Curr. Opin. Genet. Dev.1:485– 493.
23. Gorchakov R, Garmashova N, Frolova E, Frolov I.2008. Different types of nsP3-containing protein complexes in Sindbis virus-infected cells. J. Virol.82:10088 –10101.
24. Wang YF, Sawicki SG, Sawicki DL.1994. Alphavirus nsP3 functions to form replication complexes transcribing negative-strand RNA. J. Virol. 68:6466 – 6475.
25. Gorchakov R, Frolova E, Sawicki S, Atasheva S, Sawicki D, Frolov I. 2008. A new role for ns polyprotein cleavage in Sindbis virus replication. J. Virol.82:6218 – 6231.
26. LaStarza MW, Lemm JA, Rice CM.1994. Genetic analysis of the nsP3 region of Sindbis virus: evidence for roles in minus-strand and sub-genomic RNA synthesis. J. Virol.68:5781–5791.
27. Michel G, Petrakova O, Atasheva S, Frolov I. 2007. Adaptation of Venezuelan equine encephalitis virus lacking 51-nt conserved sequence element to replication in mammalian and mosquito cells. Virology362: 475– 487.
28. Vihinen H, Ahola T, Tuittila M, Merits A, Kaariainen L.2001. Elimi-nation of phosphorylation sites of Semliki Forest virus replicase protein nsP3. J. Biol. Chem.276:5745–5752.
on November 7, 2019 by guest
http://jvi.asm.org/
29. Malet H, Coutard B, Jamal S, Dutartre H, Papageorgiou N, Neuvonen M, Ahola T, Forrester N, Gould EA, Lafitte D, Ferron F, Lescar J, Gorbalenya AE, de Lamballerie X, Canard B.2009. The crystal structures of chikungunya and Venezuelan equine encephalitis virus nsP3 macro domains define a conserved adenosine binding pocket. J. Virol.83:6534 – 6545.
30. Atasheva S, Krendelchtchikova V, Liopo A, Frolova E, Frolov I.2010. Interplay of acute and persistent infections caused by Venezuelan equine encephalitis virus encoding mutated capsid protein. J. Virol.84:10004 – 10015.
31. Frolova EI, Fayzulin RZ, Cook SH, Griffin DE, Rice CM, Frolov I.2002. Roles of nonstructural protein nsP2 and alpha/beta interferons in deter-mining the outcome of Sindbis virus infection. J. Virol.76:11254 –11264. 32. Rice CM, Levis R, Strauss JH, Huang HV.1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: mapping of lethal mu-tations, rescue of a temperature-sensitive marker, andin vitromutagenesis to generate defined mutants. J. Virol.61:3809 –3819.
33. Liljeström P, Lusa S, Huylebroeck D, Garoff H.1991.In vitro mutagen-esis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol.65: 4107– 4113.
34. Lemm JA, Durbin RK, Stollar V, Rice CM.1990. Mutations which alter the level or structure of nsP4 can affect the efficiency of Sindbis virus replication in a host-dependent manner. J. Virol.64:3001–3011. 35. Spuul P, Balistreri G, Kaariainen L, Ahola T.2010. Phosphatidylinositol
3-kinase-, actin-, and microtubule-dependent transport of Semliki Forest virus replication complexes from the plasma membrane to modified lyso-somes. J. Virol.84:7543–7557.
36. Meissner JD, Huang CY, Pfeffer M, Kinney RM.1999. Sequencing of
prototype viruses in the Venezuelan equine encephalitis antigenic com-plex. Virus Res.64:43–59.
37. Oberste MS, Parker MD, Smith JF.1996. Complete sequence of Vene-zuelan equine encephalitis virus subtype IE reveals conserved and hyper-variable domains within the C terminus of nsP3. Virology219:314 –320. 38. Davis NL, Willis LV, Smith JF, Johnston RE.1989. In vitro synthesis of
infectious Venezuelan equine encephalitis virus RNA from a cDNA clone: analysis of a viable deletion mutant. Virology171:189 –204.
39. Lastarza MW, Grakoui A, Rice CM. 1994. Deletion and duplication mutations in the C-terminal nonconserved region of Sindbis virus nsP3: effects on phosphorylation and on virus replication in vertebrate and in-vertebrate cells. Virology202:224 –232.
40. Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP.2012. Chikungunya virus nsP3 blocks stress granule assem-bly by recruitment of G3BP into cytoplasmic foci. J. Virol.86:10873– 10879.
41. Panas MD, Varjak M, Lulla A, Eng KE, Merits A, Karlsson Hedestam GB, McInerney GM.2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell23:4701– 4712.
42. Neuvonen M, Kazlauskas A, Martikainen M, Hinkkanen A, Ahola T, Saksela K.2011. SH3 domain-mediated recruitment of host cell am-phiphysins by alphavirus nsP3 promotes viral RNA replication. PLoS Pathog.7:e1002383. doi:10.1371/journal.ppat.1002383.
43. Froshauer S, Kartenbeck J, Helenius A.1988. Alphavirus RNA replicase is located on the cytoplasmic surface of endosomes and lysosomes. J. Cell Biol.107:2075–2086.
44. Frolova EI, Gorchakov R, Pereboeva L, Atasheva S, Frolov I. 2010. Functional Sindbis virus replicative complexes are formed at the plasma membrane. J. Virol.84:11679 –11695.
Foy et al.