0022-538X/82/120893-07$02.00/0
Copyright01982, American Society forMicrobiology
Identification and
Characterization of a DNase Induced by
Epstein-Barr
Virus
RENG-SENGTAN,1 ALOK K.DA1TA,2ANDYUNG-CHI CHENGl*
DepartmentsofPharmacology'andBiochemistry2 and Cancer Research Center, UniversityofNorth Carolina, ChapelHill,North Carolina27514
Received 24 March 1982/Accepted 16 August 1982
The diterpene ester promoter of mouse skin tumors, 12-0-tetradecanoyl-phorbol-13-acetate, induced a DNaseactivityin theEpstein-Barr virus-producer
cell line P3HR-1. The elution patterns of the enzyme from DEAE-cellulose, phosphocellulose, andDNA-cellulosecolumns were different from virus-associat-ed DNA polymerase activity.Thepartiallypurified activity couldbe neutralized to the extentof90%obyseraof patients with nasopharyngealcarcinoma. Purified immunoglobulin G from sera ofnasopharyngeal carcinomapatientsinhibited this enzyme and thatobtainedfromsuperinfected Rajicells to the same extent. The
partially purified enzymepreferred native DNA as a substrate over denatured
DNA and 3'-terminally labeled activated calfthymus DNA. The activity was
inhibited by high ionic strength. Phosphonoformic acid did not have any effect on this enzyme activity.
Virus alkaline DNase and DNA polymerase
are induced in various mammalian cells after
infection with herpes groupviruses(3-5, 9, 10,
13-17, 20,21,26, 27).Epstein-Barrvirus(EBV),
one of the herpes group viruses, is closely
associated with nasopharyngeal carcinoma
(NPC). It has been demonstrated that
EBV-specific DNase activity can be induced from
superinfected Raji cells orfrom
iododeoxyuri-dine-treated D98/HR-1 somatic cell hybrids (2, 8). The enzyme can be neutralized by sera of
patients with NPC, but not by sera of most
normal, healthy individuals (1). The frequency
and levels ofantibody against EBV-associated DNase in sera ofpatients with NPC is much
higherthan those in seraofpatients with other
tumors. Itthus appears to bepromising to use
this criterion for aiding the early diagnosis of
NPC in NPChigh-risk populationsandfor
moni-toringtheprognosis ofpatientsbeingtreatedfor
NPC.However,amajor problemlies in the fact
that EBV-associated DNase obtained from
su-perinfected Raji cells isunstable, and the
culti-vation of virus and cells isrelatively expensive
and technically difficult. Thus, it would be
ad-vantageous to find another source of
EBV-specificDNase.
Zur Hausen et al. (29, 30) reported a very efficient induction of EBV in the virus-produc-ing cell line P3HR-1 by the tumor-promotvirus-produc-ing agent 12-O-tetradecanoyl-phorbol-13-acetate
(TPA). The sixfold increase in EBV genome
copiesper P3HR-1 cellparalleled the increase in
thepercentage ofcellssynthesizing viral capsid
antigen (VCA) (18). Datta et al. (5) took advan-tageof this effect and foundarapid induction of
EBV-associated DNA polymerase activity in
P3HR-1 cells after treatment with TPA. The presence ofEBV-associated DNase in P3HR-1 cells has beenreported byClough(3,4),andits presence in superinfected Raji cells has been found by workers in this laboratory (2). Al-though the partial purification of this enzyme from P3HR-1 cells has been reported (3), there is no evidence that the enzyme studied is indeed the same as that found in superinfected Raji cells. This paper reportsonthecharacterization
of DNase activity from TPA-induced P3HR-1 cells. The immunoglobulin G (IgG) fraction of sera taken from NPC patients could neutralize
theactivityof thispartially purifiedenzyme to a
similarextent asthatof the enzyme from super-infected cells.
MATERIALS AND METHODS
Cells. The Burkitt lymphoma-derived cell line P3HR-1 wasmaintained between 5 x 105to 1 x 106
cells per ml in RPMI 1640 medium containing 10% heat-inactivated fetal calf serum and supplemented with 100 IU ofpenicillinand 100F.gstreptomycin per ml(5). The culture andpreparationof cellularextracts ofsuperinfected RajicellsweredescribedbyChenget al. (2).
Inductionwith TPA. P3HR-1 cells were seededat
106 cells per ml in culture medium. After 24 h of cultivationat37°C,TPAwasaddedatafinal concen-tration of 30ng/ml.The culturewasincubatedat37°C for the indicatedperiodsof time. The determination of EBVearly antigen (EA)-VCA-positive cellswas car-893
on November 10, 2019 by guest
http://jvi.asm.org/
ried out on cell smears by the indirect immunofluores-cence test as described by Henle and Henle (12).,
Chemicals. TPA, calf thymus DNA, bovine serum albumin,1-mercaptoethanol(m-ME), and phenylmeth-ylsulfonyl fluoride (PMSF) were obtained from Sigma Chemical Co. Phosphocellulose and DEAE-cellulose were purchased from Whatman Inc. DNA-cellulose was prepared by the method of Potuzak and Winters-burger (24).
DNA substrates.Native double-stranded Escherich-ia coli [14C]DNA was prepared from the thymine-requiring E. coli strain ATCC 23851. The bacteria was grown in minimal medium containing 5 ,umol of unla-beledthymineand 125 ,Ciof[14C]thymine perliter. The [14C]DNA was isolated by the procedure of Marmur(19). Heated denatured [14C]DNA was pre-pared as previouslydescribed (19).
3'-Terminally labeled activated calf thymus DNA was prepared as described previously (7).
Enzyme assays. Alkaline DNase was measured by the release ofacid-soluble nucleotides from double-stranded ['4C]DNA from E. coli by the method of Hoffman and Cheng (14). One unit of enzyme was definedasthat amountwhichcatalyzedthe conversion of1,ugof double-strandedDNA toacid-soluble nucle-otide in 10 min at370C.
When 3'-3H-terminally labeled activated calf
thy-mus DNA was used as a substrate, the reaction mixture of 0.2 ml contained 20 mM Tris (pH 8.0), 2 mM MgCl2, 1 mM dithiothreitol, 0.5 mg of bovine serumalbumin perml,0.0 to 0.3 Uof DNase, and 2.4 ,gof3'-[3H]-terminallylabeled activated calf thymus DNA.Thespecific activity ofDNAwas6x103 cpm/ ,ug.After incubation for 30min,the radioactive prod-uct wasdetermined bythe samemethod,asdescribed above.
Determination of anti-EBV DNaseactivitywas as-sayed asdescribed by Chengetal. (1, 2).
DNA polymerase was assayed as described by Weissbach et al. (28), except 150 mMKClwasadded insteadof ammonium sulfate.
Preparation ofIgG. Normal serum IgG and serum IgG from patients with NPC were prepared by the method ofChengetal.(1).
PurificationofEBV-associated DNase.Alloperations werecarried out at 2 to40Cunless otherwise indicated. Step 1. Preparation of crudeextract. P3HR-1 cells (4 x 109) treated with TPA for 72 h were washed, harvested, andsuspendedin 18 ml of Tris buffer(pH 7.5)containing2 mM1-MEand 10±gof PMSFper ml. The cell suspension was frozen and thawed three times. KCIwasaddedto afinal salt concentration of 0.4M.Thesuspensionwascentrifugedat12,000rpm for 10min, and thesupernatantwascollected (super-natant I). Twelve milliliters of the above extraction buffercontaining0.4 MKCIwasaddedtothe precipi-tate to extract theremainingenzyme, and the suspen-sionwassonicated withaBransonsonifier for1min in aseries of four 15-s burstsseparated by1-mincooling periods. The suspension was then centrifuged at 12,000 rpm for 10 min, and the supernatant was collected(supernatant II). Supernatants Iand IIwere combined to obtain thefinalextract.
Step 2.DEAE-celulose chromatography. The com-binedsupernatantwasdialyzed against1liter ofbuffer A(400mMpotassiumphosphate [pH 7.5] containing
20%oglycerol,5 mM1-ME,2mMMgC12,and 10,ug of
PMSFper ml)overnight.About 22 mlof the dialyzed extract was loaded on aDEAE-cellulose column (24 by 8 cm) which had beenpreequilibratedwith 300 ml of buffer A. Fractions of 3 ml were collected, and those containing DNase activity were pooled and dialyzed against 2liters of buffer B (20 mM potassium phosphate [pH7.51-20%'oglycerol-2mMMgCl2-5mM 1-ME-10,ugof PMSF per ml) for 18 h.
About 45 ml of the above-described dialyzed frac-tion from the first column was loaded on a second DEAE-cellulose column (2.4 by 11 cm) which had been preequilibrated with 500 ml of buffer B. The column was washedwith about 45 ml of buffer B, and a 150-mlgradientof column buffers B and A wasapplied tothecolumn. Fractions of 3 ml werecollected,and those containing DNase and DNA polymerase activity were pooled separately(Fig. 1A). Fractions containing DNaseand DNA polymerase activity were dialyzed separatelyagainst1liter of buffer C (20 mM potassium phosphate [pH8.01-20%glycerol-2mMMgCl,-5mM 1-ME-10,ugof PMSF perml)for 17 h.
Step 3. Phosphocellulose chromatography. Approxi-mately 21 ml ofDEAE-cellulose-purifiedDNase frac-tion(Fig.1A,fractionIII)wasloadedon a phospho-cellulose column (1.4 by 8 cm) preequilibrated with 120 ml of buffer C. After washing the column, the elution was carried out with buffer C containing a lineargradientofpotassium phosphate(pH 8.0)from 20 to 330 mM. Fractions of 2 ml were collected.
Step 4. DNA cellulose column chromatography. A pooledphosphocellulose-purifiedDNasefraction (8to 10ml; Fig. 1B,fractionIII) wasdialyzed against500 ml ofbufferCfor2 h. Thefraction was loaded on a DNA-cellulose column(1 by8 cm)equilibratedwith buffer C. After washing the column with 15 ml of buffer C, a50-ml gradientelution from 0 to 500 mM potassiumchloride in buffer C wascarriedout. Frac-tions of 1 ml were collected. The active fracFrac-tions were pooled (Fig. 1C, fraction III) and dialyzed against buffer C for 5 h. Thepreparation wasmade40%o(wt/ vol)in glycerol and stored at -70°C.
The DNA polymerase-associated DNase fractions (Fig. 1A, fraction II) were also purified separately (Fig. 1).
RESULTS
Induction of alkaline DNaseactivity afterTPA treatment. Figure 2A shows the kinetics of
in-duction of alkalineDNaseactivityin extractsof TPA-treated and untreated P3HR-1 cells. The induction started from day 1 oftreatment and wasmaintainedatthe elevated level.Partof this alkaline DNase activity in the crude extractof P3HR-1 cellscanbe neutralized by NPCserum.
Interestingly,thereisonlyalittle change in
non-neutralizable DNase activity. Under these
con-ditions, Raji cells, a nonproducer cell line, do
notshowanyincrease inalkaline DNase activi-ty.
With the increase in alkaline DNase activity therewasalsoanincrease in the numberof EA-VCA-positive cells (Fig. 2B). The maximum numberofEA-VCA-positive cells occurred after 3days oftreatment. Incontrast, both the
on November 10, 2019 by guest
http://jvi.asm.org/
I
to 4.
'E
a-z
0
-1
n
E
0, 0
a.
vt
E
to
m 0
10%
-E
[image:3.491.105.392.71.410.2]Volume (ml)
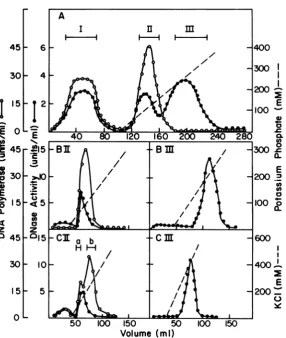
FIG. 1. Chromatography ofextractsfrom TPA-treated P3HR-1 cells. Detailsof theprocedures for DEAE cellulose (A),phosphocellulose(B),and DNA-cellulose(C)chromatographyaredescribedin thetext.
line DNase activity and the percentage of
EA-VCA-positive cells in nontreated cultures of
P3HR-1cells remainedthesamethroughout this course. Thus, theparallel increase in the alka-line DNase activityand EA-VCA-positive cells suggeststhattheenzymeinduced ismost proba-bly associated with virusreplication.
Identification ofvirus-associated DNase
activi-ty. About 35to45% of the total DNase activity
(Fig. 1A, fraction I) of crude extract was not
adsorbed to the DEAE-cellulose column. The unadsorbed fraction had a very high level of DNApolymerase activity; this nonadhering ac-tivity has been showntohave the characteristics of host,B-polymerase (28),suchas
N-ethylmalei-mide resistance and inhibition byhigh salt (data not shown). The adsorbed DNases are eluted intotwodistinctpeaks(Fig.1A, fractions II and
III). FractionIIDNase, whichwaselutedat100
mMpotassium phosphate, contained highlevels
of DNA polymerase activity. The fraction III
DNasewasboundmoretightlyand waselutedat
200to250mMpotassiumphosphate.No
detect-able DNApolymerase activity wasobserved in thepeak fractions offraction III.
When fractions II and III from the
DEAE-cellulosecolumnwererechromatographed
sepa-ratelyon aphosphocellulosecolumn, fractionII, which was eluted at 100 mM potassium phos-phate(Fig.1B,fractionII), hadboth DNase and DNApolymerase activities. However, fraction
III, which was eluted at 200 mM potassium phosphate(Fig. 1B, fraction III) didnotcontain anydetectableDNApolymerase.
Furtherrechromatography of fractionsIIand III (Fig. 1C) on the DNA-cellulose column re-sulted intheprofile shown in Fig. 1C, fractions IIandIII.Fraction CII, whichwaselutedat280 mM potassium chloride, still contained DNA polymeraseactivity,butinterestingly,peak frac-tionsof thesetwoenzymesdidnotcoincide and there was asmallreproducible shoulderon the DNApolymeraseactivity profile. Fraction CIII containedonlyDNaseactivityandwaselutedas one sharp peak at450mM potassium chloride. TheDNaseseluted infractions CII and CIII had
on November 10, 2019 by guest
http://jvi.asm.org/
lioo
A
E1
0
80
_
D60
(0-._
.' 40
_
0 30
-u
20_20
0
_
CLI0
,,20
_
>
1
2
3
4
5
Days after TPA addition
FIG. 2. Kinetics of induction of EBV-associated DNase(A) and VCA(B) inEBV-carrying lymphoblas-toid cells upon treatment with TPA. Cells grown in suspension(106cells per ml)weretreated with TPAat aconcentration of 30 ng/ml. On the indicated days, cells were processed as described in the text and assayed for DNase activity (A) and VCA (B). 0, Untreated P3HR-1 cells; 0, TPA-treated P3HR-1 cells; A, untreated Raji cells; O, TPA-treated Raji cells. Thenon-neutralizable DNase activity(U)from TPA-treated P3HR-1 cells is described asthe differ-encebetween total DNase andneutralized DNase. For theneutralization assay, theextract wastreatedwith NPCserumfor 20 min atroomtemperatureand then usedfor assay.
specificactivities of 61.5 and 480 U/mgof pro-tein, respectively.
Neutralizing activity of serum from patients withNPC on DNases. Reports from this labora-tory (1) have shown that DNase activity in
superinfectedRaji cells couldbeneutralizedby
seraof NPCpatients. About 40to45% ofDNase activity of crudeextractfrom P3HR-1 cells after TPA treatmentwasneutralizedby seraof NPC patients (more than 10 patient seratested, Fig. 3). After DNA-cellulose purification, almost 90% of the activity (fraction CIII) could be neutralized. In contrast, fractions Al and CII showedno neutralization withthe serum.
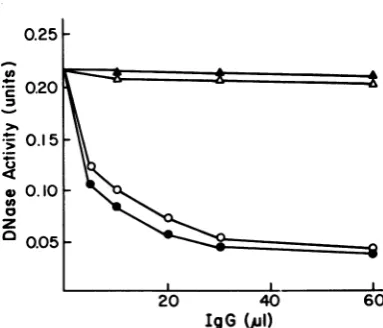
IdenticalresultswereobtainedwhenIgGwas
purified from theseraofpatientswith NPCand
.-.*
100
0
75
to
0
z
0
50
0
25
0
0-5
10
15
20
[image:4.491.54.453.64.396.2]NPC
Serum
(jil)
FIG. 3. Neutralizing activity of NPC serum on DNases.Themethodof neutralization of DNase activ-ity isdescribedin thelegend to Fig. 2. Native E. coli DNAwasusedasthesubstrate. DNasepreparations werefrom TPA-treated P3HR-1 cell crude extract(A), fraction I from DEAE cellulose (O), and DNA-cellu-lose-purified fractionII(0),and DNA-cellulose-puri-fiedfraction III(0).
putinto thesametest(Fig. 4). Fraction CIIIwas almost90%neutralized. Thisresultissimilarto those obtained with the extract from
superin-fected Raji cells, whereas IgG from normal
individualswas inactive.
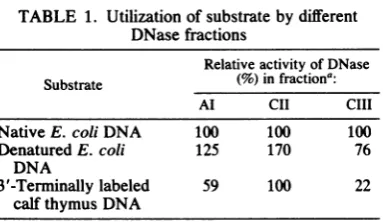
Utilization ofsubstrates.Allthree DNase
frac-tions could utilize native and denatured DNAs
fromE. coli and 3'-terminallylabeled activated DNAfrom calfthymus in differentdegrees
(Ta-ble 1). Quantitatively, DNases offractions Al
0.25
0
c 0.20
* 0.15 * 0.10
0
z aAoA
IgG
(Oil)
FIG. 4. Neutralizing activity of IgG on DNases. Preparations ofIgGand theneutralization ofDNases aredescribedin thetext.IgGfrom normalserumwas addedto the superinfected Rajicell extract(A) and DNA-cellulose-purified DNase of fraction III
(A).
IgG from sera of patients with NPC was added to the superinfected Raji cell extract (0) and theDNA-cellulose-purifiedDNaseof fraction III(0). UUZ)
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.491.55.259.70.368.2] [image:4.491.257.449.438.602.2]TABLE 1. Utilization of substrate by different DNasefractions
Relativeactivity of DNase Substrate (%)infractiona:
Al CII CIII
Native E. coli DNA 100 100 100
Denatured E. coli 125 170 76
DNA
3'-Terminallylabeled 59 100 22
calf thymus DNA
a Standard enzyme assay and the preparation of different substrates are described in thetext. Fraction II and III DNaseswere DNA-cellulose column puri-fied; 0.2 U of DNase of different fractions (Fig. 1A, fractionI, and 1C, fractions II and III)wereused.
and CII utilize denatured E. coli DNA more efficiently than fraction CIII as compared with nativeE. coliDNA. In contrast,when 3'-termi-nally labeled activated calf thymus DNA was
usedasthesubstrate,fraction CIIwasfoundto
beveryefficient, whereas fractions AlandCIII represented 50 and 22%, respectively, of the activityascompared with fraction CII. It is thus evident that fraction CIII DNase has clearly different substraterequirements thanothertwo DNasefractions.
Effect of salt.Therewerenotmanydifferential effectsof saltonthree DNasefractions when
3'-terminally labeled activated calfthymus DNA
was used as the substrate (Fig. 5A). However,
potassiumchloride exertedamarkeddifferential
effectonthreeDNaseswhen nativeE.coliDNA was used as a substrate. The
DNA-cellulose-purifiedfractionswereverysensitivetohighsalt
concentrations. Fractions II and III were 50%o inhibited at45and 100 mM salt concentrations, respectively, whereas the50% infective doseof fraction I DNases was shown to bemore than 200 mM. This characteristic, therefore, distin-guishes three fractionsfrom each other.
Effect of PFA on DNase activity. It has been reported that herpes simplex virus-specified DNApolymeraseand its associatedexonuclease are inhibited by phosphonoacetic acid (PAA) and phosphonoformic acid (PFA) (17, 22). Therefore, it was of interest tocheck the effect ofPAA and PFA on thepurifiedDNases. In our system, none of the DNases were affected by PFA.Thiswasobserved both whenE. coli DNA and 3'-terminally labeled activated calf thymus DNA wasused assubstrate (datanot shown).
Properties of DNA polymerase activity. Since reports onthe effect of salt and pyrophosphate analogs on EBV-associated DNA polymerase are conflicting (5, 10, 21), it is important to reexamine this issue in our system. We have shown(Fig. 1C, fraction II) thatareproducibly smallshoulder is observed in theDNA polymer-aseelution profile. This result led us tobelieve that the activity profile mightrepresent a mix-tureoftwoactivities. As pyrophosphate analogs andhigh salts arethetwo widely usedreagents known to differentiate between various DNA
polymerases (5, 17, 22), the effect of these
reagents were checkedon these two putatively different activities. There was a definite differ-encein theconcentration-dependentsalt activa-tion pattern withrespect tothefractions used for assay (Fig. 6A). Fractions pooled from the left side of theprofile (Fig. 1C, fractionIla) respond-ed more to salt activation than the fractions
I00
z80-C60
6
.-
40-C
a.
20-50 100 150 200 50 100 150 200
KCI
(mM)
FIG. 5. Effect ofpotassiumchloride on DNase activities fromdifferent fractions with3H-terminallylabeled activated calfthymusDNA(A)and E.coli"C-labelednative DNA(B) as substrates. FractionsI(O), CII(0),
andCIII(0)wereassayed with the indicated concentrations of salt.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.491.99.391.470.649.2]100I
-._
0.
aL
80-401
201
PFA (jjM)
FIG. 6. Effect of ammonium sulfate (A) and phos-phonoformic acid (B)onDNase-associated DNA poly-merase fractions CIIa and Cllb. DNA polymerase fromfractions CIla (0) andCIIb (0)wereassayedas
described in thetext.
pooled from the right side (Fig.1C,fractionIIb). However, when increasing amounts of PFA
were used in the reaction mixture (Fig. 6B),
fractionspooled from the left side of the profile responded moretoinhibition than the fractions pooled from the rightside. Implications of these results will be made in the discussion.
DISCUSSION
We have reported on the identification and
characterization ofa newalkaline DNase activi-tyinduced aftertreatmentoftheEBV-producing cellline P3HR-1with TPA.Theinabilityof TPA toinduceasimilar kind ofactivityin non-virus-producing Raji cellssupportsthenotion that the
enzyme is associatedwith the virus replication cycle and is not due to artifacts of chemical treatment. The partially purified activity de-scribed here is devoid ofany detectable DNA polymerase activityand is thus clearlydifferent from the viral DNA polymerase-associated
nu-clease activity, as has been reported in other systems (17). In fact, the activity never
co-chromatographed with viral DNA polymerase activityatany stepofpurification,as has been described elsewhere(3).
The DNase activity can be neutralized with sera obtained from patients with NPC. Moreover, the finding that purified activity is also neutralizable withpurified IgG preparations from the sera ofNPC patients andnotwith those from normal individuals points to the fact that the neutralizing activity of NPC serum is an antigen-antibody reactionand is notduetosome nonspecific factor(s) in the serum. The neutral-izationcurveof purifiedfraction (Fig. 1C, frac-tion III) is very similar to that of the DNase activity of superinfected Raji cells (1; Fig. 4).
There areseveral reasons to believe that the EBV-associated DNase activity described here isnotduetomycoplasma DNases (25).First, the induction of activity in P3HR-1 cells by TPA withrespect to identically subcultured P3HR-1 cells in the absence of TPA makes it unlikely (Fig. 2A). Second, the activity is similartothat found insuperinfectedRaji cellsasmeasured by neutralization with IgG. Third, the activity can beneutralizedbyseraofpatients withNPC, but notby thatofmosthealthyindividuals.
Our resultson thesubstrate specificity of the enzymediffers fromapreviousreport(3). These differences may be due to the use of different fractions from the DEAE-cellulose column. When 3'-terminally labeled activated calf
thy-mus DNA was used, the enzyme showed a
preference similartothat of the DNase found in
EBV-superinfected Raji cells (unpublished
data).
The EBV DNApolymerase-associated DNase
activity described here (Fig. 1C, fraction II) is
differentfromthatofherpessimplexvirus DNA
polymerase-associated exonuclease (17)by
vir-tueof the factthat, unlike herpes simplex virus DNA polymerase associated-exonuclease (7, 22), thisactivityis inhibited by salt(Fig. 5) and is insensitive to PFA (data not shown). Inthis respect, theactivityis somewhat similartothat previously reported in a herpes simplex virus system (14).
The other DNase activity which always co-purified with the DNA polymerase (Fig. 1C, fraction III) was insensitive to sera of NPC
patients.Atthispoint,therefore, we areunable
topredict anythingabout this nuclease. To our
surprise, however,the DNApolymerase activi-ty(s) of this peak(s)behaves
differentially
with respect tosaltandPFA(Fig. 6), probably owing tothefact that both EBV-associatedDNApoly-meraseandanother unidentifiedpolymerase
ac-tivityarecoelutedasmixtures. The left side of
thepeak (Fig.1C,fractionIa),becauseofahigh
EBV-enzymecontent, behavesmorelike EBV-associatedpolymerase intermsofsaltactivation
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.491.78.225.59.362.2]and PFA sensitivity (5, 6). Further work is obviously necessary tosettlethis issue.
Recently, Grawetal. (11) reported the isola-tion ofanATP-dependentDNase from
lympho-cyte. Though P3HR-1 is a kind oflymphocyte, we could not find any ATP-dependent DNase
activity in our preparations, possibly owing to the fact that the lymphocytes are diverse in
natureand it thus becomesdifficulttoextend the findings fromonetype of celltoanother (23).
Further studies onthe properties ofDNases from TPA-treatedP3HR-1 cells areunderway.
ACKNOWLEDGMENTS
Thisinvestigationwassupported by Public Health Service
grant I-ROI-AI17205-03VR from the National Institute of Allergy and Infectious Diseases and bygrantCH-29 from the American CancerSociety.
Wethank Kay Baldwin and BarbaraLeonard for secretarial assistance.
LITERATURE CITED
1. Cheng, Y.-C., J.Y.Chen,R.Glaser,andW. Henle. 1980. Frequency and levels of antibodiestoEpstein-Barr virus-specificDNaseareelevated inpatients with nasopharyn-geal carcinoma. Proc. Natl. Acad. Sci. U.S.A. 77:6162-6165.
2. Cheng, Y.-C., J.Y.Chen,P.J. Hoffman, and R. Gaser. 1980. Studiesontheactivityof DNaseassociated with the replication of the Epstein-Barr virus. Virology 100:334-338.
3. Clough, W. 1979. Deoxyribonuclease activity found in
Epstein-Barrvirusproducing lymphoblastoid cells. Bio-chemistry 18:4517-4521.
4. Clough,W. 1980. An endonucleaseisolated from Epstein-Barrvirusproducinghumanlymphoblastoidcells. Proc.
Natl. Acad. Sci. U.S.A. 77:6194-6198.
5. Datta, A. K., R. J. Feighny, and J. S. Pagano. 1980. Induction ofEpstein-Barr virus-associatedDNA
polymer-aseby 12-O-tetradecanoylphorbol-13-acetate. Purification and characterization.J.Biol. Chem.255:5120-5125.
6. Datta, A. K., and R. E. Hood. 1981. Mechanism of
inhibition ofEpstein-Barrvirusreplication by phosphono-formic acid.Virology114:52-59.
7. Derse, D., and Y.-C. Cheng.1981. Herpessimplexvirus typeI DNApolymerase. Kineticproperties of the associ-ated 3'-5' exonuclease activity and itsrole in AraAMP
incorporation.J. Biol. Chem. 256:8525-8530.
8. Feighny, R. J., B. E. Henry, and J. S. Pagano. 1981.
Epstein-Barr virus-induced deoxyribonuclease and the
utilization of host-cellDNA degradationproducts in viral
DNAreplication. Virology115:395-400.
9. Francke,B.H.,M.0.Moss,M.C.Timburg,andJ. Hay.
1978. Alkaline DNase activity in cells infected with a
temperature-sensitivemutantofherpes simplexvirus type
II.J. Virol. 26:209-213.
10. Goodman,S.R., C. Prezyna,and W.C. Benz. 1978. Two
Epstein-Barr virus-associated DNA polymerase
activi-ties.J.Biol.Chem.253:8617-8628.
11. Graw, J., E.-J. Schdaeger, and R. Knippers. 1981. A
lymphocyte ATP-dependentdeoxyribonuclease. J. Biol. Chem. 256:13207-13212.
12. Henle, G., and W. Henle. 1966. Immunofluorescence in cells derived from Burkitt's lymphoma. J. Bacteriol. 91:1248-1256.
13. Hoffman, P. J. 1981.Mechanism ofdegradationofduplex DNAby a DNase induced by herpes simplexvirus. J. Virol. 38:1005-1014.
14. Hoffman,P.J., and Y.-C. Cheng. 1978. The deoxyribonu-clease induced after infection of KB cells by herpes simplex virus type I or type II. J. Biol. Chem. 253:3557-3562.
15. Hoffman,P.J., and Y.-C. Cheng. 1979. DNaseinduced afterinfectionof KB cellsby herpessimplexvirus type I or II.J.Virol. 32:449-457.
16. Keir, H. M., and E. Gold. 1963.Deoxyribonucleic acid nucleotidyl-transferase anddeoxyribonuclease from cul-turedcellsinfectedwithherpessimplexvirus. Biochim. Biophys.Acta72:263-276.
17. Knopf, K. W. 1979. Properties ofherpes simplexvirus DNA polymeraseand characterization of its associated exonucleaseactivity. Eur. J.Biochem.98:231-244. 18. Lin,J. C., J. E.Shaw, M. C.Smith,andJ. S. Pagano.
1979.Effectof12-O-tetradecanoyl-phorbol-13-acetateon
thereplicationofEpstein-Barrvirus.I.Characterization of viral DNA.Virology99:183-187.
19. Marmur, J.1961.Aprocedurefor the isolation of deoxy-ribonucleic acid from microorganisms. J. Mol. Biol. 3:208-218.
20. Miller,R.L.,R.Blaser,andF.Rapp. 1977. Studies ofan
Epstein-Barr virus-induced DNA polymerase. Virology 76:494-502.
21. Ooka, T., G.Lenoir,andJ.Daillk. 1979.Characterization ofan Epstein-Barr virus-induced DNA polymerase. J. Virol. 29:1-10.
22. Ostrander, M., and Y.-C. Cheng. 1980. Properties of herpessimplex virustype 1and type 2 DNApolymerase. Biochim. Biophys.Acta609:232-245.
23. Paul, W. E., B. Sredni, and R.H.Schwartz.1981. Long-term growth and cloning of non-transformed lympho-cytes. Nature(London)294:697-699.
24. Potuzak,H., and U.Wintersburger. 1976. DNA covalent-ly linked tocarbosymethyl-celluloseanditsapplicationin affinity chromatography. FEBS Lett.63:167-170. 25. Razln, S. 1978. The microplasmas. Microbiol. Rev.
42:414-470.
26. Strobel-FIdler,M., and B. Francke.1980.Alkaline deoxy-ribonuclease induced by herpes simplex virus type I: compositionandproperties ofthepurifiedenzyme. Virol-ogy103:493-501.
27. Weissbach, A., S. C. L. Hong,J. Auker, and R.Muller. 1973. Characterization ofherpes simplexvirus-induced deoxyribonucleic acid polymerase. J. Biol. Chem. 248:6270-6277.
28. Welssbach, A.,A.Schiabach, B.Fridlender, andA.
Bol-den. 1971. DNApolymerasesfrom human cells. Nature (London)NewBiol.231:167-170.
29. ZurHausen, H., G. W.Bornkamm,R.Schmidt, andE.
Hecker. 1979. Tumor initiators and promoters in the induction ofEpstein-Barrvirus. Proc. Natl. Acad. Sci. U.S.A.76:782-785.
30. Zur Hausen, H.,F. J. O'Neill,andU.K. Freese. 1978.
Persisting oncogenic herpesvirus induced by the tumor
promoter TPA. Nature(London)272:373-375.
on November 10, 2019 by guest
http://jvi.asm.org/