The ATM and Rad3-Related (ATR) Protein Kinase Pathway Is
Activated by Herpes Simplex Virus 1 and Required for Efficient
Viral Replication
Terri G. Edwards,
aDavid C. Bloom,
aChris Fisher
aaDepartment of Molecular Genetics and Microbiology, University of Florida College of Medicine, Gainesville,
Florida, USA
ABSTRACT
The ATM and Rad3-related (ATR) protein kinase and its downstream
ef-fector Chk1 are key sensors and organizers of the DNA damage response (DDR) to a
variety of insults. Previous studies of herpes simplex virus 1 (HSV-1) showed no
evi-dence for activation of the ATR pathway. Here we demonstrate that both Chk1 and
ATR were phosphorylated by 3 h postinfection (h.p.i.). Activation of ATR and Chk1
was observed using 4 different HSV-1 strains in multiple cell types, while a specific
ATR inhibitor blocked activation. Mechanistic studies point to early viral gene
ex-pression as a key trigger for ATR activation. Both pATR and pChk1 localized to the
nucleus within viral replication centers, or associated with their periphery, by 3 h.p.i.
Significant levels of pATR and pChk1 were also detected in the cytoplasm, where
they colocalized with ICP4 and ICP0. Proximity ligation assays confirmed that pATR
and pChk1 were closely and specifically associated with ICP4 and ICP0 in both the
nucleus and cytoplasm by 3 h.p.i., but not with ICP8 or ICP27, presumably in a
multiprotein complex. Chemically distinct ATR and Chk1 inhibitors blocked HSV-1
replication and infectious virion production, while inhibitors of ATM, Chk2, and
DNA-dependent protein kinase (DNA-PK) did not. Together our data show that HSV-1
ac-tivates the ATR pathway at early stages of infection and that ATR and Chk1 kinase
activities play important roles in HSV-1 replication fitness. These findings indicate
that the ATR pathway may provide insight for therapeutic approaches.
IMPORTANCE
Viruses have evolved complex associations with cellular DNA damage
response (DDR) pathways, which sense troublesome DNA structures formed during
infection. The first evidence for activation of the ATR pathway by HSV-1 is
pre-sented. ATR is activated, and its downstream target Chk1 is robustly phosphorylated,
during early stages of infection. Both activated proteins are found in the nucleus
as-sociated with viral replication compartments and in the cytoplasm asas-sociated with
viral proteins. We also demonstrate that both ATR and Chk1 kinase activities are
im-portant for viral replication. The findings suggest that HSV-1 activates ATR and Chk1
during early stages of infection and utilizes the enzymes to promote its own
replica-tion. The observation may be exploitable for antiviral approaches.
KEYWORDS
herpes simplex virus, DNA damage response, ATM and Rad3-related
(ATR) kinase, ATR, Chk1, viral replication
T
he common human pathogen herpes simplex virus 1 (HSV-1) establishes lifelong,
persistent infection, with the primary infection usually occurring within oral or
genital epithelia and latency established in the sensory ganglia innervating these
tissues (1). HSV-1 possesses a large, linear, double-stranded genome of approximately
152 kb encoding roughly 90 unique transcriptional units (1). Productive infection is
accompanied by a highly regulated sequence of immediate early (IE), early (E), and late
(L) viral gene expression. The IE genes encode 5 proteins, including ICP4 and ICP0, that
Received30 October 2017Accepted13 December 2017
Accepted manuscript posted online20 December 2017
CitationEdwards TG, Bloom DC, Fisher C. 2018. The ATM and Rad3-related (ATR) protein kinase pathway is activated by herpes simplex virus 1 and required for efficient viral replication. J Virol 92:e01884-17.https://doi.org/10.1128/JVI .01884-17.
EditorRichard M. Longnecker, Northwestern University
Copyright© 2018 American Society for Microbiology.All Rights Reserved. Address correspondence to Chris Fisher, ctopherfis@ufl.edu.
crossm
on November 6, 2019 by guest
http://jvi.asm.org/
serve to create a favorable cellular milieu for the virus and to regulate subsequent E and
L gene expression. ICP4 is the major transcriptional regulatory protein essential for
transactivation of early and late viral gene expression. ICP0, which is nonessential for
growth in cell culture, is a multifunctional E3 ubiquitin ligase that nevertheless has
important roles in regulating gene expression and negating host cell defense functions
(2). Subsequent E and L gene expression includes at least seven proteins essential for
HSV-1 DNA replication: ICP8 (E; single-stranded DNA [ssDNA] binding protein), UL9 (E;
origin binding protein), a 3-member helicase-primase complex (UL8 [E], UL5 [E], and
UL52 [E]), and the polymerase UL30 (E) and its processivity factor UL42 (E) (3). ICP8 is
an essential ssDNA binding protein required for the establishment of nuclear
replica-tion centers (4, 5). ICP4 and ICP8 colocalize to viral replicareplica-tion compartments by 3 h
postinfection (h.p.i.), with the onset of viral DNA replication, where they support
translation and DNA replication (6, 7).
DNA viruses have a complex relationship with host DNA damage response (DDR)
pathways that serve to protect the cellular genome from point mutations, deletions,
insertions, and other forms of damage. Most viruses have evolved the means to evade
DDR surveillance and to use repair pathways to promote their own program (8–10).
Three DDR pathways, defined by their proximal phosphoinositide-3-kinase-related
kinases, serve as the principal mediators of the DDR. The ataxia-telangiectasia mutated
(ATM) kinase senses and organizes the cellular response to double-stranded DNA
(dsDNA) break repair, while the ATM and Rad3-related (ATR) kinase organizes the
cellular response to an array of DNA insults resulting in exposure of ssDNA, including
stalled replication forks (11). ATR, like ATM, also responds to dsDNA breaks where
resection of the DNA ends exposes ssDNA due to the nuclease activity of the dsDNA
break-sensing Mre11-Rad50-Nbs1 (MRN) complex and associated nucleases, including
CtIP, Exo1, and Dna2 (12–15). The Chk2 and Chk1 effector kinases act downstream of
ATM and ATR, respectively, to help orchestrate and integrate the DDR (16). While ATM
and ATR generally organize DNA repair via homologous recombination (HR) pathways,
the DNA-dependent protein kinase (DNA-PK) defines a third arm of the DDR that
organizes repair primarily by way of nonhomologous end joining (NHEJ) (11).
HSV-1 has significant connections with all three DDR pathways (17). ICP0 is
recog-nized to block DNA-PK function by targeting its catalytic subunit for proteasomal
degradation (2, 18, 19). ATM, on the other hand, is activated following HSV-1 infection
(20, 21). Activation of ATM accompanies the recruitment of numerous HR elements to
viral replication centers, including members of the dsDNA break-sensing MRN complex
(Mre11, Rad50, and Nbs1) and RAD51 (20–22). However, ICP0 targeting of the histone
ubiquitin ligases RNF8 and RNF168 hinders events downstream of ATM signaling, which
results in increased HSV-1 fitness (23). Thus, it appears that HSV-1 successfully attracts
and maintains numerous HR elements in replication centers in order to promote virus
replication while minimizing the antiviral properties of the pathway. Many large and
small DNA viruses employ similar strategies to promote their replication (9, 24).
Previous investigations of the other apical kinase, ATR, failed to detect activation of
this pathway, with multiple reports asserting that ATR is not activated following HSV-1
infection. The negative data include lack of phosphorylation of well-known ATR
sub-strates, including Chk1 (21, 25) and replication protein A (RPA) (22, 26). Other reports
have claimed that ATR is actually inactivated by HSV-1 because infection was found to
block hyperphosphorylation of RPA and Chk1 induced by either hydroxyurea (HU) or
UV irradiation (27, 28). Studies have attempted to mechanistically explain why ATR
would not be activated following infection. It was reported that HSV-1 infection results
in sequestration of ATRIP to virus-induced, chaperone-enriched (VICE) domains away
from ATR, which localized to nucleoli in both infected and uninfected cells, thus
preventing ATR activation (26). However, this model was abandoned with the
realiza-tion that the ATR and ATRIP antibodies originally employed lacked the appropriate
specificity (28). Another model asserts that the HSV-1 single-stranded DNA binding
protein (ICP8) colocalizes with the helicase/primase complex (UL8/UL5/UL52) to ssDNA,
where they prevent access of the 9-1-1 complex, thus blocking ATR activation (27).
on November 6, 2019 by guest
http://jvi.asm.org/
In spite of findings suggesting that ATR is not activated by HSV-1, a recent study
found that ATR-deficient fibroblasts are defective in their ability to fully support HSV-1
replication, suggesting a potential role for ATR in HSV-1 infection (29). We previously
reported on the activation and control of human papillomavirus maintenance by ATR
(30) and therefore decided to examine the ATR pathway during early stages of HSV-1
infection. We found that HSV-1 infection results in the robust activation of the ATR
pathway, as indicated by measurement of phosphorylation of both ATR and Chk1.
Furthermore, we found that ATR and Chk1 kinase activities are both required for
efficient viral replication. Multiple, chemically distinct ATR and Chk1 inhibitors
attenu-ate the maturation of HSV-1 replication centers, inhibiting viral replication and virus
production. HSV-1 replication is also significantly decreased in ATR-deficient, Seckel
patient fibroblasts, providing further support for an ATR role in early stages of HSV-1
infection. Mechanistic studies point to E gene expression as important for ATR pathway
activation. These findings have important implications for early events during HSV-1
infection and may provide opportunities for new HSV-1 therapeutic approaches.
RESULTS
U2OS cells infected with HSV-1 (17
syn
⫹
strain; multiplicity of infection [MOI]
⫽
10)
were initially examined by Western blotting (WB) (Fig. 1) using phospho-specific
antibodies to probe a 6-h time course of infection. Total ATM, ATR, and Chk1 levels,
detected with pan-specific antibodies, did not change over the course of the
experi-ments (Fig. 1). U2OS cells infected with HSV-1 (17
syn
⫹
; MOI
⫽
10) showed a single
band reacting with the phospho-specific Chk1 antibody (Ab), migrating at 56 kDa and
first appearing at 3 h.p.i. (Fig. 1A). All subsequent time points continued to show robust
Chk1 phosphorylation suggesting activation of ATR. Infection of Vero cells for the same
times and at the same MOIs confirmed the results from the U2OS cells, with clear
HSV-1-induced activation of Chk1 (Fig. 1A). Quantification of pChk1 expression in U2OS
and Vero cells relative to that in mock-infected cells showed 96- and 72-fold peak
increases, respectively. ATM and ATR phosphorylation was also detected following
M 2h 3h 4h 5h 6h
A
pChk1S345
Chk1Pan
pChk1S345
Chk1Pan
B
pATMS1981
ATMPan
pATRS428
ATRPan
M 0h 3h 6h
U2OS
Vero
U2OS
FIG 1Western blots demonstrating phosphorylation of pChk1S345and pATRS428over time following infection. (A) U2OS (top) or Vero (bottom) cells were infected with HSV-1 17syn⫹ (MOI⫽10) and prepared for Western blotting at the indicated times p.i. with antibodies recognizing pChk1S345or Chk1Pan. Chk1 activation is first detected at 3 h.p.i. for both cell types. (B) U2OS cells were infected with HSV-1 17syn⫹(MOI⫽10) and prepared for Western blotting at the indicated h.p.i. with antibodies recognizing ATMS1981, ATMPan, ATRS428, and ATRPan. ATM activation is first detectable at the end of the preincubation period (0 h.p.i.), while evidence of ATR phosphorylation is first visible at 3 h.p.i.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:3.585.135.276.73.297.2]infection. pATR levels were increased by 3 h.p.i., while pATM levels were found to be
increased by 0 h.p.i., at the time of removal of HSV-1 inoculum (Fig. 1B). The detected
changes in ATR phosphorylation during activation, compared to that in mock-infected
cells, were comparable to changes detected in several cell lines following DNA damage
according to antibody manufacturers’ data, and as established in the literature (31).
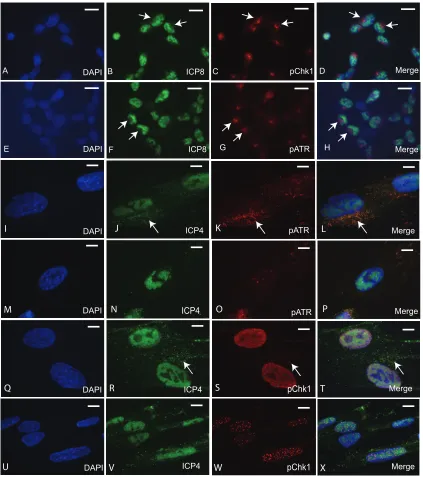
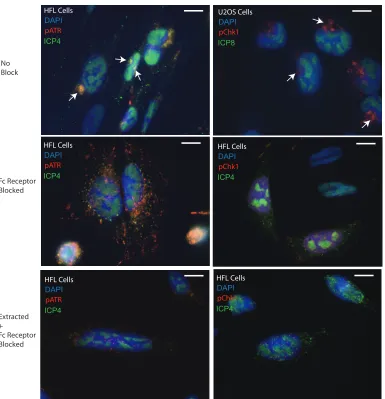
In initial immunofluorescence (IF) experiments, infected cells exhibited strong pATR
and pChk1 fluorescence at 3 h.p.i. that was primarily concentrated in juxtanuclear
domains (Fig. 2A to H). We suspected for several reasons that this finding was an
artifact. Earlier time points, such as 1 or 2 h.p.i., showed a similar result and therefore
were not in agreement with our Western blot time course of ATR activation (data not
shown). The perinuclear localization of pChk1 and pATR was also found in close
association with the microtubule organizing center (MTOC) and disrupted by colchicine
(data not shown). The MTOC is also the site of formation of the human cytomegalovirus
(hCMV) “assembly compartment,” which was previously shown to artifactually bind
rabbit IgG antibodies (32) due to the presence of CMV-encoded Fc receptor-like protein
(33). HSV-1 has also been shown to encode and express a similar Fc receptor-binding
complex (34, 35).
We therefore optimized our immunofluorescence protocol to include an Fc receptor
blocking step and, in some cases, a modification of a widely used procedure for
studying DDR dynamics by preextraction of live cells (
in situ
fractionation) (36). When
applied to a time course of HSV-1-infected U2OS cells, the results reproduced the
kinetics of ATR and Chk1 activation seen in Western blots (data not shown). Both pATR
(Fig. 2I to P) and pChk1 (Fig. 2Q to X) were found to be localized within the nuclei of
infected human fetal lung (HFL) cells by 5 h.p.i. HSV-1 replication centers were well
established by 5 h.p.i. as seen by ICP4 labeling. Punctate foci of both pATR (Fig. 2I to
P) and pChk1 (Fig. 2Q to X) were found to be associated with these structures within
the nucleus. In addition, cytoplasmic accumulation of both pATR and pChk1 was noted
especially in unextracted cells (Fig. 2I to L and Q to T), where they often appeared to
colocalize with ICP4 (arrows). The nuclear pATR and pChk1 foci were particularly
evident in cells prepared by
in situ
fractionation (Fig. 2M to P and U to X). Additional
examples of preextracted cells are provided in Fig. 3A. Both pATR and pChk1 puncta
were closely associated with replication centers, often on their periphery (Fig. 2M to P
and U to X and 3A). The foci surviving
in situ
extraction represent insoluble,
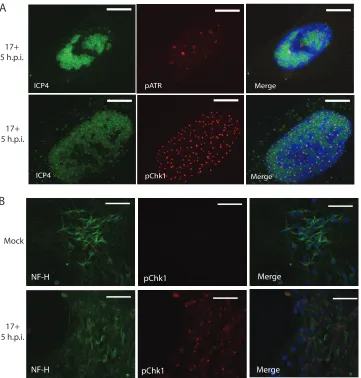
chromatin-associated DDR complexes (36). Three additional HSV-1 strains—KOS/M, McKrae, and
McIntyre—were also found to be capable of activating the ATR pathway (data not
shown). Dopamine-like neurons differentiated from the Lund human mesencephalic
(LUHMES) cell line likewise activated the ATR pathway in response to HSV-1 infection
(Fig. 3B). Thus, the ATR pathway is activated in numerous cell types (Vero, HFL, U2OS,
and differentiated LUHMES cells) of broad tissue and species origins in response to
HSV-1 infection (Fig. 1 to 3). Micrographs comparing the 3 different
immunofluores-cence protocols in HSV-1-infected cells are provided in Fig. 4. Immunofluoresimmunofluores-cence
experiments were regularly conducted without primary antibody and were routinely
negative (data not shown).
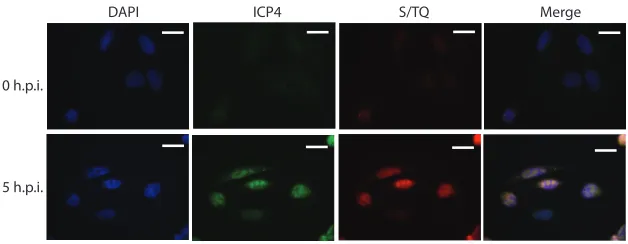
An ATR/ATM substrate antibody recognizing phospho-SQ/TQ sites reacted strongly
with HSV-1 replication centers, consistent with the ATR pathway being highly active
within these centers (Fig. 5). The specific ATR kinase inhibitor VE-822 was next used to
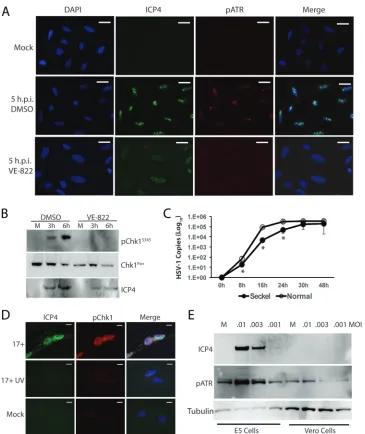
examine ATR/Chk1 activation. U2OS cells receiving vehicle (0.1% dimethyl sulfoxide
[DMSO]) or ATR inhibitor (10
M) were infected with HSV-1 (17
syn
⫹
; MOI
⫽
10) and
examined by immunofluorescence for pATR or by Western blotting for pChk1 (Fig. 6A
and B). VE-822 blocked ATR activation, as observed by immunofluorescence (Fig. 6A),
while Chk1 activation was also blocked, as observed in Western blots (Fig. 6B).
Equiv-alent results were also obtained using Vero and HFL cells (data not shown).
Interest-ingly, this series of experiments also showed that ATR inhibition by VE-822 blocked
maturation of HSV-1 replication centers and inhibited expression of ICP4 (Fig. 6A and
B), suggesting that ATR kinase activity is required for maturation of HSV-1 replication
compartments and accumulation of ICP4. It is noteworthy that ICP4-positive nuclear
on November 6, 2019 by guest
http://jvi.asm.org/
compartments of VE-822-treated cells appear to be similar to the prereplicative sites
observed following inhibition of HSV-1 polymerase activity (37). These results are
suggestive of and consistent with VE-822 either indirectly or directly inhibiting HSV-1
DNA replication. Consistent with this notion, inhibition of HSV-1 polymerase activity
with phosphonoacetic acid (PAA) results in accumulation of prereplication
compart-ments with similar morphology (data not shown) (1, 37).
We next asked if Seckel patient fibroblasts, which carry an ATR hypomorphic
DAPI ICP8 pChk1 Merge
A B C D
pChk1
DAPI ICP4 Merge
U
V
W
X
DAPI
M
K
pATR
DAPI ICP8 Merge
E F G H
ICP4
I
J
L
ICP4
pChk1
DAPI Merge
Merge
N
O
P
Q
R
ICP4S
T
pATR
Merge pATR
DAPI
FIG 2Localization of pATR and pChk1 following HSV-1 17syn⫹infection (MOI⫽10) of U2OS cells (A to H) and HFL cells (I to X). (A to D) Localization of DAPI, ICP8, and pChk1S345in formalin-fixed cells at 3 h.p.i. without Fc receptor block shows artifactual binding of rabbit pChk1 antibody to a juxtanuclear position. (E to H) Localization of DAPI, ICP8, and pATRS428in formalin-fixed cells at 3 h.p.i. without Fc receptor block shows artifactual binding of rabbit pATR antibody to juxtanuclear position. (I to L) Localization of DAPI, ICP4, and pATRS428at 5 h.p.i. with HSV-1 17syn⫹in Fc receptor-blocked, unextracted HFL cells. Arrows indicate colocalization of ICP4 and pATR in cytoplasm. (M to P) Localization of DAPI, ICP4, and pATRS428at 5 h.p.i. with HSV-1 17syn⫹in Fc receptor-blocked, preextracted HFL cells. (Q to T) Localization of DAPI, ICP4, and pChk1S345at 5 h.p.i. with HSV-1 17syn⫹in Fc receptor-blocked, unextracted cells. Arrows indicate colocalization of ICP4 and pChk1 in cytoplasm. (U to X) Localization of DAPI, ICP4, and pChk1S345at 5 h.p.i. with HSV-1 17syn⫹in Fc receptor-blocked, preextracted cells. Scale bars⫽20m in panels A to H and 10m in panels I to X.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:5.585.41.464.70.547.2]mutation resulting in impaired ATR function (38), would support HSV-1 replication.
Following HSV-1 infection (17
syn
⫹
; MOI
⫽
0.01), the production of viral genomes was
significantly inhibited in the ATR-deficient cells relative to that in normal patient
fibroblasts at the 8-, 16-, and 24-h time points (Fig. 6C). Inhibition of protein synthesis
with cycloheximide completely blocked ATR activation, while the viral polymerase
inhibitor PAA partially suppressed ATR activation (data not shown). HSV-1 (17
syn
⫹
)
virions that had been inactivated by UV irradiation were likewise found to not activate
the ATR pathway (Fig. 6D). A range of factors, including stalled replication forks,
replication stress, and exposure of ssDNA, are known to activate the ATR pathway. KD6,
a nonreplicating (ICP4
⫺) HSV-1 recombinant virus (39), was employed to further
address requirements for ATR activation. KD6 activated the ATR pathway in the
complementing Vero-derived cell line E5, which is transformed with ICP4 (Fig. 6E). The
E5 cells also supported expression of ICP4 following infection. Wild-type Vero cells, on
the other hand, which are unable to support KD6 replication, showed no evidence of
ICP4 expression and did not activate the ATR pathway (Fig. 6E). Together these results
suggested that the incoming viral inoculum was not responsible for ATR activation.
ICP4-dependent events, such as viral E gene expression, are likely key to activation of
ICP4 pChk1 Merge
Merge pATR
ICP4
NF-H
Mock
17+
5 h.p.i.
B
NF-H
NF-H
pChk1
pChk1
Merge
Merge
A
B
17+
5 h.p.i.
17+
5 h.p.i.
FIG 3Additional micrographs of pATR and pChk1 distribution in HFL and LUHMES cells. (A) High-magnification micrographs of extracted, Fc receptor-blocked HFL cells show association of both pATR and pChk1 in puncta closely associated with HSV-1 replication centers. Scale bars⫽10m. (B) Neurofilament heavy subunit (NF-H)-positive neurons differentiated from LUHMES cells show activation of Chk1 following infection by HSV-1 17syn⫹. Cells are unextracted. Scale bars⫽50m.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:6.585.43.404.71.449.2]the ATR pathway. However, viral DNA synthesis cannot be ruled out in playing a
cooperative role in ATR pathway activation.
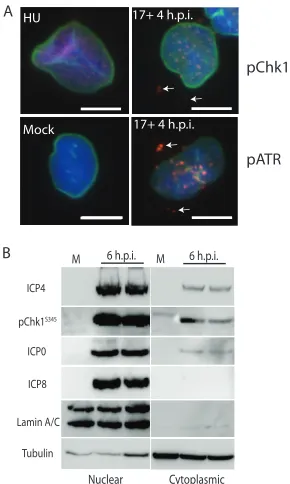
As mentioned above, ICP4, pATR, and pChk1 were routinely found in the cytoplasm
of infected cells following infection. In many cases, especially in unextracted cells,
colocalization of ICP4 with pATR and pChk1 was observed (arrows in Fig. 2J to L and R
to T). Coimmunofluorescence staining of pChk1 or pATR with lamin A/B also revealed
nuclear and cytoplasmic activation of both proteins following HSV-1 infection (Fig. 7A).
By comparison, U2OS cells treated with hydroxyurea (HU) to stall DNA replication forks
exhibited pChk1 labeling in the form of small puncta throughout the nuclei (Fig. 7A).
Infection with HSV-1 resulted in formation of larger, more pronounced pATR and pChk1
puncta at 4 h.p.i., while mock-infected cells showed no evidence of ATR activation (Fig.
7A). The infected cells, but not HU-treated cells, also showed evidence of pATR and
pChk1 outside the bounds of the nuclear envelope. In order to confirm the cytoplasmic
distribution of these proteins, we fractionated, in duplicate, both mock-infected cells
and cells infected for 6 h with HSV-1 17
syn
⫹
(MOI
⫽
10) and subjected the fractions to
pATR ICP4 DAPI HFL Cells
pATR ICP4 DAPI HFL Cells pATR ICP4 DAPI HFL Cells
pChk1 ICP4 DAPI HFL Cells
pChk1 ICP4 DAPI HFL Cells
pChk1 ICP8 DAPI U2OS Cells
No Block
Fc Receptor Blocked
Extracted + Fc Receptor Blocked
FIG 4Comparison of the 3 different immunofluorescence protocols used in this study. The top micrographs were captured of cells prepared with no block of the Fc receptor. The positions of aggresomes that artifactually bind primary antibodies are indicated with arrows. The middle-row micrographs were captured from cells in which Fc receptor was blocked prior to immunolabeling. The distribution of pATR and pChk1 in the nucleus is more easily ascertained without camera exposures shortened by bright aggresome labeling. The bottom micrographs show immunolabeling of cells that were preextracted prior to blocking Fc receptor. Nuclear labeling associated with the HSV-1 replication centers is clearer in these cells. Scale bars⫽10m.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:7.585.43.428.68.467.2]Western blotting (Fig. 7B). Lamin A/C and tubulin separated into their appropriate
fractions. Most pChk1 was recovered with the nuclear fraction, but the protein was also
detected in the cytoplasmic fraction of infected cells. ICP4 and ICP0 were also found in
the cytoplasmic fractions, while ICP8 was detected only in the nuclear fraction (Fig. 7B).
Mock-infected cells were negative for viral proteins and lacked detectable Chk1
phos-phorylation.
Because of their close cytoplasmic association following infection (Fig. 2I to L and Q
to T and 4), we next asked if a proximity ligation assay (PLA) could detect a direct
association between ICP4 and pATR or pChk1 in infected cells. A robust PLA signal was
generated so long as both ICP4 and either pATR or pChk1 antibodies were present (Fig.
8A). PLA of mock-infected cells, or PLA with a single antibody, gave only background
signal (Fig. 8A). The PLA signals were predominantly cytoplasmic, although nuclear
amplification events were also noted and judged significant because they were absent
from controls or mock-infected cells. Quantification of the PLA amplification events
revealed close interactions of both pATR and pChk1 with both ICP4 and ICP0 (Fig. 8B).
On the other hand, probing for interaction of either pATR or pChk1 with ICP8 or ICP27
detected only background levels of signal (Fig. 8B). The results showed good
agree-ment with the cell fractionation results (Fig. 7B), indicating that ICP4 and ICP0, but not
ICP8, are found in the cytoplasm following infection, where they form close associations
with both pATR and pChk1 (Fig. 8A and B).
We next asked whether pharmacological inhibition of ATR or Chk1 kinase activity
has antiviral effects upon HSV-1. U2OS cells infected with HSV-1 17
syn
⫹
(MOI
⫽
0.1)
were treated with one of four chemically distinct ATR inhibitors (VE-822, VE-821,
AZD-6738, and AZ20) or two unique Chk1 inhibitors (Chir124 and PF477736), or
inhibitors of ATM, Chk2, or DNA-PK (Fig. 9). All ATR and Chk1 inhibitors, as well as the
positive control, aphidicolin, which was expected to inhibit both viral and cellular DNA
replication (40), significantly inhibited progeny virion production, as measured by titer
assays (
P
⬍
0.01) (Fig. 9A). Notably, the ATR inhibitors VE-822 and AZ20, and the Chk1
inhibitors CHIR124 and PF47736, resulted in
ⱖ
10-fold reduction of infectious viral
progeny. Inhibition of ATM and Chk2, on the other hand, did not significantly inhibit
viral titers (Fig. 9A). Inhibition of DNA-PK activity resulted in a modest increase in
infectious progeny that did not achieve statistical significance. An increase in response
to inhibition of DNA-PK might be expected given the function of ICP0 in disabling this
pathway (19, 41). Viral genome copy numbers measured by quantitative PCR (qPCR)
were in good agreement with the viral titer results (Fig. 9B).
DISCUSSION
This is the first report of ATR pathway activation by HSV-1. ATR and Chk1 are
activated shortly after infection of multiple cell lines, including HFL, Vero, and U2OS
cells, as well as neurons differentiated from LUHMES cells. Four different HSV-1 strains
were observed to activate the ATR pathway. Several lines of evidence are presented to
DAPI ICP4 S/TQ Merge
0 h.p.i.
5 h.p.i.
FIG 5ATR/ATM substrate antibody (S/TQ) shows little nuclear reactivity at 0 h.p.i. by 17syn⫹ of unextracted U2OS cells but is highly reactive with HSV-1 replication centers where it colocalizes with ICP4 at 5 h.p.i. Scale bars⫽20m.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:8.585.49.363.71.193.2]demonstrate and confirm ATR pathway activation. (i) Chk1 is phosphorylated on S345,
a site known to be phosphorylated by ATR (42, 43), shortly after infection by HSV-1. (ii)
Chk1 phosphorylation is inhibited by the ATR-specific inhibitor VE-822. (iii) ATR is itself
phosphorylated on S428 following HSV-1 infection at the time of Chk1 activation. While
S428 is not an autophosphorylation site, its phosphorylation has been shown by many
labs to correlate with ATR activation following DNA damage (31). (iv) pATR and pChk1
complexes interact closely with HSV-1 replication centers while these structures acquire
strong reactivity with an ATR/ATM substrate antibody recognizing phospho-SQ/TQ
sites. (v) Finally, multiple, chemically distinct ATR and Chk1 kinase inhibitors block the
FIG 6The ATR kinase inhibitor VE-822 blocks activation of ATR and Chk1, while HSV-1 replication is deficient in Seckel patient fibroblasts hypomorphic for ATR. (A) Immunofluorescence images of ICP4 and pATR at 5 h.p.i. with HSV-1 17syn⫹(MOI⫽10) and treatment of U2OS cells with either a vehicle (DMSO) or VE-822 (10M). Cells are unextracted. Scale bars⫽20m. (B) Western blot demonstrating that VE-822 (10M) also blocks phosphorylation of Chk1 in HSV-1-infected U2OS cells while suppressing ICP4 expression. (C) Seckel syndrome patient fibroblasts, hypomorphic for ATR, are defective for HSV-1 replication relative to control patient (IBR3) fibroblasts. (D) UV-inactivated HSV-1 17syn⫹(17⫹UV; MOI⫽10) does not significantly activate the ATR pathway. Cells are unextracted. Scale bars⫽10m. (E) An ICP4⫺, replication-incompetent mutant HSV-1 virus (KD6) does not express ICP4 and does not activate the ATR pathway in nonpermissive (Vero) cells following 24 h of infection with a low MOI. The permissive cell line E5, which supplies ICP4, shows expression of ICP4 and activation of the ATR pathway.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:9.585.40.405.67.501.2]maturation of HSV-1 replication centers and inhibit HSV-1 genome copy number and
progeny virus production.
We detected HSV-1 activation of the ATR pathway by 3 h.p.i., which correlates well
with the onset of E gene expression and viral DNA replication (44). Both pATR and
pChk1 form insoluble nuclear complexes closely associated with HSV-1 replication
centers, primarily on their periphery. Phosphonoacetic acid (PAA) is well known for its
ability to block the maturation of HSV-1 replication centers, resulting in the
accumu-lation of small ICP4 and ICP8 prereplication centers (1, 37). We observed that PAA
blocks replication center maturation and that this is not sufficient for blocking ATR
activation. We therefore hypothesize that ICP4-dependent E gene expression is the
primary cause of ATR activation. Several observations are consistent with this notion: (i)
inhibition of protein synthesis with cycloheximide blocks ATR activation completely; (ii)
the specific ATR inhibitor VE-822 blocks activation of both ATR and Chk1 during
infection, and it also blocks maturation of replication centers; (iii) ICP4- and
ICP8-positive nuclear centers that form in the presence of VE-822 are very similar to those
formed in the presence of PAA; (iv) UV-inactivated HSV-1 fails to activate the ATR
pathway; and (v) a mutant HSV-1 virus lacking ICP4 (KD6) fails to activate the ATR
pathway in wild-type Vero cells. The findings suggest that the ATR pathway is activated
by HSV-1 E gene expression and that both ATR and Chk1 kinase activities play an
important role in promoting viral DNA synthesis.
With these findings in mind, we tested multiple, chemically distinct inhibitors of ATR
and Chk1 for antiviral effects. All tested ATR inhibitors and Chk1 inhibitors showed
significant antiviral activity, while inhibitors of ATM, Chk2, or DNA-PK did not. The
antiviral effects of the most potent of these ATR and Chk1 inhibitors reduce infectious
viral progeny greater than 10-fold, thus approaching the antiviral activity of the
eukaryotic and herpesvirus polymerase inhibitor aphidicolin. Furthermore, we found
that HSV-1 replication is also impaired in Seckel patient fibroblasts that are
hypomor-phic for ATR. Together these findings demonstrate that HSV-1 activates ATR and Chk1
Mock
A HU 17+ 4 h.p.i.
pChk1
17+ 4 h.p.i.
pATR
B
ICP4
pChk1S345
Lamin A/C
Tubulin
Nuclear Cytoplasmic M 6 h.p.i. M 6 h.p.i.
ICP0
ICP8
FIG 7Activated Chk1 and ATR are found in the nucleus and also in the cytoplasm where they sometimes colocalize with ICP4. (A) Merged images of DAPI (blue), pChk1 or pATR (red), and lamin A/C (green)-labeled unextracted U2OS cells. Activated Chk1 is found within the nucleus in hydroxyurea (HU)-treated cells. Cells infected with 17syn⫹show pChk1 and pATR distribution as defined by colocalization with lamin A/C. Arrows point to pATR or pChk1 outside the nucleus. Scale bars⫽10m. (B) Western blots of fractionated samples for the indicated proteins. Shown are two separately prepared but identical fractionations conducted at 6 h.p.i. with HSV-1 17syn⫹(MOI⫽10).
on November 6, 2019 by guest
http://jvi.asm.org/
[image:10.585.133.278.72.314.2]during infection and that the activated kinases significantly contribute to HSV-1
repli-cation fitness.
The observation that pATR and pChk1 are found in the cytoplasm following HSV-1
infection was surprising; however, significant precedence exists for the cytoplasmic
localization and function of activated ATR and Chk1. For instance, ATR regulates
hundreds of downstream targets, including antiapoptotic mitochondrial elements, and
its distribution in the cytoplasm following DNA damage is well documented (45).
Likewise, Chk1 has nuclear export sequences and is mobilized to control cytoplasmic
targets following DNA damage (46). Precedence also exists for exploitation of these
DDR elements in the cytoplasm by viruses. Vaccinia virus, a poxvirus that replicates
exclusively in the cytoplasm, activates cytoplasmic ATR, ATM, and DNA-PK and utilizes
ATR and Chk1 to promote viral genome replication (47, 48). Interestingly, we show here
that activated ATR and Chk1 appear in the cytoplasm at early stages of HSV-1 infection,
where they associate with ICP4 and ICP0. This is the first report of the cytoplasmic
localization and association of activated ATR and Chk1 with ICP4 and ICP0 during HSV-1
infection. Cell fractionation, immunofluorescence colocalization, and PLA data all
sup-port this novel finding. We are at present unsure of what role this cytoplasmic
association plays in the early stages of HSV-1 infection. The cytoplasmic localization of
20 40 60 80 100 120
E
v
en
ts / C
ell
pATR + ICP4pATR + ICP0pATR + ICP8
pChk1 + ICP4pChk1 + ICP0pCHK1 + ICP27
pATRpChk1ICP4 ICP0 ICP8ICP27
pATR + ICP4pATR + ICP0pATR + ICP8pChk1 + I CP4
pChk1 + ICP0
Mock ICP4 + pATR
ICP4 + pATR ICP4 + pChk1ICP4 + pChk1
ICP4 alone ICP4 alone ICP4 + pATR
ICP4 + pATR 17syn+
17syn+ 17syn+
Mock A
B
FIG 8Proximity ligation assay (PLA) demonstrates the close association of pATR and pChk1 with ICP0 and ICP4 at 3 h.p.i of U2OS cells infected with HSV-1 17syn⫹. (A) Representative micrographs of the indicated PLAs. Positive results were obtained with infected cells when both ICP4 and pATR or pChk1 antibodies were present (top micrographs). Note prominent cytoplasmic signals and also sporadic nuclear signals. Mock-infected cells or assays conducted with a single antibody gave negative results (bottom micrographs). Scale bars⫽10m. (B) Bar graph shows the number of positive amplification events calculated per cell for phospho-ATRS428or phospho-Chk1S345 interaction with the indicated proteins. No significant interactions were found in mock-infected cells, while infected cells showed significant pATR and pChk1 interactions with ICP0 and ICP4 but not with ICP8 or ICP27. PLA for individual proteins established background levels.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:11.585.98.315.74.400.2]both ICP4 and ICP0 is well documented (49, 50), as is the cooperative interaction among
ICP4, ICP0, and ICP27 to regulate viral gene expression (see, for example, reference 51
and references therein). We establish that both pATR and pChk1 interact specifically
with ICP0 and ICP4, but not ICP27 or ICP8, primarily in the cytoplasm. Although nuclear
interactions among these proteins are also detected, the events are far fewer than
detected in the cytoplasm. The significance of these interactions remains to be
deter-mined. One possibility is that the viral proteins act to sequester or redirect the kinase
activities away from cellular or DNA repair functions toward viral targets for promotion
of DNA replication.
The data presented here clearly demonstrate that ATR is activated to robustly
phosphorylate Chk1 during early stages of HSV-1 infection. ATR and Chk1 kinase
activities are both required for efficient HSV-1 replication. These data do not support
previous reports concluding that the ATR pathway is not activated during HSV-1
infection (21, 26), nor are they consistent with ATR being inactivated by the virus (27,
28). To the best of our knowledge, pATR or pChk1 localization by immunological
techniques using phospho-specific antibodies was not pursued in those prior studies.
Previous reports of ATR inactivation by HSV-1 also rested heavily upon the observation
that HSV-1 infection blocks phosphorylation of the nuclear ATR target RPA (28). Perhaps
one function of the association of ICP4 and ICP0 with activated ATR and Chk1 is to
redirect their kinase activity away from cellular targets to viral targets that promote viral
DNA replication.
Our studies reveal that HSV-1 activates the ATR DDR pathway to promote its life
0 20 40 60 80 100 120 140
DMSO VE-822 VE-821AZD6738 AZ 20
CHIR124 PF 477736 AT M-I
Chk2-I
DNA-PK-I APH
B
HSV
-1 C
opies (% c
on
tr
ol)
R
educ
tion HSV
-1 tit
er (% DMSO
)
0 20 40 60 80 100 120 140 160 180
DMSO VE-822 VE-821 AZD6738 AZ20 CHIR124 PF 477
736 ATM-I Chk2-I
DNA-PK-I APH
A
*
*
*
*
*
*
*
FIG 9Inhibition of either ATR or Chk1 kinase activity significantly inhibits HSV-1 replication and virus production. (A) Quantification of HSV-1 PFU following inhibitor treatment shows that all ATR and Chk1 inhibitors tested, and the control polymerase inhibitor aphidicolin, significantly impact virus production. Inhibitors of ATM, Chk2, or DNA-PK were not found to significantly inhibit virus production. Asterisks indicate statistical significance (Student’sttest):Pⱕ0.005 in all cases except for PF477736, for which the
Pvalue was 0.006. (B) qPCR measurements of HSV-1 genome copies show good agreement with the viral titer assays.
on November 6, 2019 by guest
http://jvi.asm.org/
[image:12.585.104.310.75.386.2]cycle and that inhibitors of this pathway are deleterious to viral replication. Multiple
ATR and Chk1 inhibitors show potent antiviral effects against HSV-1, exceeding 10-fold
reductions in infectious virus progeny, when used at concentrations that are reported
to significantly inhibit their target protein and spare closely related kinases such as ATM
and Chk2. These findings show that ATR and Chk1 kinase activities are utilized by HSV-1
to promote important aspects of the viral life cycle. While the mechanism by which ATR
pathway activation enhances HSV-1 infection is currently not clear, we hypothesize that
it is through downstream effects on viral and/or host processes. It will be useful to
identify those viral or host targets of ATR and Chk1 phosphorylation that are required
to promote viral replication. Importantly, the understanding that ATR and Chk1 are
activated by HSV-1 to promote viral replication will enable future studies designed to
understand this important process and to perhaps design new therapeutic approaches.
MATERIALS AND METHODS
Viruses and cell lines.Several HSV-1 viral strains were used in this study: 17syn⫹, KOS/M, McIntyre and McKrae. KD6, a replication-defective, ICP4⫺KOS/M mutant virus, was employed along with the permissive Vero cell line E5, which expresses ICP4 (39). Low-passage stocks of these viruses were obtained from J. Stevens at UCLA. U2OS cells were obtained from the ATCC and maintained in McCoy’s 5A medium (Corning, catalog no. 10-050-CV) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Atlanta Biological) and 1⫻penicillin-streptomycin-glutamine (PSG; Gibco; catalog no. 10378). Vero and human fetal lung (HFL) cells were obtained from the ATCC and maintained in Dulbecco modified Eagle medium (DMEM; Corning; catalog no. 10-013-CV) with 10% FBS and 1⫻PSG. Rabbit skin cells (RSC), obtained from B. Roizman (52), were maintained in minimum essential medium (MEM; Gibco; catalog no. 11700077) plus 5% heat-inactivated bovine serum and 1⫻PSG. Seckel patient (ATR⫺) (38) and normal patient (1BR3) fibroblast lines immortalized with hTert (kindly provided by Peggy Jeggo, University of Sussex) were maintained in Eagle’s MEM supplemented with 10% FBS and 1⫻PSG using a 1:2 split ratio. Seckel/ATR⫺or 1BR3 cells were plated in 24-well dishes at a density of 1.5⫻105per well and the following day infected with 17syn⫹at an MOI of 0.01. Following incubation for the desired periods, cells were harvested and DNA was isolated using DNAzol by following the manufacturer’s recommendation. Twenty nanograms of total DNA was analyzed for HSV-1 genomes by qPCR (see below). Lund human mesencephalic (LUHMES) neuronal cells were purchased from the ATCC (catalog no. CRL-2927) and cultured on poly-L-ornithine hydrobromide (Sigma; catalog no. P3655)-plus-fibronectin (Sigma; catalog no. F2006)-coated flasks, plates, and coverslips in proliferation medium (DMEM–F-12 [ATCC; catalog no. 30-2006], 1⫻N-2 supplement [Thermo Fisher Scientific; catalog no. 17502048], 40 ng/ml of recombinant human FGF-basic [Peprotech; catalog no. 100-18B-100UG]) as described previously (53). Experiments were conducted following at least 5 days of differentiation in proliferation medium supplemented with tetracycline hydrochloride at 1g/ml (Sigma; catalog no. T7660), N6,2=-O -dibutyryladenosine 3=,5=-cyclic monophosphate sodium salt at a final concentration of 1 mM (Sigma; catalog no. D0627), and recombinant human glial cell line-derived neurotrophic factor (GDNF) at 2 ng/ml (R&D Systems; catalog no. 212-GD-010) (53).
Inhibitors. The following ATR inhibitors were purchased from Selleckchem and used at a final concentration of 10M: VE-822 (catalog no. S7102; batch no. 1), VE-821 (catalog no. S8007), AZD6738 (catalog no. S7693), and AZ20 (catalog no. S7050). Chk1 inhibitors were purchased from Axon Medchem and used at a final concentration of 250 nM (CHIR 124 [catalog no. Axon 1636] and PF 477736 [catalog no. Axon 1379]). Other chemical inhibitors and their final concentrations described in this study are as follows: Chk2 inhibitor II hydrate, 5M (Sigma; catalog no. C3742); ATM inhibitor KU55933, 10M (Tocris; catalog no. 3544); DNA-PK inhibitor NU7441, 1M (Tocris; catalog no. 3712); hydroxyurea, 3 mM (Sigma; catalog no. H8627); and aphidicolin, 4M (Sigma; catalog no. A0781).
Antibodies. The following antibodies and dilutions were used in this study: phospho-Chk1S345 (pChk1S345; 133D3) rabbit monoclonal antibody (MAb) (Cell Signaling; catalog no. 2348; 1:1,000 in Western blotting [WB]), phospho-Chk1S345rabbit polyclonal antibody (Thermo Fisher Scientific; catalog no. PA5-34625; 1:2,000 in immunofluorescence [IF]), Chk1 (2G1D5) mouse MAb (Cell Signaling; catalog no. 2360; 1:1,000 in WB), anti-HSV-1 ICP4 hybridoma (1:100 in IF and 1:500 in WB), tubulin (␣plus) mouse MAb (Abcam; catalog no. ab44928; 1:10,000 in WB), anti-HSV-1 ICP8 mouse MAb 11E2 (Abcam; catalog no. ab20194; 1:200 in IF and 1:10,000 in WB), anti-HSV-1 ICP0 mouse MAb 5H7 (Abcam; catalog no. ab6513; 1:10,000 in IF and 1:5,000 in WB), phospho-ATRS428rabbit polyclonal Ab (Cell Signaling; catalog no. 2853; 1:1,000 in WB), phospho-ATRS428rabbit polyclonal Ab (Thermo Fisher Scientific; catalog no. 720107; 1:500 in IF), ATR(H300) rabbit polyclonal Ab (Santa Cruz; catalog no. sc-28901; 1:1,000 in WB), phospho-ATMS1981rabbit MAb EP1890Y (Abcam; catalog no. ab81292; 1:5,000 in WB), ATM mouse MAb 2C1(1A1) (Abcam; catalog no. ab78; 1:2,000 in WB), phospho-(S/TQ) ATM/ATR substrate (4F7) rabbit MAb (Cell Signaling; catalog no. 2909; 1:50 in IF); lamin A/C (4C11) mouse MAb (Cell Signaling; catalog no. 4777; 1:200 in IF), and anti-neurofilament 200 mouse MAb (Sigma; catalog no. N0142; 1:2,000 in IF). Secondary antibodies included Alexa Fluor 594 goat anti-rabbit (Thermo Fisher Scientific; catalog no. A11072) and Alexa Fluor 488 goat anti-mouse (Thermo Fisher Scientific; catalog no. A11029).
Antiviral assays, HSV-1 titer determination, and qPCR.Cells were plated at 5⫻105per well in 6-well plates and the following morning were incubated with inhibitors. Following 1 h of preincubation with inhibitors, cells were infected with HSV-1 at an MOI of 0.1 in a volume of 200l for 1 h, viral
on November 6, 2019 by guest
http://jvi.asm.org/
inoculum was removed, and cells were overlaid with medium containing fresh inhibitors and cultured for 48 h. After 48 h, cells and supernatant were harvested by scraping, separated into two Eppendorf tubes, and centrifuged at 10,000⫻gfor 40 min. The cell pellet from one tube was used for extraction of DNA (DNazol, according to the manufacturer’s recommendation; Thermo Fisher Scientific; catalog no. 10503027), the second pellet was resuspended in 200l of serum-free MEM and freeze-thawed 3 times, and virus titers were determined on RSC. For HSV-1 titer determination, RSC were plated at 150,000/well of 24-well plates and the following day infected with 10-fold serial dilutions of virus harvested from antiviral experiments. Cells were incubated for 48 h and stained with 1% crystal violet solution, and plaques were counted from three separate wells for each experimental sample. qPCR was conducted using 20 ng of input DNA, TaqMan Fast Universal PCR 2⫻master mix (Applied Biosystems; catalog no. 4352041) along with TaqMan probes and target-specific primers (Applied Biosystems; Assays by Design part no. 4331348) against HSV-1 polymerase. Samples were run on a StepOnePlus real-time PCR system (Applied Biosystems) using fast cycling: denaturation at 95°C for 20 s followed by 40 cycles at 95°C for 1 s and 60°C for 20 s.
Additional experimental compounds were also tested as follows. (i) Cycloheximide (Sigma; catalog no. C7698) or DMSO vehicle was added to the overlay media at 50g/ml and the infection allowed to proceed for 5 h. (ii) For HSV-1 replication inhibition, phosphonoacetic acid (PAA) was added at the time of virus adsorption and included in overlay medium at 400g/ml for 5 h. (iii) A final concentration of 3 mM hydroxyurea was added at the time of cell plating and incubated for 18 h. (iv) Colchicine (Sigma; catalog no. C9754) was added to overlay medium at 10M following 1 h of virus adsorption.
Western blotting.A total of 5⫻105cells were plated on 6-well plates and infected the following day with 17syn⫹(MOI⫽10). At the desired times, cells were lysed in buffer containing 1% NP-40, 150 mM sodium chloride, 20 mM Tris-HCl (pH 8.0), 2 mM EDTA, and 5% glycerol supplemented with phosphatase and protease inhibitor cocktails (PhosSTOP tablets; catalog no. 04906837001 [Roche] or Halt protease inhibitor cocktail; catalog no. 1862209; Thermo Fisher Scientific). Subcellular fractionation (SF) Western blots were processed as follows: U2OS cells were plated and infected as described above, washed twice with cold PBS, lysed, and scraped on ice in SF buffer (250 mM sucrose, 20 mM HEPES [pH 7.4], 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, and 1 mM EGTA, plus 1 mM dithiothreitol [DTT] and protease/phosphatase inhibitors), transferred to 1.5-ml Eppendorf tubes, and agitated for 30 min at 4°C. The lysates were then centrifuged at 720⫻gat 4°C for 5 min, and the supernatant (cytoplasmic fraction) was transferred to a new Eppendorf tube. The pellet (nuclear fraction) was washed with SF buffer, centrifuged at 720⫻gfor 10 min at 4°C, and resuspended in nuclear lysis (NL) buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS, plus 10% glycerol and protease/phosphatase inhibitors). Twenty microliters of each fraction was prepared for SDS-PAGE. For SDS-PAGE, 20g of protein was mixed with NuPAGE 4⫻LDS sample buffer (Thermo Fisher Scientific; catalog no. NP0007) along with NuPAGE sample reducing agent (Thermo Fisher Scientific; catalog no. NP0004) and run on 4 to 12% bis-Tris NuPAGE gels (Thermo Fisher Scientific) with NuPAGE morpho-linepropanesulfonic acid (MOPS)-SDS running buffer (Thermo Fisher Scientific; catalog no. NP000102). Gels were transferred onto a 0.22-m polyvinylidene difluoride (PVDF) membrane in Towbin buffer (0.2 M glycine, 25 mM Tris base) containing 20% methanol (MeOH) for 2 h under a constant voltage (30 V); the membrane was blocked with 5% nonfat dry milk (NFDM)-TBST (20 mM Tris-HCl [pH 7.4],150 mM NaCl, 0.1% Tween 20) and probed with antibodies diluted in 1% NFDM-TBST overnight at 4°C. For ATR and ATM Western blots, samples were processed as described above, except that DTT (50 mM; Cell Signaling; catalog no. 7016) was used as the reducing agent and samples were run on NuPAGE Novex 3 to 8% Tris-acetate gels (Thermo Fisher Scientific) with NuPAGE Tris-acetate SDS running buffer (Thermo Fisher Scientific; catalog no. LA0041). Gels were transferred under a constant current (7.5 mA/cm2) for 24 h at 4°C with Towbin buffer containing 0.1% SDS for ATR blotting or 0.05% SDS plus 20% MeOH for ATM blotting onto PVDF membranes (0.45m); the membranes were then allowed to dry at room temper-ature (RT), reactivated with MeOH, blocked in 5% NFDM-TBST containing phosphatase inhibitors (Roche; PhoSTOP tablets), and incubated overnight at 4°C with the specified antibody. Secondary antibody detection was performed by incubating blots with Pierce goat anti-rabbit poly-horseradish peroxidase (HRP) (Thermo Fisher Scientific; catalog no. 32260) or Pierce goat anti-mouse poly-HRP (Thermo Fisher Scientific; catalog no. 32230) at 1:5,000 diluted in 5% NFDM-TBST. Blots were developed with chemilu-minescent substrate (Pierce ECL Western blotting substrate; catalog no. 32209) and imaged with a GE ImageQuant LAS4000 instrument. For reprobing, membranes were stripped with 6 M guanidine hydro-chloride, 0.2% NP-40, 0.1 M-mercaptoethanol, and 20 mM Tris-HCl (pH 7.4) (twice for 5 min at RT, followed by 4 3-min washes with TBST).
Immunofluorescence and microscopy.Cells were plated onto coverslips at 75,000 per well of 24-well plates and the following day were infected at an MOI of 10 for the desired times. Initial immunofluorescence experiments with paraformaldehyde-fixed cells found that rabbit phospho-specific antibodies bound nonspecifically to a juxtanuclear domain associated with the microtubule-organizing center (MTOC) (see Results). Thereafter, cells were processed for immunofluorescence in one of two ways that incorporated an Fc receptor blocking step. (i) Cells were fractionated in situ by preextraction following previously published procedures; briefly, cells were incubated in cytoskeleton buffer (CB; 100 mM PIPES [pH 6.8], 0.5% Triton X-100, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 1 mM EGTA) for 5 min on ice, followed by incubation in cytoskeleton stripping buffer (CSK; 10 mM Tri-HCl [pH 7.4], 10 mM NaCl, 3 mM MgCl2, 1% Tween 40, 1% Tween 20, 0.5% sodium deoxycholate) for 5 min on ice. Cells were then washed 3 times with PBS and fixed with Streck fixative (150 mM 2-bromo-2-nitro-1,3-propanediol, 108 mM diazolidinyl urea, 10 mM sodium citrate, 50 mM EDTA) for 30 min at RT, washed with PBS, and equilibrated with 0.5% Triton X-100 in 100 mM Tris-HCl (pH 7.4) plus 50 mM EDTA for 15 min at RT. Cells
on November 6, 2019 by guest
http://jvi.asm.org/
were then incubated sequentially with Fc receptor blocker (Innovex Biosciences, catalog no. NB309) for 1 h at RT followed by blocking buffer (5% goat serum, 0.1% NP-40, 20 mM Tris-HCl [pH 7.4], 150 mM NaCl) for 30 min at RT. (ii) In some cases, cells were fixed in 3.7% paraformaldehyde in PBS for 5 min at RT and permeabilized with Triton buffer as described above, followed by blocking with Fc receptor blocker and blocking buffer as above. Cells were incubated with primary antibodies at the dilutions indicated for 1 h at RT, followed by incubation with a secondary antibody at RT for 1 h. Antibody dilution buffer and wash buffer consisted of 20 mM Tris-HCl (pH 7.4) buffer containing 150 mM NaCl and 0.1% NP-40. Coverslips were mounted onto slides with ProLong Diamond antifade mountant with 4= ,6-diamidino-2-phenylindole (DAPI) (Thermo Fisher Scientific; catalog no. P36962). All immunofluorescence was con-ducted using a Nikon Eclipse E600 microscope with epifluorescence and photographed with a Qimaging Exi Aqua monochrome digital camera.
PLA.Experiments were conducted utilizing the Duolink In Situ Orange kit (mouse/rabbit; Sigma; catalog no. DUO92102) according to the manufacturer’s recommendations, with some modifications. U2OS cells were plated onto coverslips at 100,000 per well in 24-well plates and incubated overnight in a 37°C humidified incubator. The following day, cells were infected with 17syn⫹at an MOI of 10 as follows: virus was adsorbed, cells were washed with PBS after 1 h, and the infection allowed to proceed for 3 h postadsorption. Cells were fixed in 3.7% paraformaldehyde in PBS for 5 min at RT and processed as described previously for immunofluorescence. Following permeabilization and blocking, cells were incubated with primary antibodies (same antibodies and dilutions as described for IF) for 1 h at RT, washed 3 times for 5 min with PBS containing 1% bovine serum albumin (BSA) and 0.1% NP-40 (WDB), and incubated with the diluted proximity ligation assay (PLA) probes (diluted in WDB) per the manu-facturer’s recommendation for 1 h at 37°C. Cells were washed twice for 5 min with wash buffer A (from kit), followed by incubation with the ligation reaction mix for 30 min at 37°C. Cells were washed twice for 2 min with wash buffer A, and amplification was performed by incubating cells with polymerase reaction mix for 100 min at 37°C. Cells were washed twice for 10 min with wash buffer B (from kit) and once for 10 min with 0.01⫻wash buffer B. Coverslips were mounted onto slides with ProLong Gold mounting medium containing DAPI. PLA analysis of amplification events was carried out with the aid of Nikon’s NIS-Elements basic research (BR3.2) software interfaced with a Nikon Eclipse E600 microscope using edge detection and object count dialog options.
ACKNOWLEDGMENTS
This work was supported in part by NIH grants R42 AI062182 and R42 AI068159 (for
reagents from NanoVir) and R01 AI097376.
REFERENCES
1. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601.InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed, vol 2. Lippincott Williams & Wilkins, Philadelphia, PA.
2. Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465– 481.https://doi.org/10 .1099/vir.0.048900-0.
3. Weller SK, Coen DM. 2012. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol 4(9):a013011.https://doi.org/ 10.1101/cshperspect.a013011.
4. McNamee EE, Taylor TJ, Knipe DM. 2000. A dominant-negative herpes-virus protein inhibits intranuclear targeting of viral proteins: effects on DNA replication and late gene expression. J Virol 74:10122–10131.
https://doi.org/10.1128/JVI.74.21.10122-10131.2000.
5. de Bruyn Kops A, Knipe DM. 1988. Formation of DNA replication struc-tures in herpes virus-infected cells requires a viral DNA binding protein. Cell 55:857– 868.https://doi.org/10.1016/0092-8674(88)90141-9. 6. Knipe DM, Senechek D, Rice SA, Smith JL. 1987. Stages in the nuclear
association of the herpes-simplex virus transcriptional activator protein ICP4. J Virol 61:276 –284.
7. Quinlan MP, Chen LB, Knipe DM. 1984. The intranuclear location of a herpes-simplex virus DNA-binding protein is determined by the status of viral DNA replication. Cell 36:857– 868. https://doi.org/10.1016/0092 -8674(84)90035-7.
8. Weitzman MD, Lilley CE, Chaurushiya MS. 2010. Genomes in conflict: maintaining genome integrity during virus infection. Annu Rev Microbiol 64:61– 81.https://doi.org/10.1146/annurev.micro.112408.134016. 9. Turnell AS, Grand RJ. 2012. DNA viruses and the cellular DNA-damage
response. J Gen Virol 93:2076 –2097. https://doi.org/10.1099/vir.0 .044412-0.
10. Luftig M. 2014. Viruses and the DNA damage response: activation and antagonism. Annu Rev Virol 1:20. https://doi.org/10.1146/annurev -virology-031413-085548.
11. Harper JW, Elledge SJ. 2007. The DNA damage response: ten years after. Mol Cell 28:739 –745.https://doi.org/10.1016/j.molcel.2007.11.015. 12. You Z, Shi LZ, Zhu Q, Wu P, Zhang Y-W, Basilio A, Tonnu N, Verma IM,
Berns MW, Hunter T. 2009. CTiP links DNA double-strand break sensing to resection. Mol Cell 36:954 –969.https://doi.org/10.1016/j.molcel.2009 .12.002.
13. Tomimatsu N, Mukherjee B, Deland K, Kurimasa A, Bolderson E, Khanna KK, Burma S. 2012. Exo1 plays a major role in DNA end resection in humans and influences double-strand break repair and damage signal-ing decisions. DNA Repair (Amst) 11:441– 448.https://doi.org/10.1016/j .dnarep.2012.01.006.
14. Peng G, Dai H, Zhang W, Hsieh H-J, Pan M-R, Park Y-Y, Tsai RY-L, Bedrosian I, Lee J-S, Ira G, Lin S-Y. 2012. Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res 72:2802–2813.https://doi.org/10.1158/0008-5472.CAN-11-3152. 15. Brown EJ, Baltimore D. 2003. Essential and dispensable roles of ATR in
cell cycle arrest and genome maintenance. Genes Dev 17:615– 628.
https://doi.org/10.1101/gad.1067403.
16. Stracker TH, Usui T, Petrini JH. 2009. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst) 8:1047–1054.https://doi.org/10 .1016/j.dnarep.2009.04.012.
17. Smith S, Weller SK. 2015. HSV-I and the cellular DNA damage response. Future Virol 10:383–397.https://doi.org/10.2217/fvl.15.18.
18. Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. 1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J Virol 70:7471–7477.
19. Parkinson J, Lees-Miller SP, Everett RD. 1999. Herpes simplex virus type 1 immediate-early protein Vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J Virol 73:650 – 657.
20. Lilley CE, Carson CT, Muotri AR, Gage FH, Weitzman MD. 2005. DNA repair
on November 6, 2019 by guest
http://jvi.asm.org/
proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102:5844 –5849.https://doi.org/10.1073/pnas.0501916102. 21. Shirata N, Kudoh A, Daikoku T, Tatsumi Y, Fujita M, Kiyono T, Sugaya Y,
Isomura H, Ishizaki K, Tsurumi T. 2005. Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J Biol Chem 280:30336 –30341.https://doi.org/ 10.1074/jbc.M500976200.
22. Wilkinson DE, Weller SK. 2004. Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replica-tion is dependent on the composireplica-tion of viral proteins within prerepli-cative sites and correlates with the induction of the DNA damage response. J Virol 78:4783– 4796. https://doi.org/10.1128/JVI.78.9.4783 -4796.2004.
23. Lilley CE, Chaurushiya MS, Boutell C, Landry S, Suh J, Panier S, Everett RD, Stewart GS, Durocher D, Weitzman MD. 2010. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J 29:943–955.https://doi.org/10.1038/emboj.2009.400. 24. Lo Piano A, Martinez-Jimenez MI, Zecchi L, Ayora S. 2011. Recombination-dependent concatemeric viral DNA replication. Virus Res 160:1–14.https:// doi.org/10.1016/j.virusres.2011.06.009.
25. Mohni KN, Dee AR, Smith S, Schumacher AJ, Weller SK. 2013. Efficient herpes simplex virus 1 replication requires cellular ATR pathway pro-teins. J Virol 87:531–542.https://doi.org/10.1128/JVI.02504-12. 26. Wilkinson DE, Weller SK. 2006. Herpes simplex virus type I disrupts the
ATR-dependent DNA-damage response during lytic infection. J Cell Sci 119:2695–2703.https://doi.org/10.1242/jcs.02981.
27. Mohni KN, Smith S, Dee AR, Schumacher AJ, Weller SK. 2013. Herpes simplex virus type 1 single strand DNA binding protein and helicase/ primase complex disable cellular ATR signaling. PLoS Pathog 9(10): e1003652.https://doi.org/10.1371/journal.ppat.1003652.
28. Mohni KN, Livingston CM, Cortez D, Weller SK. 2010. ATR and ATRIP are recruited to herpes simplex virus type 1 replication compartments even though ATR signaling is disabled. J Virol 84:12152–12164.https://doi .org/10.1128/JVI.01643-10.
29. Botting C, Lu X, Triezenberg SJ. 2016. H2AX phosphorylation and DNA damage kinase activity are dispensable for herpes simplex virus replica-tion. Virol J 13:15.https://doi.org/10.1186/s12985-016-0470-1. 30. Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. 2013. Human
papillomavirus episome stability is reduced by aphidicolin and con-trolled by DNA damage response pathways. J Virol 87:3979 –3989.
https://doi.org/10.1128/JVI.03473-12.
31. Liu S, Shiotani B, Lahiri M, Marechal A, Tse A, Leung CCY, Glover JNM, Yang XH, Zou L. 2011. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol Cell 43:192–202.https://doi.org/10.1016/ j.molcel.2011.06.019.
32. Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:4.https://doi.org/10.1371/journal .ppat.1002878.
33. Murayama T, Natsuumesakai S, Shimokawa K, Furukawa T. 1986. Fc receptor(s) induced by human cytomegalovirus bind differentially with human immunoglobulin G subclasses. J Gen Virol 67:1475–1478.https:// doi.org/10.1099/0022-1317-67-7-1475.
34. Hanke T, Graham FL, Lulitanond V, Johnson DC. 1990. Herpes simplex virus IgG Fc receptors induced using recombinant adenovirus vectors expressing glycoprotein-E and glycoprotein I. Virology 177:437– 444. 35. Sprague ER, Martin WL, Bjorkman PJ. 2004. pH dependence and
stoichi-ometry of binding to the Fc region of IgG by the herpes simplex virus Fc receptor gE-gI. J Biol Chem 279:14184 –14193.https://doi.org/10.1074/ jbc.M313281200.
36. Mirzoeva OK, Petrini JHJ. 2003. DNA replication-dependent nuclear dy-namics of the Mre11 complex. Mol Cancer Res 1:207–218.
37. Taylor TJ, McNamee EE, Day C, Knipe DM. 2003. Herpes simplex virus replication compartments can form by coalescence of smaller compart-ments. Virology 309:232–247. https://doi.org/10.1016/S0042-6822(03) 00107-7.
38. O’Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. 2003. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33:497–501.
https://doi.org/10.1038/ng1129.
39. Dobson AT, Margolis TP, Sedarati F, Stevens JG, Feldman LT. 1990. A latent, nonpathogenic HSV-1-derived vector stably expresses beta-galactosidase in mouse neurons. Neuron 5:353–360.https://doi.org/10 .1016/0896-6273(90)90171-B.
40. Larsson A, Wraak M, Oberg B. 1983. Effect of aphidicolin on DNA synthesis in HSV-1 infected and uninfected Vero cells. Antiviral Res 3:87–91.https://doi.org/10.1016/0166-3542(83)90029-3.
41. Abraham RT. 2004. PI 3-kinase related kinases: ‘big’ players in stress-induced signaling pathways. DNA Repair (Amst) 3:883– 887.https://doi .org/10.1016/j.dnarep.2004.04.002.
42. Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ. 2000. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 14:1448 –1459.https://doi .org/10.1101/gad.840500.
43. Zhao H, Piwnica-Worms H. 2001. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21:4129 – 4139.https://doi.org/10.1128/MCB.21.13.4129-4139.2001. 44. Boehmer PE, Lehman IR. 1997. Herpes simplex virus DNA replication. Annu
Rev Biochem 66:347–384. https://doi.org/10.1146/annurev.biochem.66.1 .347.
45. Hilton BA, Li ZK, Musich PR, Wang H, Cartwright BM, Serrano M, Zhou XZ, Lu KP, Zou Y. 2015. ATR plays a direct antiapoptotic role at mitochondria, which is regulated by prolyl isomerase Pin1. Mol Cell 60:35– 46.https:// doi.org/10.1016/j.molcel.2015.08.008.
46. Wang JN, Han XZ, Feng XJ, Wang ZH, Zhang YW. 2012. Coupling cellular localization and function of checkpoint kinase 1 (Chk1) in checkpoints and cell viability. J Biol Chem 287:25501–25509.https://doi.org/10.1074/ jbc.M112.350397.
47. Ferguson BJ, Mansur DS, Peters NE, Ren H, Smith GL. 2012. DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1:e00047. 48. Postigo A, Ramsden AE, Howell M, Way M. 2017. Cytoplasmic ATR
activation promotes vaccinia virus genome replication. Cell Rep 19: 1022–1032.https://doi.org/10.1016/j.celrep.2017.04.025.
49. Zhu ZM, Schaffer PA. 1995. Intracellular localization of the herpes sim-plex virus type 1 major transcriptional regulatory protein, ICP4, is af-fected by ICP27. J Virol 69:49 –59.
50. Kawaguchi Y, VanSant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle reg-ulator cyclin D3. J Virol 71:7328 –7336.
51. Zhu ZM, Cai WZ, Schaffer PA. 1994. Cooperativity among herpes-simplex virus type-1 immediate-early regulatory proteins: ICP4 and ICP27 affect the intracellular localization of ICP0. J Virol 68:3027–3040.
52. Tran RK, Lieu PT, Aguilar S, Wagner EK, Bloom DC. 2002. Altering the expression kinetics of VP5 results in altered virulence and pathogenesis of herpes simplex virus type 1 in mice. J Virol 76:2199 –2205.https://doi .org/10.1128/jvi.76.5.2199-2205.2002.
53. Scholz D, Poltl D, Genewsky A, Weng M, Waldmann T, Schildknecht S, Leist M. 2011. Rapid, complete and large-scale generation of post-mitotic neurons from the human LUHMES cell line. J Neurochem 119: 957–971.https://doi.org/10.1111/j.1471-4159.2011.07255.x.
on November 6, 2019 by guest
http://jvi.asm.org/