0022-538X/09/$12.00 doi:10.1128/JVI.01238-09
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Adaptive Mutations in a Human Immunodeficiency Virus Type 1
Envelope Protein with a Truncated V3 Loop Restore
Function by Improving Interactions with CD4
䌤
Caroline Agrawal-Gamse,
1Fang-Hua Lee,
1Beth Haggarty,
2Andrea P. O. Jordan,
2Yanjie Yi,
2Benhur Lee,
3Ronald G. Collman,
2James A. Hoxie,
2Robert W. Doms,
1* and Meg M. Laakso
1 Departments of Microbiology1and Medicine,2University of Pennsylvania, Philadelphia, Pennsylvania 19104, and Department ofMicrobiology, Immunology, and Molecular Genetics, University of California at Los Angeles, Los Angeles, California 900953
Received 15 June 2009/Accepted 6 August 2009
We previously reported that a human immunodeficiency virus type 1 (HIV-1) clade B envelope protein with a severely truncated V3 loop regained function after passage in tissue culture. The adapted virus, termed TA1, retained the V3 truncation, was exquisitely sensitive to neutralization by the CD4 binding site monoclonal antibody b12 and by HIV-positive human sera, used CCR5 to enter cells, and was completely resistant to small molecule CCR5 antagonists. To examine the mechanistic basis for these properties, we singly and in combi-nation introduced each of the 5 mutations from the adapted clone TA1 into the unadapted envelope. We found that single amino acid changes in the C3 region, the V3 loop, and in the fusion peptide were responsible for imparting near-normal levels of envelope function to TA1. T342A, which resulted in the loss of a highly conserved glycosylation site in C3, played the primary role. The adaptive amino acid changes had no impact on CCR5 antagonist resistance but made virus more sensitive to neutralization by antibodies to the CD4 binding site, modestly enhanced affinity for CD4, and made TA1 more responsive to CD4 binding. Specifically, TA1 was triggered by soluble CD4 more readily than the parental Env and, unlike the parental Env, could mediate entry on cells that express low levels of CD4. In contrast, TA1 interacted with CCR5 less efficiently and was highly sensitive to antibodies that bind to the CCR5 N terminus and ECL2. Therefore, enhanced utilization of CD4 is one mechanism by which HIV-1 can overcome mutations in the V3 region that negatively affect CCR5 interactions.
The human immunodeficiency virus type 1 (HIV-1) enve-lope protein (Env) mediates sequential binding to CD4 and a coreceptor, with these interactions triggering conformational changes in Env that result in fusion between the viral and cellular membranes (2, 12, 66). The V3 loop in the gp120 subunit of the Env protein is thought to interact with the extracellular loops (ECLs) of the seven-transmembrane do-main HIV-1 coreceptors, CCR5 and CXCR4 (9, 10, 28, 45, 51), while the base of the V3 loop and the bridging sheet region of gp120 are thought to engage the amino-terminal domains of the coreceptors (23). In addition, the V3 loop plays a major role in determining whether a given virus strain utilizes CCR5, CXCR4, or both coreceptors subsequent to CD4 binding (6, 7, 57). Perhaps because of its role in coreceptor engagement, the overall length of the V3 loop is highly conserved, as are specific residues that may play key roles in receptor binding (11, 33, 70). However, the V3 loop is also a target for neutralizing antibodies, making it subject to immune selection (20, 25, 26, 44, 47). In addition, the V3 loop as well as the highly variable V1/V2 region shield more conserved regions of Env that are also involved in receptor binding (16, 20, 33, 58, 59).
The importance of the V3 loop for Env function is shown by the fact that genetic deletion of residues in V3 typically results
in a nonfunctional Env protein (5, 19, 67). While V3 loop-deleted Envs appear to fold normally and retain the ability to bind CD4, coreceptor interactions are apparently lost (5, 19, 27, 54, 65, 67, 69). This loss of function complicates immuno-gen design approaches that are predicated upon removing vari-able loops in gp120 in the hopes of focusing the humoral immune response on more conserved regions of Env (22). To overcome this limitation, we introduced partial V3-loop trun-cations into a series of HIV-1 Env proteins and identified an R5X4 HIV-1 Env, termed R3A, that could tolerate partial loss of its V3 loop (31). When 15 residues were removed from the center of the V3 loop, leaving the first 9 and last 9 residues of the region intact, the resulting virus [termed V3(9,9)] was poorly functional. However, after passage in vitro, function was enhanced via the acquisition of five mutations in theenvgene. An Env cloned from the tissue culture-adapted virus, termed TA1, used CCR5 to infect cells but lost the ability to use CXCR4, was completely resistant to CCR5 antagonists by be-ing able to recognize the drug-bound conformation of the coreceptor, and was exquisitely sensitive to neutralization by HIV-1-positive human sera and by a broadly neutralizing an-tibody to the CD4 binding site (31). Whether these attributes were due to the V3 loop truncation, the adaptive mutations, or some combination of the two was unclear. In addition, it is not apparent how an Env can function efficiently despite the loss of a domain that plays an important role in coreceptor engage-ment.
In the present study, we investigated the roles played by the
* Corresponding author. Mailing address: 3610 Hamilton Walk, 225A Johnson Pavilion, University of Pennsylvania, Philadelphia, PA 19104. Phone: (215) 573-6780. Fax: (215) 898-9557. E-mail: doms @mail.med.upenn.edu.
䌤Published ahead of print on 19 August 2009.
11005
on November 8, 2019 by guest
http://jvi.asm.org/
adaptive mutations in TA1 function. We found that the V3 loop truncation alone accounted for resistance to CCR5 an-tagonists. A subset of the adaptive mutations played a major role in restoring function to V3(9,9), doing so via improved utilization of CD4. Compared to the parental R3A Env, TA1 bound to CD4 with slightly higher affinity, was more easily induced to cause membrane fusion by incubation with soluble CD4 (sCD4), and was able to mediate infection of cells ex-pressing low levels of CD4. In contrast, utilization of CCR5 was less efficient. Thus, in this instance, loss of a region of the V3 loop that is important for coreceptor interactions was com-pensated for, unexpectedly, not by restoring efficient use of CCR5 but rather by enhanced use of CD4. Improved interac-tions with CD4 might make induction and exposure of the coreceptor binding site in the bridging sheet region of gp120 more efficient, enabling Env to overcome its impaired ability to interact with CCR5.
MATERIALS AND METHODS
Cells.QT6 Japanese quail fibrosarcoma and 293T/17 human embryonic kidney cells were cultured in Dulbecco modified Eagle medium (DMEM) supplemented
with 10% fetal bovine serum, 60g of penicillin/ml, and 100g of
streptomy-cin/ml (complete DMEM). Complete DMEM was supplemented with 2 mM
L-glutamine for QT6 cells. HEK293-derived Affinofile cells (34, 49) were
cul-tured in complete DMEM supplemented with 50g of blasticidin/ml and 200g
of G418/ml. Human glioma NP2.CD4.CCR5 cells were cultured in complete
DMEM supplemented with 1 mg of G418/ml and 1 g of puromycin/ml.
SupT1.CCR5 and SupT1.CCR5.DCSIGNR cells were cultured in RPMI 1640
supplemented with 10% fetal bovine serum, 60g of penicillin/ml, and 100g of
streptomycin/ml.
Plasmids.All viralenvgenes used for production of viral pseudotypes,
includ-ing 400 nucleotides upstream of theenvinitiation codon with the complete
coding sequences oftat,rev, andvpu, were cloned into the EcoRI and XhoI sites
of pHSPG (41). Overlap PCR was used to construct Envs containing single or multiple adaptive mutations. The dualtropic Env R3A (42) allows entry into both macrophages and T cells, as well as into both CXCR4- and CCR5-expressing cell lines. V3(9,9) and TA1 have been described previously in detail (31). The reporter plasmid encoding luciferase under the control of a T7 promoter (pT7.luciferase) used in cell-cell fusion assays has been described previously (52). Replication-competent molecular clones were constructed by subcloning the EcoRI-XhoI fragment from pHSPG into pNL, which contains the proviral se-quence of NL4-3. All viral genes are intact, with the exception of a small deletion
innef.
Cell-cell fusion assay.This assay has been previously described (52). Briefly, “target” QT6 cells cotransfected with expression plasmids encoding a luciferase reporter gene (pGEM2 T7-luc; Promega), CD4, and either CCR5 or a control expression plasmid, were incubated with QT6 “effector” cells transfected with
Env expression plasmids (1). Effector cells were added to target cells⬃18 h
posttransfection, and the cells were allowed to interact at 37°C for 7 to 8 h. The cells were lysed, and the luciferase activity was measured with a luminometer (3). To determine the ability of sCD4 to induce cell-cell fusion, serial dilutions of four-domain sCD4 (IAVI; Neutralizing Antibody Consortium repository) were added to CD4-negative, CCR5-positive target cells immediately prior to the addition of Env-expressing effector cells.
Viral infection assays.Luciferase reporter viral pseudotypes were produced in 293T/17 cells by cotransfection of the NL4-3-based luciferase vector, pNL-luc
(Env⫺Vpr⫹), with plasmids expressing the desired Env protein, as previously
described (4, 8). Replication-competent viruses were produced in 293T/17 cells
by transfection of a NL4-3-based proviral vector with the HIVenvcloned into the
XhoI and EcoRI sites. The virus titer was normalized by p24 content (Cell BioLabs, Inc., San Diego, CA). For neutralization sensitivity assays, 5 ng of virus was incubated with serial dilutions of monoclonal antibody (MAb) or sCD4 for 1 h at 37°C and then spinoculated onto SupT1.CCR5 cells (46). The cells were assayed for luciferase expression 3 days postinfection. MAbs 17b, immunoglob-ulin G (IgG) b12, and sCD4 were obtained from the IAVI Neutralizing Antibody Consortium repository. MAb 19 binds to CD4 and blocks HIV infection (17). For neutralization assays with MAbs 45531 and 45529 (R&D Systems, Minneapolis, MN) and 2D7 (BD Biosciences, San Jose, CA), which bind CCR5 ECL2, the
target cells were NP2.CD4.CCR5. For drug inhibition assays, the CCR5 antag-onist maraviroc was incubated with SupT1.CCR5 cells in the presence of 100 nM AMD3100 (NIH AIDS Research and Reference Reagent Program) for 1 h at 37°C before addition of 5 ng of virus, followed by spinoculation. Cells were incubated with virus and drug for 72 h and subsequently analyzed for luciferase expression.
For kinetic assays with CD4, CCR5, and fusion blockers, NP2.CD4.CCR5 cells
were plated in a 96-well format at 1.5⫻104cells per well. The following day, a
1:10 dilution of MAb 19 ascites, MAbs 2D7 and CTC5 (R&D Systems) at 50
g/ml each, or enfuvirtide at 90g/ml was added to pretreatment wells and
incubated for 1 h before 5 ng of virus/well was spinoculated onto cells for 2 h at 4°C. For all other wells, virus was first spinoculated onto cells at 4°C, followed by
transfer to 37°C, with blocking agents added immediately (T⫽0 min) or at 15,
30, 60, 120, or 240 min after spinoculation. Infected cells were incubated for 72 h at 37°C and then assayed for luciferase activity. Infection levels were normalized against control well infections performed in the absence of antibodies or drug. To monitor growth of replication-competent virus, SupT1.CCR5.DCSIGNR cells were infected with 5 or 50 ng of replication-competent virus, and the reverse
transcriptase activity measured by using a [3
H]thymidine incorporation assay on culture supernatants pelleted by ultracentrifugation (32).
Affinofile assays.Infection of the HEK293-based CD4/CCR5 dual-inducible cell line (293-Affinofile) with luciferase reporter viral pseudotypes was used to determine the CD4 and CCR5 usage efficiencies as previously described (34, 49). Please see accompanying report (27a) for full details. Briefly, 96-well plates were
seeded with 104
inducible cells per well 2 days prior to CD4 and CCR5 induction.
Cells were induced in a 6⫻8 matrix format using twofold serial dilutions from
0.156 to 5 ng of minocycline/ml (to induce CD4) and 0.0156 to 2M ponasterone
A (to induce CCR5) and allowed to incubate for 18 h at 37°C. Induction medium was removed, and the cells were then infected with viral pseudotypes normalized by p24 content. For dualtropic virus infection, the induced cells were treated with 10 nM AMD3100 at 37°C for 1 h prior to infection to inhibit use of endogenous CXCR4 on 293-Affinofile cells. Infection was measured by luciferase expression 3 days postinfection.
The CD4 and CCR5 surface expression levels of induced cells were analyzed by flow cytometry using phycoerythrin-conjugated mouse human CD4 anti-body (Invitrogen, Carlsbad, CA) or phycoerythrin-conjugated mouse anti-human CCR5 antibody (BD Biosciences) and quantified with standardized beads from QuantiBRITE system (BD Biosciences). The sensitivity vector describing the relative sensitivity of viral entry to CD4 and CCR5 surface expression levels was derived by using the VERSA computational platform (Viral Entry Receptor Sensitivity Analysis [http://versa.biomath.ucla.edu/]) (27a).
ELISAs.Enzyme-linked immunosorbent assay (ELISA) was used to
deter-mine the 50% effective concentration (EC50) of four-domain sCD4 binding to
gp120. gp120 proteins of R3A, TA1, and V3(9,9) were expressed in 293T cells
using recombinant vaccinia virus vectors and purified byGalanthus nivalislectin
chromatography (Vector Laboratory, Burlingame, CA) as previously described (56). Purified gp120 at 100 ng per well was adsorbed onto Immunolon 2HB 96-well plates (Thermo Labsystems, Waltham, MA) in ELISA capture buffer (0.15 M sodium carbonate, 0.35 M sodium bicarbonate in phosphate-buffered saline (PBS; pH 9.6) overnight at 4°C. Wells were washed with PBS–0.05% Tween and then blocked with 2% bovine serum albumin in PBS for 1 h at 4°C.
Starting at 25g/ml, fivefold dilutions of sCD4 were made in blocking solution,
added to the plate at 100l/well, and allowed to bind for 2 h at 4°C. After a
washing step, OKT4 antibody at 1g/ml was added and allowed to bind for 2 h
at 4°C. OKT4 was detected with horseradish peroxidase-conjugated goat anti-mouse IgG. The optical density at 450 nm was measured by using an MRX
Revelation microplate reader (Dynex Technologies, Chantilly, VA). EC50s were
calculated and analyzed by using a two-sided Studentttest in GraphPad Prism
4.0 (GraphPad Software, Inc.).
RESULTS
Mutations responsible for restoring function to a V3-trun-cated Env protein.We previously reported that replacing the central 15 amino acids from the V3 loop of the R5X4 HIV-1 strain R3A with a three-residue GAG linker resulted in a poorly functional Env, termed V3(9,9), with this designation describing a V3 loop containing its first and last nine residues in addition to the cysteine residues at the base of the loop (31). However, function was eventually restored upon passaging this
on November 8, 2019 by guest
http://jvi.asm.org/
virus on SupT1 cells expressing high levels of CCR5. The adapted virus, termed TA1 (for tissue culture adapted 1), re-tained the V3 loop truncation but exhibited greatly enhanced membrane fusion activity compared to the unpassaged V3(9,9) virus. In addition, TA1 used CCR5 but not CXCR4 to mediate infection, was highly resistant to CCR5 antagonists by virtue of the ability to utilize drug-bound conformations of CCR5, and was highly sensitive to neutralization by b12, a broadly neu-tralizing antibody targeting the CD4 binding site (31). Relative to the poorly functional V3(9,9) Env, TA1 contained five mu-tations, including deletion of residues TTKN in the V2 loop, and a single amino acid change, T342A, in C3 near the base of V3, both of which result in the loss of N-linked glycosylation sites (Table 1). The other adaptive mutations consisted of an alanine-to-valine change at the tip of the truncated V3 loop, R254K in the C2 region, and an A509V mutation at the N terminus of the gp41 subunit. To identify the mutations re-sponsible for the enhanced function of TA1 and, more impor-tantly, the mechanism by which an Env protein can mediate entry despite a significant deletion in the functionally impor-tant V3 loop, we singly and in combination introduced each of the five mutations from the adapted clone TA1 into the un-adapted V3(9,9) background (Table 1). In addition, we pro-duced the reciprocal mutants and, subsequently, propro-duced a series of combination mutants.
The panel of Env mutants was used to produce viral pseudotypes that were normalized for p24 content, titrated on SupT1 cells expressing CCR5 to define the linear range of the assay, and then used for comparative infection assays using equivalent amounts of p24. Western blot analyses indicated that all Envs were incorporated onto virions with approxi-mately the same efficiency (data not shown). We found that the T342A mutation in the V3(9,9) background restored infection to 40% of TA1 levels (Fig. 1B), whereas the GVG mutation in
the V3 linker and the A509V mutation in gp41 enhanced infection efficiency by a modest amount. The ⌬TTKN and R254K mutations in the V2 loop and C2, respectively, did not increase viral entry in this assay. Experiments with various combinations of TA1 mutations revealed that T342A, in com-bination with either the GVG mutation in V3 or the A509V mutation in gp41 restored viral entry in this single-cycle infec-tion assay to the same levels as TA1. Conversely, restoring these mutations to wt sequences in TA1 reduced infectivity to levels near that of V3(9,9) (Fig. 1C). Similar results were obtained when using human 293T or NP2 cells expressing CD4 and CCR5, though overall infection levels were not as great as those obtained with SupT1.CCR5 cells.
[image:3.594.48.540.79.305.2]Growth kinetics of reengineered envelopes. Based on the results of the single cycle infection assays, we expected that introduction of the T342A mutation along with either the GVG or A509V mutation into replication competent V3(9,9) virus would enhance replication to levels similar to that of the TA1 virus. However, while the double mutants T342A/GVG and T342A/A509V, as well as the triple mutant T342A/GVG/ A509V, replicated efficiently on SupT1 cells that expressed CCR5 and the C-type lectin DC-SIGNR, they did so with delayed kinetics (Fig. 2). Consistent with this, TA1-infected cultures were characterized by extensive cytopathic effects and very large syncytia during the first 3 to 5 days postinfection, while few syncytia were observed in cultures infected with the triple mutant until more than 7 days postinfection. For all viruses, the pattern of replication was similar for virus inputs of 5 ng (Fig. 2) or 50 ng (data not shown), with the only difference being the time to peak RT levels. Therefore, while the T342A mutation, along with either the GVG or A509V mutations, played the greatest role in restoring function to the V3(9,9) Env, the remaining two adaptive mutations make functional
TABLE 1. Schematic representation of envelope constructsa
Envelope
Region关R3A amino acids(s)兴
Function V1/V2
(185–188) C2 (254) V3 (308-322) C3 (342)
Peptide (509)
Parental envelopes
R3A TTKN R RVTLGPGRVYYTTGQ T A ⫹⫹⫹⫹
V3(9,9) TTKN R ----GAG--- T A ⫹/⫺
TA1 ----* K ----GVG--- A* V ⫹⫹⫹
Adaptive mutations placed in a V3(9,9) background
⌬TTKN ----* R ----GAG--- T A ⫺
R254K TTKN K ----GAG--- T A ⫹/⫺
GVG TTKN R ----GVG--- T A ⫹
T342A TTKN R ----GAG--- A* A ⫹⫹
A509V TTKN R ----GAG--- T V ⫹
Adaptive mutations removed from a TA1 background
⫹TTKN TTKN K ----GVG--- A* V ⫹⫹⫹
K254R ----* R ----GVG--- A* V ⫹⫹
GAG ----* K ----GAG--- A* V ⫹⫹
A342T ----* K ----GVG--- T V ⫹⫹
V509A ----* K ----GVG--- A* A ⫹⫹
a
Amino acid changes associated with adaptation of the V3 truncated virus are indicated in boldface. Asterisks indicate amino acid sequences that result in loss of an N-linked glycosylation site relative to V3(9,9).
on November 8, 2019 by guest
http://jvi.asm.org/
contributions that are evident in the context of a spreading infection, but not in a single-cycle infection assay.
Impact of TA1 adaptive mutations on sensitivity to CCR5 inhibitors.One of the most striking features of the TA1 virus is its complete resistance to CCR5 antagonists. Previous ex-periments using the unadapted V3(9,9) Env in cell-cell fusion assays suggested that the V3 truncation alone was able to impart some level of drug resistance. However, for TA1 it was not clear whether simple truncation of the V3 loop was solely responsible for this phenotype or whether the adaptive muta-tions contributed to CCR5 antagonist resistance. To examine
this, we performed infection assays using viral pseudotypes bearing R3A or TA1 Envs or Envs containing combinations of the most important adaptive mutations in the V3(9,9) back-ground. Unfortunately, infection levels obtained with the un-adapted V3(9,9) Env were too low to measure in this assay.
[image:4.594.113.473.69.262.2]Infection assays were performed on SupT1.CCR5 cells in the presence of increasing concentrations of the CCR5 antag-onist maraviroc (Fig. 3). As expected, viral pseudotypes with the R3A Env were fully inhibited by maraviroc, whereas TA1 pseudotypes were completely resistant. We found that all vi-ruses bearing the V3 loop truncation were fully resistant to
FIG. 1. A subset of adaptive mutations impart function to a V3-truncated Env. SupT1.CCR5 cells were infected with 5 ng of luciferase reporter viral pseudotypes bearing the indicated Env, and luciferase expression was measured 72 h postinfection. Entry levels were normalized against the parental Env TA1. (A) Viral pseudotypes bearing R3A, TA1, or V3(9,9) parental Envs. (B) Adaptive mutations present in TA1, introduced into the V3(9,9) Env singly and in combination, as indicated. Asterisks indicate entry levels that are significantly higher than entry by V3(9,9) (P⬍
0.028) (C) Viral pseudotypes with reciprocal mutations introduced into TA1 Env. The average entry⫾the standard error of the mean is indicated.
[image:4.594.73.253.478.659.2]FIG. 2. Adaptive mutations that impart envelope function do not confer wild-type growth kinetics. SupT1.CCR5.DC-SIGNR cells were infected with 5 ng of replication competent virus bearing the indicated Env on a pNL4-3 proviral background. Culture supernatants were collected at the indicated time points and assayed for reverse trans-criptase (RT) activity. The data are representative of two independent experiments.
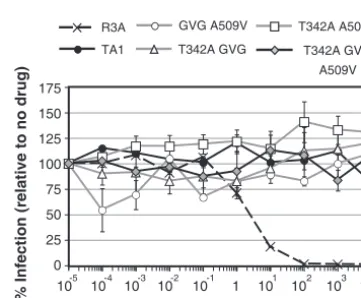
FIG. 3. Adaptive mutations in TA1 do not modulate resistance to the CCR5 inhibitor maraviroc. SupT1.CCR5 cells were incubated with various concentrations of maraviroc for 1 h prior to the addition of 5 ng of luciferase reporter viral pseudotypes bearing the indicated Envs. Saturating levels of AMD3100 were also included to block any entry through CXCR4 by the R5X4 parental R3A virus. Luciferase activity was read 72 h postinfection and graphed as the percentage of infection relative to infection in the absence of drug. The data represent the average of three independent experiments⫾the standard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:4.594.329.510.483.632.2]maraviroc regardless of the presence or absence of any adap-tive mutations. When two of the most important adapadap-tive mu-tations (T342A and A509V) were introduced into the parental Env R3A, full sensitivity to maraviroc was retained (data not shown). Taken together, these results indicate that the V3 loop truncation introduced into the R3A Env protein is largely or entirely responsible for CCR5 antagonist resistance and is not significantly modified by adaptive mutations that arose during passaging in tissue culture.
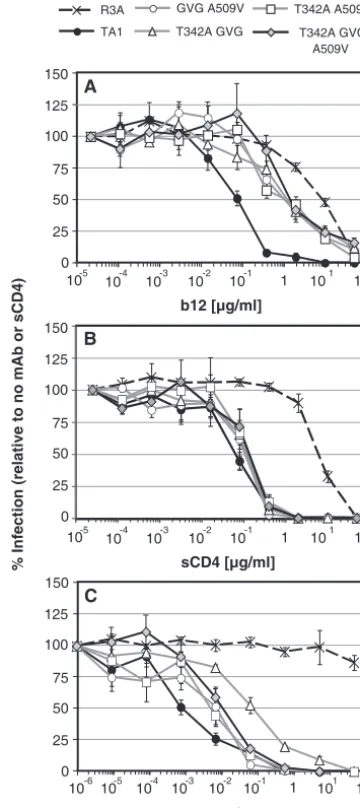
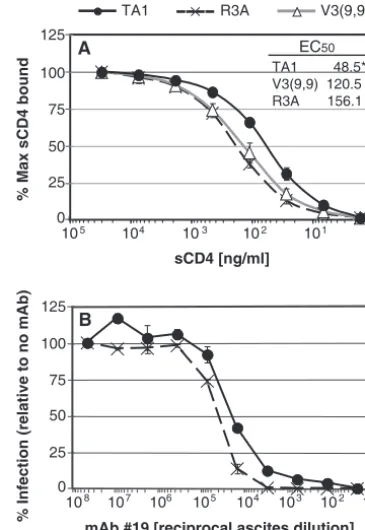
Impact of TA1 adaptive mutations on sensitivity to MAbs and sCD4.TA1 is exquisitely sensitive to neutralization by the broadly neutralizing MAb b12, which engages the CD4 binding site on gp120 (31), and is even more sensitive to MAb 17b, which binds to the bridging sheet region, probably due to enhanced exposure of this functionally important domain. Al-though the adaptive mutations GVG, T342A and A509V did not obviously contribute to the CCR5 antagonist resistance phenotype, we found that they did play a role in the pro-nounced neutralization sensitivity of TA1. Viral pseudotypes were preincubated with MAb b12 prior to incubation with SupT1.CCR5 cells. Three days later, cells were lysed and the amount of luciferase activity determined. As we have reported previously, TA1 was more than 100-fold more sensitive to neutralization by b12 than parental R3A (Fig. 4A). In com-parison, V3(9,9) Envs containing two or three of the five adap-tive mutations in TA1 exhibited intermediate neutralization phenotypes (Fig. 4A). Viruses containing the unadapted V3(9,9) Env did not infect cells sufficiently well to determine accurate 50% inhibitory concentrations (IC50s). These data argue that while the V3 truncation by itself largely accounts for the resistance of TA1 to CCR5 antagonists, it is the combina-tion of the V3 loop truncacombina-tion and the adaptive changes that increase sensitivity to b12, perhaps by increasing exposure of the CD4 binding site. Indeed, TA1 was approximately two-logs more sensitive to neutralization by sCD4 than R3A (Fig. 4B). However, the adaptive changes did not contribute strongly to this phenotype. Instead, the V3 truncation alone was primarily responsible for the enhanced sensitivity of TA1 to sCD4, whose binding site on gp120 overlaps but is not identical to the b12 epitope.
The adaptive mutations found in TA1 also played a role in enhancing sensitivity to neutralization by MAb 17b, which binds to the CD4-induced bridging sheet region of gp120 (30). Although R3A is resistant to MAb 17b, all V3 truncation-containing Envs were sensitive to the antibody. However, V3(9,9) Envs bearing two or three adaptive mutations were not as sensitive to neutralization by MAb 17b as was the fully adapted TA1 Env (Fig. 4C). If the V3 truncation were solely responsible for enhanced sensitivity to 17b, then we would expect the double and triple mutants to be neutralized by 17b as efficiently as TA1, but this was not seen. This suggests that that the V3 truncation likely plays a central role in sensitivity to MAb 17b but that the adaptive mutations that confer greater replication capacity on the truncated Env substantially increase sensitivity to this bridging sheet-directed MAb. In summary, the adaptive mutations in TA1 result in increased sensitivity to neutralization by MAbs b12 and 17b (but not to sCD4), which bind to regions in gp120 that are involved in CD4 and core-ceptor binding, respectively.
[image:5.594.331.511.70.474.2]Mechanism of enhanced function via altered interactions with CD4 and CCR5. One mechanism to account for the en-hanced sensitivity of TA1 to antibodies that bind to the CD4 and coreceptor binding sites would be if these epitopes on gp120 were more exposed as a result of the adaptive mutations. If so, TA1 might be able to more efficiently engage CD4 and/or CCR5 and thus be able to infect cells expressing low levels of receptor more efficiently. To investigate this, we used the dual-inducible 293-Affinofile cell line, in which the expression levels of both CD4 and CCR5 are independently modulated via sep-arate, inducible promoters (27a, 34, 49). In this cell line, in-creasing concentrations of the inducers minocycline and pon-asterone A result in corresponding increases in cell surface
FIG. 4. Adaptive mutations in TA1 modulate sensitivity to neutral-ization by MAbs b12 and 17b, and by sCD4. Five nanograms of lucif-erase reporter viral pseudotypes bearing the indicated Envs was incu-bated with various concentrations of MAb b12 (A), sCD4 (B), or MAb 17b (C) for 1 h prior to infection of SupT1.CCR5 cells. The luciferase activity was measured 72 h postinfection and graphed as the percent-age of infection relative to infection in the absence of sCD4 or anti-body. The data represent the average of three (sCD4) or four (MAbs) independent experiments⫾the standard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
expression levels of CD4 and CCR5, respectively. By using six concentrations of minocycline and eight concentrations of pon-asterone A, a matrix of 48 different expression levels of CD4 and CCR5 can be assayed. Quantitative flow cytometry was used to confirm that the expression levels of receptor and coreceptor were reproducible and consistent among experi-ments. Expression levels in response to graded doses of in-ducer varied from⬃2,000 to⬃110,000 copies of CD4 per cell, while CCR5 varied from⬃1,200 to⬃22,000 copies per cell. Primary human CD4⫹T cells typically express between 50,000 and 70,000 copies of CD4 per cell, while CCR5 expression levels tend to be quite variable between individuals, though are typically less than 14,000 copies per cell (36). Thus, expression levels of both CD4 and CCR5 spanned a physiological range in response to induction.
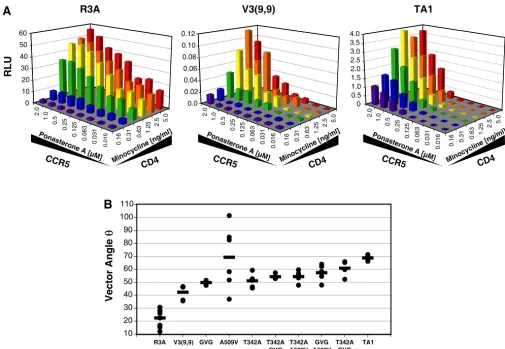
[image:6.594.42.547.66.415.2]Pseudotyped viruses bearing the R3A, TA1 or panel of V3(9,9) Envs containing key adaptive mutations were used to infect the panel of variably induced 293-Affinofile cells, with each condition being performed in duplicate within each ex-periment, and each experiment being replicated at least four times. We found that the parental R3A virus was able to infect cells expressing low levels of CCR5, but only if a threshold level of CD4 was expressed (Fig. 5A). If CD4 levels were low, R3A was unable to infect, even if CCR5 levels were high. In contrast, TA1 was able to infect cells expressing very low levels of CD4 but was much more sensitive than R3A to CCR5 expression levels. Infection mediated by the V3(9,9) Env was both inefficient [note the scale of the y axis: V3(9,9) exhibited a signal-to-noise ratio of approximately 10:1] and required high levels of both CD4 and CCR5. In comparing the infection
FIG. 5. V3 truncation results in inefficient use of CCR5, and adaptive mutations restore envelope function by increasing efficiency of CD4 use. The 293-Affinofile cell line, which can be induced to express graded levels of CD4 and CCR5 in response to minocycline and ponasterone A, respectively, was used to measure infection of viral pseudotypes at 48 combinations of CD4 and CCR5 expression levels. Luciferase reporter viral pseudotypes (10 to 20 ng) bearing the indicated Envs were added to cells at 18 h postinduction, and the luciferase levels were measured at 72 h postinfection. For infection with viral pseudotypes bearing the dualtropic Env R3A, induced cells were treated with 10 nM AMD3100 for 1 h prior to infection. (A) Infection levels of viral pseudotypes bearing the three parental Envs are shown, with each bar representing the level of infection (relative light units [RLUs]) at a particular combination of CD4 and CCR5 expression. The data shown are from a single representative experiment for each Env. Note that the differenty-axis scales reflect differences in overall infection efficiency. The infection levels of viral pseudotypes bearing adaptive mutations in the V3(9,9) Env were measured and used for vector angle analysis, but their plots are not shown. (B) Using the VERSA computational platform, sensitivity vectors were fit to surface plots of the infection data, and the vector anglecalculated for each Env. The CD4/CCR5 usage pattern for each isolate is reflected by its vector angle, with small angles representing efficient use of CCR5 and large angles representing efficient use of CD4. Dots represent vector angles calculated from individual experiments, with the average vector angle from four to eight experiments indicated by a dash.
on November 8, 2019 by guest
http://jvi.asm.org/
pattern of V3(9,9) to that of TA1, it is evident that the adaptive mutations acquired by TA1 improved its infection efficiency by greater than 1 log, enhanced the ability of the virus to utilize low levels of CD4, but had no obvious impact on CCR5 utili-zation. To quantify this, the complex infection data generated by the 293-Affinofile cells were mathematically fitted to three-dimensional surface plots and used to derive a sensitivity vec-tor that describes the relative dependence of virus entry on CD4 and CCR5 levels (see accompanying report by Johnston et al. [27a]). The angle of this vector () is a numerical de-scription of whether a virus is most sensitive to changes in CD4 or CCR5 levels. The greater the angle, the greater the depen-dence on CCR5 levels for virus entry. Thus, TA1 had a vector angle of 69°, reflecting its greater dependence on CCR5 levels while being able to use low CD4 levels, while R3A had a vector angle of 22°, reflecting its greater dependence on CD4 levels and ability to utilize low CCR5 levels (Fig. 5B). When various adaptive mutations were introduced into V3(9,9) singly and in combination, it was evident that the adaptive mutations pro-gressively increased the vector angle, indicating that A509V, T342A, and the V3-linker GVG mutations each improved CD4 utilization, with double and triple mutants showing a combinatorial effect. For reasons that we do not understand, results with V3(9,9) containing the A509V mutation were highly variable.
The results with the Affinofile cells were confirmed in part by performing inhibition studies with antibodies to CCR5 (data not shown). We found that TA1 entry was more sensitive than R3A to antibodies that bind to the second extracellular loop (ECL2) of CCR5, a domain that is important for gp120 binding (13, 14, 60, 64), as well as by antibodies to the N-terminal domain of CCR5. The IC50for TA1 was fivefold lower than that of R3A for MAb 2D7, whose epitope includes residues at the base of ECL2 (35, 64). Similarly, the IC50for TA1 was
15-to 20-fold lower than that of R3A for MAbs 45529 and 45531, which recognize the C-terminal portion of ECL2 (35). Thus, antibodies that block CCR5 domains important for gp120 binding and thereby reduce the levels of available CCR5 on the target cell have a greater effect on TA1 entry, confirming that TA1 is more sensitive than R3A to levels of CCR5 on target cells.
TA1 adaptations confer slightly increased affinity for CD4.
The ability of TA1 to more efficiently utilize low levels of CD4 to infect cells could be due to enhanced affinity. To test this, monomeric R3A, V3(9,9) or TA1 gp120 proteins were cap-tured directly onto 96-well ELISA plates. Increasing concen-trations of 4-domain sCD4 were added to the captured gp120 and detected with the MAb OKT4, which recognizes a mem-brane-proximal epitope of CD4 (53). The EC50for TA1 was 48.5 ng of sCD4/ml, compared to R3A at 156.1 ng/ml and V3(9,9) at 120.5 ng of sCD4/ml (Fig. 6A). In addition, we found that TA1 was slightly less sensitive than R3A to inhibi-tion by MAbs directed against CD4, suggesting that TA1 is able to bind CD4 with higher affinity (Fig. 6B and data not shown). Finally, these differences were relatively slight, though statistically significant and highly reproducible. Besides affinity, the ability of TA1 to utilize low levels of CD4 for infection could be due to postbinding effects, such as the induction of conformational changes needed for coreceptor engagement. One way that this can be determined is to measure the
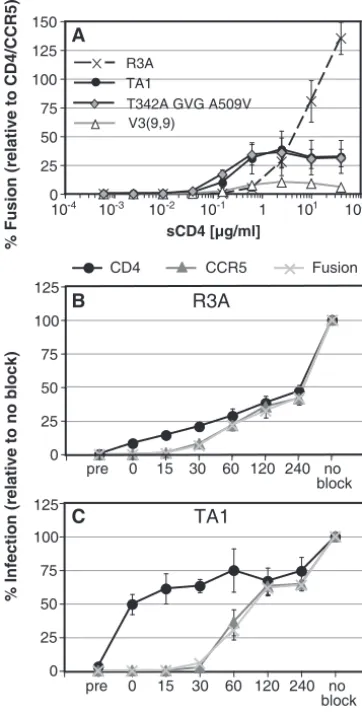
effi-ciency with which a viral Env protein can be triggered by sCD4 to elicit fusion with cells expressing coreceptor alone. We ex-pressed the R3A, TA1, V3(9,9), or the triple mutant Env proteins in QT6 cells and incubated them with QT6 cells ex-pressing high levels of CCR5 in the presence of increasing concentrations of sCD4 (Fig. 7A). Under these conditions, all Env proteins were able to elicit cell-cell fusion, with R3A doing so up to threefold more efficiently than TA1 or the triple mutant. However, both TA1 and the triple mutant were trig-gered by lower concentrations of sCD4 than was R3A, suggest-ing that the adaptive mutations enhance the sensitivity of these V3-loop deleted Envs to sCD4. However, the inability of TA1 or the triple-mutant Env to achieve high levels of fusion even with high concentrations of sCD4 suggests that there are ob-stacles to efficient entry mediated by these Envs after CD4 has been bound in this context, most likely attributed to inefficient use of CCR5 (Fig. 5).
[image:7.594.328.511.69.334.2]TA1 entry is delayed at a step after CD4 engagement.If the adaptive changes in TA1 enhance its ability to both bind CD4 and to undergo subsequent conformational changes more ef-ficiently, then virions bearing the TA1 Env protein should
FIG. 6. TA1 has increased affinity for CD4. (A) Equilibrium bind-ing of sCD4 to immobilized monomeric R3A, V3(9,9), or TA1 gp120 at 4°C was measured by ELISA. Bound CD4 was detected by using MAb OKT4 and horseradish peroxidase-conjugated anti-mouse IgG. The percent binding relative to the maximum for each gp120 is shown, representing the average of four independent experiments. An asterisk indicates that the EC50of TA1 for sCD4 is significantly lower than that of R3A and V3(9,9) (P⬍0.039). (B) Sensitivity of viral pseudotypes bearing R3A or TA1 Env to inhibition by MAb 19, which targets CD4. SupT1.CCR5 cells were incubated with various dilutions of MAb 19 for 1 h prior to the addition of 5 ng of luciferase reporter virus. Luciferase activity was read 72 h postinfection. The percent infection relative to infection in the absence of antibody is shown. The data represent the average of three independent experiments⫾the stan-dard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
become resistant to antibodies that bind to CD4 more quickly than virions bearing the parental R3A Env protein. However, the diminished ability of TA1 to utilize CCR5 could translate into a kinetic delay between CD4 binding and subsequent CCR5 interactions, which could account for the enhanced sen-sitivity of TA1 to enfuvirtide (31, 50). To test this, we modified a single-cycle replication assay such that inhibitors of CD4 binding, CCR5 binding, or virus-cell fusion were added at
intervals after infection. In this assay, virus is spinoculated onto target NP2.CD4.CCR5 cells at 4°C, which allows the virus to contact the cell surface but prevents fusion and entry. NP2.CD4.CCR5 cells were used due to their adherence to substrate, making time of addition experiments with associated medium changes more efficient. At time zero, the spinoculated cells are transferred to 37°C with or without saturating con-centrations of various inhibitors being added at different times. Since each inhibitor targets a specific step in viral entry, it is possible to assess the relative rates at which virus binds to CD4 or CCR5 or undergoes membrane fusion.
Under these conditions on NP2.CD4.CCR5 cells, MAbs to CD4 or CCR5, or the membrane fusion inhibitor enfuvirtide, completely prevented infection by either R3A or TA1 if added prior to the addition of virus. When the various inhibitors were added at different times after virus binding, we found that R3A became progressively resistant to the neutralizing antibody to CD4 (Fig. 7B). After CD4 engagement, however, coreceptor binding and fusion occurred in relatively quick succession, as indicated by the nearly overlapping curves of R3A infection in the presence of antibodies to CCR5 or enfuvirtide. Thus, for R3A, CD4 binding appears to be the rate-limiting step in the entry process, a result that is consistent with fact that R3A required relatively high levels of CD4 to support virus infection (Fig. 5). In contrast, TA1 became resistant to the anti-CD4 MAb very quickly, indicating that it binds to CD4 in a func-tionally irreversible fashion soon after binding to the cell sur-face (Fig. 7C). However, there was an extensive lag phase before TA1 became resistant to anti-CCR5 MAbs. Like R3A, there was a short lag between CCR5 binding and membrane fusion as judged by resistance to enfuvirtide. This result indi-cates that TA1 engages CD4 quickly, a finding consistent with the Affinofile cell data, as well as with the ease with which TA1 membrane fusion activity can be triggered by sCD4. However, after binding to CD4, it is evident that TA1 binds to CCR5 inefficiently, which is consistent with the finding that high levels of CCR5 are needed to support infection by this virus (Fig. 5) and by the fact that TA1 is about 1 log more sensitive to enfuvirtide than is R3A (31).
DISCUSSION
[image:8.594.72.253.72.430.2]The central goals of this study were to examine how Env function can be restored via adaptive mutations after deletion of a significant portion of the V3 loop, and to determine whether mutations that restore Env function contribute to CCR5 inhibitor resistance or the marked sensitivity exhibited by TA1 to neutralization by the CD4 binding site MAb b12. Functionally, the V3 loop plays the major role in determining whether Env uses CCR5, CXCR4 or both coreceptors to infect cells (6, 7, 57). Structural studies indicate that in the CD4-bound conformation the V3 loop extends from the surface of gp120 by⬃30 Å (24), a distance thought to be sufficient for the tip of the V3 loop to engage the ECLs of the coreceptor, while the base of the V3 loop and the adjoining bridging sheet engage the N-terminal domain of the coreceptor (9, 10, 21, 23, 24, 51). Since coreceptor binding is critically important for triggering the conformational changes needed for virus-mem-brane fusion, disruption of V3 loop-coreceptor interactions can prevent virus infection. Small molecule CCR5 and CXCR4
FIG. 7. TA1-mediated entry is limited by inefficient engagement of CCR5. (A) QT6 cells expressing R3A, TA1, V3(9,9), or the triple-mutant Env protein were incubated with QT6 cells expressing cell-surface CCR5, in the presence of increasing concentrations of sCD4. Fusion between Env-expressing cells and CD4⫺/CCR5⫹cells is shown as the percentage of fusion relative to fusion with CD4⫹/CCR5⫹cells. The data represent the average of three independent experiments⫾ the standard error of the mean. (B and C) Kinetic analysis of CD4 engagement, CCR5 engagement, and fusion of viral pseudotypes bearing R3A (B) or TA1 (C) Env proteins during infection of NP2.CD4.CCR5 cells. At each time point in independent wells, CD4 binding was blocked using MAb 19, CCR5 binding was blocked using MAbs 2D7 and CTC5, and virus-cell fusion was blocked using enfu-virtide. The “pre” time point indicates addition of blocker prior to spinoculation (4°C) of virus onto cells; time zero and onward indicate addition of blocker postspinoculation and transfer to 37°C. Infection mediated by 5 ng of luciferase reporter viral pseudotypes bearing the indicated Env was measured at 72 h postinfection. The percentage of infection relative to infection in the absence of any blocker is shown⫾ the standard error of the mean.
on November 8, 2019 by guest
http://jvi.asm.org/
antagonists disrupt these interactions via an allosteric mecha-nism in which coreceptor conformation is altered (15, 39, 55, 61, 62), while genetic truncation of the V3 loop directly im-pacts Env-coreceptor interactions. TA1 provides a useful model to study the plasticity of Env-coreceptor interactions as it efficiently utilizes CCR5 even in the presence of saturating concentrations of potent CCR5 antagonists.
Theoretically, the adaptive changes in TA1 Env could en-hance function via several mechanisms. The most obvious, perhaps, would be for mutations in TA1 to enhance CCR5 interactions by increasing binding affinity or by making Env more susceptible to the conformational changes that are in-duced by CCR5 binding. Our results are not consistent with either of these models, although the assays available to assess these functions are indirect and not as quantitative as we would like. That TA1 interacts with CCR5 less efficiently than R3A is suggested by the fact that its fusion activity is reduced in most contexts and by the finding that while R3A can mediate infec-tion of cells expressing low levels of CCR5, TA1 cannot. Sim-ilarly, our time of addition studies with various HIV entry inhibitors revealed that after binding CD4, R3A quickly en-gages CCR5. In contrast, a lag phase of⬃60 min follows CD4 binding for TA1 before it irreversibly engages CCR5. How-ever, do the mutations present in TA1 enable it to interact with CCR5 more efficiently than the unadapted V3(9,9) Env? Due to the poor function exhibited by V3(9,9), our data on this point are not conclusive. If the adaptive changes present in TA1 improved its ability to utilize CCR5, we would anticipate that TA1 would be able to infect cells that express low levels of CCR5 more efficiently than the unadapted V3(9,9) Env. Our results with the Affinofile cell system suggest that this is not the case: TA1 and V3(9,9) were similarly dependent upon CCR5 expression levels under conditions in which CD4 was not lim-iting, with neither being able to mediate infection of cells expressing low levels of CCR5 regardless of CD4 expression levels.
While our results argue that the adaptive changes in TA1 do not enhance CCR5 utilization, we found clear evidence that, relative to R3A and V3(9,9), TA1 exhibits enhanced ability to bind CD4 and to undergo CD4-induced conformational changes. This conclusion is supported by several lines of evi-dence. First, provided that sufficiently high levels of CCR5 were expressed, TA1 was able to mediate infection of cells expressing low levels of CD4 more efficiently than either R3A or V3(9,9). Second, TA1 gp120 bound to sCD4 with higher affinity than R3A or V3(9,9) gp120, although this does not necessarily mean that trimeric TA1 Env binds CD4 with greater affinity than R3A. However, time of addition studies with antibodies that bind CD4 and block infection indicated that virions expressing the TA1 Env engaged cell surface CD4 more quickly than did the parental R3A virus, which is con-sistent with enhanced affinity. Finally, the membrane fusion activity of TA1 could be triggered by low levels of sCD4. Taken together, these results indicate that the adaptive mutations that arose in V3(9,9) upon passaging on SupT1 cells, ultimately giving rise to TA1, did so at least in part by not only enhancing the ability of Env to bind to CD4, but to undergo CD4-induced conformational changes.
In addition to the fact that TA1 functions relatively well despite lacking much of its V3 loop, it exhibits two other
unusual phenotypes: complete resistance to CCR5 inhibitors and markedly enhanced sensitivity to neutralization by the CD4-binding site MAb b12. In the case of CCR5 inhibitor resistance, our results suggest that the V3 loop truncation alone accounts for this phenotype. If so, by what mechanism does this occur, and might V3 truncation always result in re-sistance to coreceptor inhibitors? The model we propose for this phenotype has as its basis the well-known plasticity of Env-coreceptor interactions. A large number of structure-func-tion studies are consistent with a two-step binding model, in which the V3 loop engages the ECLs of CCR5 (with the sec-ond ECL being particularly important) while the N terminus of CCR5 binds to the base of the V3 loop and the bridging sheet, with sulfated tyrosine residues in CCR5 playing a central role (9, 10, 18, 23, 24, 38, 45). Nonetheless, this model is subject to variability: some Envs are more sensitive to mutations in the N terminus of CCR5, while others are more sensitive to muta-tions in the ECLs (14). The picture that emerges is a two-step binding model in which the contributions of each binding event (N terminus versus ECLs) to inducing membrane fusion vary to some degree between virus strains. We propose that R3A is better able to utilize the N-terminal domain of CCR5 than many other viruses. Thus, loss of the distal portion of the V3 loop, with the concomitant loss of interactions with the CCR5 ECLs, is tolerated to some degree. In contrast, similar dele-tions in other virus strains usually result in a complete loss of function (19, 54, 65, 67, 69). Since small molecule CCR5 in-hibitors bind to a hydrophobic pocket in the transmembrane helices of the coreceptor, they may preferentially disrupt the conformation of the ECLs (15, 39, 55, 61–63). If this model is correct, then viruses that can engage the N-terminal domain of the coreceptor more efficiently than other virus strains may continue to engage CCR5 even in its drug-bound conforma-tion. Our limited structure-function studies with CCR5 mu-tants and TA1 are consistent with this model. Thus, provided that a virus can tolerate loss of the distal portion of the V3 loop, it may very well exhibit resistance to CCR5 inhibitors in the context of reduced overall function. Consistent with this, an HIV-2 strain that functions well despite loss of its V3 loop also exhibits complete resistance to CCR5 inhibitors (37).
Although the adaptive mutations in TA1 made little or no contribution to CCR5 inhibitor resistance, they clearly contrib-uted to the sensitivity of TA1 to neutralization by the CD4 binding site MAb b12. It is tempting to speculate that the enhanced ability of TA1 to both bind and respond to CD4 is linked to its enhanced sensitivity to a CD4 binding site anti-body. If the CD4 binding site becomes more exposed as a consequence of partial V3 loop truncation coupled with adap-tive mutations, including the loss of two N-linked glycosylation sites, then enhanced sensitivity to neutralization would likely result. A crystal structure of an unliganded HIV-1 gp120 would be needed to more fully understand the contributions of the adaptive mutations to increased sensitivity to MAb b12.
In summary, we find that deletions in the V3 loop that abrogate important interactions with CCR5 are partially com-pensated for by mutations that enhance interactions with CD4, the step in the virus entry pathway that is immediately up-stream of coreceptor binding. How enhanced CD4 binding can compensate for less efficient CCR5 interactions is not clear, but might be related to the cooperative nature of the
on November 8, 2019 by guest
http://jvi.asm.org/
brane fusion reaction. It is likely that for membrane fusion to occur, an Env trimer will have to bind multiple CD4 and coreceptor molecules (29, 43, 48). In addition, membrane fu-sion may also require the concerted action of several Env trimers (40), though some models suggest that a single trimer might be sufficient to form a fusion pore (68). By binding to CD4 more quickly, the coreceptor binding sites in TA1 Env trimers should be induced more efficiently, making subsequent interactions with CCR5 possible. Associated with this pheno-type is exquisite sensitivity to the broadly neutralizing MAb b12. If this property is due to enhanced exposure of the CD4 binding site, V3-truncated and adapted Envs could be explored for their abilities to induce antibodies against this conserved and functionally important region.
ACKNOWLEDGMENTS
C.A.-G. and M.M.L. were supported in part by NIH T32 AI007632. R.W.D. was supported by NIH R01 AI 040880 and by the Interna-tional AIDS Vaccine Initiative. R.G.C. was supported by NIH R01 AI 035572. J.A.H. and R.W.D. were also supported by NIH RO1 AI45378.
REFERENCES
1.Alexander, W. A., B. Moss, and T. R. Fuerst.1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA
polymerase and theEscherichia colilac repressor. J. Virol.66:2934–2942.
2.Berger, E. A., P. M. Murphy, and J. M. Farber.1999. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu. Rev.
Immunol.17:657–700.
3.Biscone, M. J., J. L. Miamidian, J. M. Muchiri, S. S. Baik, F. H. Lee, R. W. Doms, and J. D. Reeves.2006. Functional impact of HIV coreceptor-binding
site mutations. Virology351:226–236.
4.Chen, B. K., K. Saksela, R. Andino, and D. Baltimore.1994. Distinct modes of human immunodeficiency virus type 1 proviral latency revealed by super-infection of nonproductively infected cell lines with recombinant
luciferase-encoding viruses. J. Virol.68:654–660.
5.Chiou, S. H., E. O. Freed, A. T. Panganiban, and W. R. Kenealy.1992. Studies on the role of the V3 loop in human immunodeficiency virus type 1
envelope glycoprotein function. AIDS Res. Hum. Retrovir.8:1611–1618.
6.Choe, H., M. Farzan, Y. Sun, N. Sullivan, B. Rollins, P. D. Ponath, L. Wu, C. R. Mackay, G. LaRosa, W. Newman, N. Gerard, C. Gerard, and J. Sodroski.1996. The beta-chemokine receptors CCR3 and CCR5 facilitate
infection by primary HIV-1 isolates. Cell85:1135–1148.
7.Cocchi, F., A. L. DeVico, A. Garzino-Demo, A. Cara, R. C. Gallo, and P. Lusso.1996. The V3 domain of the HIV-1 gp120 envelope glycoprotein is
critical for chemokine-mediated blockade of infection. Nat. Med.2:1244–
1247.
8.Connor, R. I., B. K. Chen, S. Choe, and N. R. Landau.1995. Vpr is required for efficient replication of human immunodeficiency virus type-1 in
mono-nuclear phagocytes. Virology206:935–944.
9.Cormier, E. G., and T. Dragic.2002. The crown and stem of the V3 loop play distinct roles in human immunodeficiency virus type 1 envelope glycoprotein
interactions with the CCR5 coreceptor. J. Virol.76:8953–8957.
10.Cormier, E. G., D. N. Tran, L. Yukhayeva, W. C. Olson, and T. Dragic.2001. Mapping the determinants of the CCR5 amino-terminal sulfopeptide inter-action with soluble human immunodeficiency virus type 1 gp120-CD4
com-plexes. J. Virol.75:5541–5549.
11.De Wolf, F., E. Hogervorst, J. Goudsmit, E. M. Fenyo, H. Rubsamen-Waig-mann, H. Holmes, B. Galvao-Castro, E. Karita, C. Wasi, S. D. Sempala, et
al.1994. Syncytium-inducing and non-syncytium-inducing capacity of human
immunodeficiency virus type 1 subtypes other than B: phenotypic and
geno-typic characteristics. AIDS Res. Hum. Retrovir.10:1387–1400.
12.Doms, R. W., and J. P. Moore.2000. HIV-1 membrane fusion: targets of
opportunity. J. Cell Biol.151:F9–F14.
13.Doranz, B. J., Z. H. Lu, J. Rucker, T. Y. Zhang, M. Sharron, Y. H. Cen, Z. X. Wang, H. H. Guo, J. G. Du, M. A. Accavitti, R. W. Doms, and S. C. Peiper.
1997. Two distinct CCR5 domains can mediate coreceptor usage by human
immunodeficiency virus type 1. J. Virol.71:6305–6314.
14.Dragic, T.2001. An overview of the determinants of CCR5 and CXCR4
coreceptor function. J. Gen. Virol.82:1807–1814.
15.Dragic, T., A. Trkola, D. A. Thompson, E. G. Cormier, F. A. Kajumo, E. Maxwell, S. W. Lin, W. Ying, S. O. Smith, T. P. Sakmar, and J. P. Moore.
2000. A binding pocket for a small molecule inhibitor of HIV-1 entry within
the transmembrane helices of CCR5. Proc. Natl. Acad. Sci. USA97:5639–
5644.
16.Edwards, T. G., T. L. Hoffman, F. Baribaud, S. Wyss, C. C. LaBranche, J. Romano, J. Adkinson, M. Sharron, J. A. Hoxie, and R. W. Doms.2001. Relationships between CD4 independence, neutralization sensitivity, and exposure of a CD4-induced epitope in a human immunodeficiency virus type
1 envelope protein. J. Virol.75:5230–5239.
17.Endres, M. J., P. R. Clapham, M. Marsh, M. Ahuja, J. D. Turner, A. McKnight, J. F. Thomas, B. Stoebenau-Haggarty, S. Choe, P. J. Vance, T. N. Wells, C. A. Power, S. S. Sutterwala, R. W. Doms, N. R. Landau, and J. A. Hoxie.1996. CD4-independent infection by HIV-2 is mediated by fusin/
CXCR4. Cell87:745–756.
18.Farzan, M., N. Vasilieva, C. E. Schnitzler, S. Chung, J. Robinson, N. P. Gerard, C. Gerard, H. Choe, and J. Sodroski.2000. A tyrosine-sulfated peptide based on the N terminus of CCR5 interacts with a CD4-enhanced epitope of the HIV-1 gp120 envelope glycoprotein and inhibits HIV-1 entry.
J. Biol. Chem.275:33516–33521.
19.Grimaila, R. J., B. A. Fuller, P. D. Rennert, M. B. Nelson, M. L. Hammar-skjold, B. Potts, M. Murray, S. D. Putney, and G. Gray.1992. Mutations in the principal neutralization determinant of human immunodeficiency virus type 1 affect syncytium formation, virus infectivity, growth kinetics, and
neutralization. J. Virol.66:1875–1883.
20.Hartley, O., P. J. Klasse, Q. J. Sattentau, and J. P. Moore.2005. V3: HIV’s
switch-hitter. AIDS Res. Hum. Retrovir.21:171–189.
21.Hu, Q., K. B. Napier, J. O. Trent, Z. Wang, S. Taylor, G. E. Griffin, S. C. Peiper, and R. J. Shattock.2005. Restricted variable residues in the C-terminal segment of HIV-1 V3 loop regulate the molecular anatomy of
CCR5 utilization. J. Mol. Biol.350:699–712.
22.Hu, S. L., and L. Stamatatos.2007. Prospects of HIV Env modification as an
approach to HIV vaccine design. Curr. HIV Res.5:507–513.
23.Huang, C. C., S. N. Lam, P. Acharya, M. Tang, S. H. Xiang, S. S. Hussan, R. L. Stanfield, J. Robinson, J. Sodroski, I. A. Wilson, R. Wyatt, C. A. Bewley, and P. D. Kwong.2007. Structures of the CCR5 N terminus and of a
tyrosine-sulfated antibody with HIV-1 gp120 and CD4. Science317:1930–
1934.
24.Huang, C. C., M. Tang, M. Y. Zhang, S. Majeed, E. Montabana, R. L. Stanfield, D. S. Dimitrov, B. Korber, J. Sodroski, I. A. Wilson, R. Wyatt, and P. D. Kwong.2005. Structure of a V3-containing HIV-1 gp120 core. Science
310:1025–1028.
25.Javaherian, K., A. J. Langlois, G. J. LaRosa, A. T. Profy, D. P. Bolognesi, W. C. Herlihy, S. D. Putney, and T. J. Matthews.1990. Broadly neutralizing antibodies elicited by the hypervariable neutralizing determinant of HIV-1.
Science250:1590–1593.
26.Javaherian, K., A. J. Langlois, C. McDanal, K. L. Ross, L. I. Eckler, C. L. Jellis, A. T. Profy, J. R. Rusche, D. P. Bolognesi, S. D. Putney, et al.1989. Principal neutralizing domain of the human immunodeficiency virus type 1
envelope protein. Proc. Natl. Acad. Sci. USA86:6768–6772.
27.Jeffs, S. A., J. McKeating, S. Lewis, H. Craft, D. Biram, P. E. Stephens, and R. L. Brady.1996. Antigenicity of truncated forms of the human
immuno-deficiency virus type 1 envelope glycoprotein. J. Gen. Virol.77(Pt. 7):1403–
1410.
27a.Johnston, S. H., M. A. Lobritz, S. Nguyen, K. Lassen, S. Delair, F. Posta, Y. J. Bryson, E. J. Arts, T. Chou, and B. Lee.2009. A quantitative affinity-profiling system that reveals distinct CD4/CCR5 usage patterns among hu-man immunodeficiency virus type 1 and simian immunodeficiency virus
strains. J. Virol.83:11016–11026.
28.Kajumo, F., D. A. Thompson, Y. Guo, and T. Dragic.2000. Entry of R5X4 and X4 human immunodeficiency virus type 1 strains is mediated by nega-tively charged and tyrosine residues in the amino-terminal domain and the
second extracellular loop of CXCR4. Virology271:240–247.
29.Kuhmann, S. E., E. J. Platt, S. L. Kozak, and D. Kabat.2000. Cooperation of multiple CCR5 coreceptors is required for infections by human
immuno-deficiency virus type 1. J. Virol.74:7005–7015.
30.Kwong, P. D., R. Wyatt, J. Robinson, R. W. Sweet, J. Sodroski, and W. A. Hendrickson.1998. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature
393:648–659.
31.Laakso, M. M., F. H. Lee, B. Haggarty, C. Agrawal, K. M. Nolan, M. Biscone, J. Romano, A. P. Jordan, G. J. Leslie, E. G. Meissner, L. Su, J. A. Hoxie, and R. W. Doms.2007. V3 loop truncations in HIV-1 envelope impart resistance to coreceptor inhibitors and enhanced sensitivity to neutralizing antibodies.
PLoS Pathog.3:e117.
32.LaBranche, C. C., M. M. Sauter, B. S. Haggarty, P. J. Vance, J. Romano, T. K. Hart, P. J. Bugelski, and J. A. Hoxie.1994. Biological, molecular, and structural analysis of a cytopathic variant from a molecularly cloned simian
immunodeficiency virus. J. Virol.68:5509–5522.
33.Labrijn, A. F., P. Poignard, A. Raja, M. B. Zwick, K. Delgado, M. Franti, J. Binley, V. Vivona, C. Grundner, C. C. Huang, M. Venturi, C. J. Petropoulos, T. Wrin, D. S. Dimitrov, J. Robinson, P. D. Kwong, R. T. Wyatt, J. Sodroski, and D. R. Burton.2003. Access of antibody molecules to the conserved coreceptor binding site on glycoprotein gp120 is sterically restricted on
primary human immunodeficiency virus type 1. J. Virol.77:10557–10565.
34.Lassen, K. G., M. A. Lobritz, J. R. Bailey, S. Johnston, S. Nguyen, B. Lee, T. Chou, R. F. Siliciano, M. Markowitz, and E. J. Arts.2009. Elite
on November 8, 2019 by guest
http://jvi.asm.org/
derived HIV-1 envelope glycoproteins exhibit reduced entry efficiency and
kinetics. PLoS Pathog.5:e1000377.
35.Lee, B., M. Sharron, C. Blanpain, B. J. Doranz, J. Vakili, P. Setoh, E. Berg, G. Liu, H. R. Guy, S. R. Durell, M. Parmentier, C. N. Chang, K. Price, M. Tsang, and R. W. Doms.1999. Epitope mapping of CCR5 reveals multiple conformational states and distinct but overlapping structures involved in
chemokine and coreceptor function. J. Biol. Chem.274:9617–9626.
36.Lee, B., M. Sharron, L. J. Montaner, D. Weissman, and R. W. Doms.1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived
macro-phages. Proc. Natl. Acad. Sci. USA96:5215–5220.
37.Lin, G., A. Bertolotti-Ciarlet, B. Haggarty, J. Romano, K. M. Nolan, G. J. Leslie, A. P. Jordan, C. C. Huang, P. D. Kwong, R. W. Doms, and J. A. Hoxie.
2007. Replication-competent variants of human immunodeficiency virus type 2 lacking the V3 loop exhibit resistance to chemokine receptor antagonists.
J. Virol.81:9956–9966.
38.Lin, G., B. Lee, B. S. Haggarty, R. W. Doms, and J. A. Hoxie.2001. CD4-independent use of Rhesus CCR5 by human immunodeficiency virus type 2 implicates an electrostatic interaction between the CCR5 N terminus and the
gp120 C4 domain. J. Virol.75:10766–10778.
39.Maeda, K., D. Das, H. Ogata-Aoki, H. Nakata, T. Miyakawa, Y. Tojo, R. Norman, Y. Takaoka, J. Ding, G. F. Arnold, E. Arnold, and H. Mitsuya.
2006. Structural and molecular interactions of CCR5 inhibitors with CCR5.
J. Biol. Chem.281:12688–12698.
40.Magnus, C., P. Rusert, S. Bonhoeffer, A. Trkola, and R. R. Regoes.2009. Estimating the stoichiometry of human immunodeficiency virus entry. J.
Vi-rol.83:1523–1531.
41.Meissner, E. G., V. M. Coffield, and L. Su.2005. Thymic pathogenicity of an HIV-1 envelope is associated with increased CXCR4 binding efficiency and V5-gp41-dependent activity, but not V1/V2-associated CD4 binding
effi-ciency and viral entry. Virology336:184–197.
42.Meissner, E. G., K. M. Duus, F. Gao, X. F. Yu, and L. Su.2004. Character-ization of a thymus-tropic HIV-1 isolate from a rapid progressor: role of the
envelope. Virology328:74–88.
43.Mkrtchyan, S. R., R. M. Markosyan, M. T. Eadon, J. P. Moore, G. B. Melikyan, and F. S. Cohen.2005. Ternary complex formation of human immunodeficiency virus type 1 Env, CD4, and chemokine receptor captured
as an intermediate of membrane fusion. J. Virol.79:11161–11169.
44.Nara, P., L. Smit, N. Dunlop, W. Hatch, M. Merges, D. Waters, J. Kelliher, W. Krone, and J. Goudsmit.1990. Evidence for rapid selection and deletion of HIV-1 subpopulations in vivo by V3-specific neutralizing antibody: a
model of humoral-associated selection. Dev. Biol. Stand.72:315–341.
45.Nolan, K. M., A. P. Jordan, and J. A. Hoxie.2008. Effects of partial deletions within the human immunodeficiency virus type 1 V3 loop on coreceptor
tropism and sensitivity to entry inhibitors. J. Virol.82:664–673.
46.O’Doherty, U., W. J. Swiggard, and M. H. Malim.2000. Human immuno-deficiency virus type 1 spinoculation enhances infection through virus
bind-ing. J. Virol.74:10074–10080.
47.Palker, T. J., M. E. Clark, A. J. Langlois, T. J. Matthews, K. J. Weinhold, R. R. Randall, D. P. Bolognesi, and B. F. Haynes.1988. Type-specific
neu-tralization of the human immunodeficiency virus with antibodies to env
-encoded synthetic peptides. Proc. Natl. Acad. Sci. USA85:1932–1936.
48.Platt, E. J., J. P. Durnin, U. Shinde, and D. Kabat.2007. An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for
mem-brane fusion and overcomes diverse entry limitations. J. Mol. Biol.374:
64–79.
49.Pugach, P., N. Ray, P. J. Klasse, T. J. Ketas, E. Michael, R. W. Doms, B. Lee, and J. P. Moore.2009. Inefficient entry of vicriviroc-resistant HIV-1 via the
inhibitor-CCR5 complex at low cell surface CCR5 densities. Virology387:
296–302.
50.Reeves, J. D., S. A. Gallo, N. Ahmad, J. L. Miamidian, P. E. Harvey, M. Sharron, S. Pohlmann, J. N. Sfakianos, C. A. Derdeyn, R. Blumenthal, E. Hunter, and R. W. Doms.2002. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion
kinetics. Proc. Natl. Acad. Sci. USA99:16249–16254.
51.Rizzuto, C., and J. Sodroski.2000. Fine definition of a conserved CCR5-binding region on the human immunodeficiency virus type 1 glycoprotein
120. AIDS Res. Hum. Retrovir.16:741–749.
52.Rucker, J., B. J. Doranz, A. L. Edinger, D. Long, J. F. Berson, and R. W. Doms.1997. Cell-cell fusion assay to study role of chemokine receptors in
human immunodeficiency virus type 1 entry. Methods Enzymol.288:118–
133.
53.Sattentau, Q. J., A. G. Dalgleish, R. A. Weiss, and P. C. Beverley.1986.
Epitopes of the CD4 antigen and HIV infection. Science234:1120–1123.
54.Saunders, C. J., R. A. McCaffrey, I. Zharkikh, Z. Kraft, S. E. Malenbaum, B. Burke, C. Cheng-Mayer, and L. Stamatatos.2005. The V1, V2, and V3 regions of the human immunodeficiency virus type 1 envelope differentially
affect the viral phenotype in an isolate-dependent manner. J. Virol.79:9069–
9080.
55.Seibert, C., W. Ying, S. Gavrilov, F. Tsamis, S. E. Kuhmann, A. Palani, J. R. Tagat, J. W. Clader, S. W. McCombie, B. M. Baroudy, S. O. Smith, T. Dragic, J. P. Moore, and T. P. Sakmar.2006. Interaction of small molecule
inhibitors of HIV-1 entry with CCR5. Virology349:41–54.
56.Selvarajah, S., B. Puffer, R. Pantophlet, M. Law, R. W. Doms, and D. R. Burton.2005. Comparing antigenicity and immunogenicity of engineered
gp120. J. Virol.79:12148–12163.
57.Speck, R. F., K. Wehrly, E. J. Platt, R. E. Atchison, I. F. Charo, D. Kabat, B. Chesebro, and M. A. Goldsmith.1997. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined
by individual amino acids within the envelope V3 loop. J. Virol.71:7136–
7139.
58.Sullivan, N., Y. Sun, J. Binley, J. Lee, C. F. Barbas III, P. W. Parren, D. R. Burton, and J. Sodroski.1998. Determinants of human immunodeficiency virus type 1 envelope glycoprotein activation by soluble CD4 and monoclonal
antibodies. J. Virol.72:6332–6338.
59.Thali, M., J. P. Moore, C. Furman, M. Charles, D. D. Ho, J. Robinson, and J. Sodroski.1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding.
J. Virol.67:3978–3988.
60.Thompson, D. A., E. G. Cormier, and T. Dragic.2002. CCR5 and CXCR4 usage by non-clade B human immunodeficiency virus type 1 primary isolates.
J. Virol.76:3059–3064.
61.Tsamis, F., S. Gavrilov, F. Kajumo, C. Seibert, S. Kuhmann, T. Ketas, A. Trkola, A. Palani, J. W. Clader, J. R. Tagat, S. McCombie, B. Baroudy, J. P. Moore, T. P. Sakmar, and T. Dragic.2003. Analysis of the mechanism by which the small-molecule CCR5 antagonists SCH-351125 and SCH-350581
inhibit human immunodeficiency virus type 1 entry. J. Virol.77:5201–5208.
62.Watson, C., S. Jenkinson, W. Kazmierski, and T. Kenakin.2005. The CCR5 receptor-based mechanism of action of 873140, a potent allosteric
noncom-petitive HIV entry inhibitor. Mol. Pharmacol.67:1268–1282.
63.Westby, M., C. Smith-Burchnell, J. Mori, M. Lewis, M. Mosley, M. Stock-dale, P. Dorr, G. Ciaramella, and M. Perros.2007. Reduced maximal inhi-bition in phenotypic susceptibility assays indicates that viral strains resistant to the CCR5 antagonist maraviroc utilize inhibitor-bound receptor for entry.
J. Virol.81:2359–2371.
64.Wu, L., G. LaRosa, N. Kassam, C. J. Gordon, H. Heath, N. Ruffing, H. Chen, J. Humblias, M. Samson, M. Parmentier, J. P. Moore, and C. R. Mackay.
1997. Interaction of chemokine receptor CCR5 with its ligands: multiple domains for HIV-1 gp120 binding and a single domain for chemokine
bind-ing. J. Exp. Med.186:1373–1381.
65.Wyatt, R., J. Moore, M. Accola, E. Desjardin, J. Robinson, and J. Sodroski.
1995. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor
binding. J. Virol.69:5723–5733.
66.Wyatt, R., and J. Sodroski.1998. The HIV-1 envelope glycoproteins:
fuso-gens, antifuso-gens, and immunogens. Science280:1884–1888.
67.Wyatt, R., N. Sullivan, M. Thali, H. Repke, D. Ho, J. Robinson, M. Posner, and J. Sodroski.1993. Functional and immunologic characterization of hu-man immunodeficiency virus type 1 envelope glycoproteins containing
dele-tions of the major variable regions. J. Virol.67:4557–4565.
68.Yang, X., S. Kurteva, S. Lee, and J. Sodroski.2005. Stoichiometry of
anti-body neutralization of human immunodeficiency virus type 1. J. Virol.79:
3500–3508.
69.Yang, Z. Y., B. K. Chakrabarti, L. Xu, B. Welcher, W. P. Kong, K. Leung, A. Panet, J. R. Mascola, and G. J. Nabel.2004. Selective modification of variable loops alters tropism and enhances immunogenicity of human
im-munodeficiency virus type 1 envelope. J. Virol.78:4029–4036.
70.Zhong, P., M. Peeters, W. Janssens, K. Fransen, L. Heyndrickx, G. Vanham, B. Willems, P. Piot, and G. van der Groen. 1995. Correlation between genetic and biological properties of biologically cloned HIV type 1 viruses
representing subtypes A, B, and D. AIDS Res. Hum. Retrovir.11:239–248.