0022-538X/87/010167-10$02.00/0
Copyright © 1987, AmericanSocietyforMicrobiology
Characterization of
the Simian Virus 40 Late Promoter: Relative
Importance of Sequences within the 72-Base-Pair Repeats Differs
before and after Viral DNA Replication
MICHELE ERNOULT-LANGE, FRANCIS OMILLI, DAVID R. O'REILLY, ANDEVELYNEMAY*
Institutde RecherchesScientifiques surle Cancer, Laboratoire d'OncologieMoleculaire, 94802 Villejuif Cedex, France Received 10 April 1986/Accepted 22 September 1986
We examinedsequences involved in the simian virus 40 (SV40) late promoter in vivo, by using quantitative
Si
nuclease analysisof a series of deletion mutants within the SV40 regulatory region. These mutants were constructed so as to place the alteredpromoter region in its normal position relative to the SV40 late genes. Theeffectsof the deletions on latetranscriptional activity were analyzed before and after viral DNAreplication,by
omitting orincludingSV40 large Tantigen.The data show that(i) in the absence of large T antigen, the deletion ofthe21-base-pair (bp) repeats results in a fourfold increase in latetranscription,and(ii)thesequences within the 72-bp repeats are a component of theSV40 late promoter, acting not only before, but also after viral DNA replication. We identified two domains which contain sequences important for efficient late transcription. Domain I, at the late proximal end of each 72-bp repeat, was found to function before replication and was
possiblyalso involved after replication. The contribution of domain II, at the late distal end of each 72-bp
repeat, was much more significant after replication but only of minor importance before replication.
The regulatory sequences of simian virus 40 (SV40) are
contained in the418-base-pair (bp) HindIII-HpaII fragment
(nucleotides [nt] 5171 to 346). This region includes the
controlelements requiredforearlyand latetranscriptionas
well as sequences required for viral DNAreplication. The
importance
of control elementsfor earlygeneexpression hasbeen thesubjectofanumber ofinvestigations. At least three
spatially distinct elements compose the SV40 early
pro-moter.Theterm"promoter" isused here in abroadsense to coverany DNA sequencerequired foraccurateandefficient
initiation of transcription. The first element contains the
earlystartsites andanA-T-richsequence(17bp) referredto
asthe TATAhomology sequence (TATAbox). The TATA
box plays a role in fixing the sites used for initiation of
transcription(3, 14).The secondelement, located upstream
of the TATA box, is a G-C-rich region comprising two
perfect 21-bp directrepeatsandathird, degeneraterepeatof
22 bp. Each repeat contains two
copies
of the sequence5'-CCGCCC-3'. This transcriptional control element is
es-sential for invivo and in vitro
transcription
from the SV40earlypromoter(13, 14, 19, 38). Itmay functionin a
bidirec-tional mannerand is known tocontainthebinding sites for
thecellular transcription factor Spl(10). The third element,
locatedupstreamof the G-C-rich sequences, comprises two
perfect 72-bp directrepeats and 20bp upstreamfromthese
repeats. The deletion of this element, referred to as an
enhancer, has adramatic effecton earlygeneexpression in
short-termexperiments invivo (3, 14, 17, 29). Inaddition,
the 72-bp repeat enhancer, acting in cis, can stimulate
transcription in vivo from heterologous promoters ineither
orientation (2, 29, 39).
SV40latetranscriptionoccursin theoppositedirection to
that of early transcription. Far less is known about the
transcriptional control signals ofthe SV40 late genes. The
late mRNAs are known to have heterogeneous 5' ends (15, 18, 24). However, in contrast to observations for the
major-ity of eucaryotic mRNAs (8), there is no recognizable
*Correspondingauthor.
A-T-rich sequence (TATA box) approximately 25 nt
up-streamfromany of these5' termini.
Inabortively infectedorintransformed cells, 10 to20% of
theviral transcripts correspondtoSV40 lategeneexpression
(11,25). Afterreplication in lytically infected cells, there isa
dramatic increase in late gene expression attributable to
template amplification and to a switch in the relative
strengths oftheearly and latepromoters, with late mRNAs
now accounting for up to 95% of viral transcripts (1).
Recently,twogroups haveproposedthat trans activationby
T antigen is a factor in the stimulation of late promoter
activity(4,5, 22, 23). However,it ispossible that this effect
is indirect and that other factors are also involved, as
suggested by the results ofBrady et al. (6) and Tack and
Beard(36).
Previously we have studied the structure of the late
promoter operational in the absence of the SV40 origin,
which is contained withinsequenceslocated fromnt 332to
113 (12). Thislatepromoterregion comprisestwoelements.
The firstelement, whichwe call the +7 to -53 element, is
located from nt 332 to 273 and includes the major late cap
site (nt325) and thesequence5'-GGTACCTAACC-3'(nt294
to 304), which has been shown to be important for the
efficientin vitro initiation oftranscription from this site (7).
The secondelement,locatedfromnt272 to113,includesthe enhancersequences,whicharethus acomponentof boththe
early and late promoters in vivo. We have subsequently
shown that the
principal
late promoterelementbefore viralDNAreplication is in fact the enhancer elementand thatthis
elementcanfunction in the absenceofTantigen (30). Shaw
et al. (34) have also found that enhancer sequences contrib-ute tolatepromoteractivity bothin vivoand in vitro in the absenceofreplication. However,these authorsreport thatT
antigen is required for significant late
transcription
in vivowhen replication isprevented.
Inthiswork,wenowfocusourattention on the sequence
elements operating within the promoter
region
before and after replication and late promoter activation. The data obtained show thatsequences within the72-bprepeatsare a167
on November 10, 2019 by guest
http://jvi.asm.org/
A
B
C
SV401pBR322L
ISV40 pBR322
SV401 pBR328[27]
[2770]![70 [32
SV40 i pBR322
[27701/ [375]
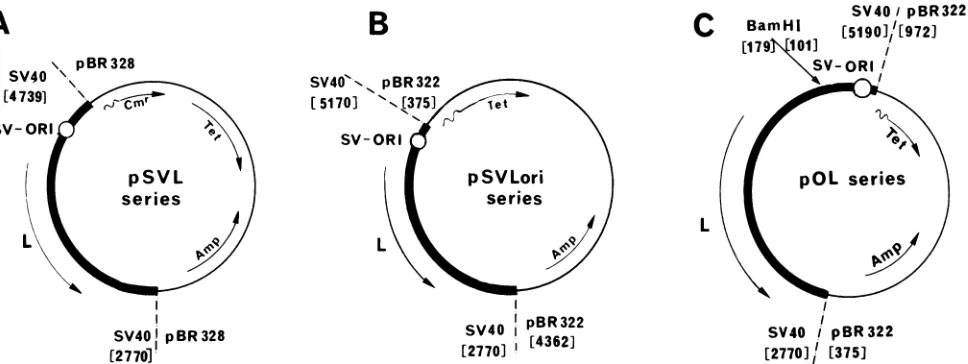
FIG. 1. (A) General structure of the pSVL series plasmids: the thick line represents SV40 sequences (the circle shows the location of the viral origin of replication, thearrowmarked L shows the direction of the late transcription). pBR328 sequences are indicated by the thin line. (B) General structure of the pSVLori series plasmids: The thick line represents the SV40 sequences (symbols as in panel A). pBR322 sequences are indicated by the thin line.(C) General structure of pOL series plasmids: The thick line represents the SV40 sequences (symbols as inpanel A). Thenucleotides at the junction of the 21-bp repeats and the single 72-bp sequencesareindicated(BamHI site). The thin line represents pBR322 sequences.
componentoftheSV40 latepromoter,actingnotonlybefore but also after viral DNAreplication. Inaddition, we show
that the relative importance for late transcription of
se-quenceswithinthe enhancerregionchangesafter replication
ofthetemplateDNAmolecule.
MATERIALS AND METHODS
SV40 nucleotide sequence numbering system. Nucleotides arenumberedaccordingtothe systemofBuchman et al. (9). Bacteria, plasmids, and cells.Thebacterialhoststrain used was Escherichia coliHB101 (27). The plasmids pAS,TB101,
and TB208 have been described by Benoist and Chambon(3)
and by Moreau et al. (29). DNA sequence analysis has
shown that: (i) SV40 nt 5236 to 1 are deleted in pAS; (ii)
SV40nt162 to265aredeleted in TB101;and(iii) SV40nt137 to232 aredeletedinTB208(seeFig.7A). TheplasmidpLP,
which contains the complete SV40 genome, has been
de-scribedpreviously (12).
Theeucaryotic cellsused wereCV-1PandCOS-7 monkey
cells and HeLa human cells. COS-7 cells are a line of
permissive CV-1cellstransformed byorigin-defectiveSV40
DNA, which synthesize a viral A gene product capable of
complementing SV40 A gene mutants (16). CV-1P and
COS-7 cells were grownin Dulbeccomodified Eagle
essen-tial medium (GIBCO Laboratories, Grand Island, N.Y.)
supplemented with 7% fetal calf serum. HeLa cells were growninDulbecco modified Eagle essential medium supple-mented with2.5% fetal and2.5% newborncalfserum.
Construction of recombinant plasmids: pLP series. The pLPseriesincludes pLAS,pLTB1l1,pLTB208, pLBK3 and pLBS6.
(i) pLAS,
pLTBlO1,
andpLTB208. pLAS,pLTB101,andpLTB208 were constructed from pAS, TB11, and TB208,
respectively, by the addition of the late region of SV40 as
described previously for pLP (12). These plasmids contain the complete SV40 genome with various deletions in the
SV40regulatory sequences(see Fig. 7A).
(ii) pLBK3. SV40DNAwas cutwith
SphI
andKpnI.Thelarge fragment (from nt 294 to 128) was treated with the
Klenow fragment of E. coli DNA polymerase I (Klenow
fragmentDNApol
l),
circularized by blunt-end ligation, andrecut withPstI. The fragment fromnt 1988 to 3204, which
includestheoriginof replication,wasligatedtoapartialPstI
digest ofpLP. The resultantplasmid isthe sameaspLPbut
containsadeletionofnt 130to 300 (seeFig. 7A).
(iii) pLBS6. SV40DNAwas cutwith SphI, digested with BAL31nuclease, repaired withKlenowfragmentDNApol
I, andcircularized by blunt-end ligation. The deleted DNA
was inserted into pLP by using the same construction as
described for pLBK3. Thisplasmid containsadeletionofnt
66to204 (seeFig. 7A).
pSVL series. The pSVL series of plasmids includes
wild-typepSVL pSVLAS, pSVL101, pSVL208, pSVLBK3, and
pSVLBS6. Plasmids ofthepLPseries,including pLP, were
digested with TaqI, and the fragment containing the SV40
lateregionandreplication origin (extendingfrom nt4739in
SV40to nt 651 inpBR322) wasisolated from eachplasmid. Thesewererepaired with KlenowfragmentDNApol Iand
inserted intotheuniquePvuIIsiteofpBR328 (Fig. 1A). The
deletions within the SV40 regulatory sequences in these
plasmids areshown in Fig. 7A.
pSVoriand pSVLori series. These series include
pSVori,
pSVoriA21, pSVoriA72, pSVLori, pSVLoriA21, and
pSVLoriA72.
(i) pSVori and pSVLori. pSV1 (3), which contains the
wild-type SV40 earlyregion (extending from theHpaII site [nt346] totheBamHI site [nt2533]), was cutwithHindIII,
repaired with Klenow fragmentDNA pol I, andrecut with
EcoRI(at thepBR322 [nt
2]-SV40
[nt 346]junction).pBR322wascutwithBamHI(nt375),repairedby Klenow fragment
DNApol
l,
andrecutwithEcoRI (nt2). ThepSV1 HindIII-EcoRI fragment, which contains the SV40 regulatory se-quences, was then ligated to the large pBR322EcoRI-BamHI fragment, to givepSVori. The SV40 late region (nt
2770to294)wasisolated from SV40DNAbyBclIcleavage, Klenow fragment DNA pol I repair, and KpnI cleavage.
pSVori was then cut with EcoRI, repaired, and recut with
KpnI. The large fragment thus obtained was ligated to the SV40 lateregion togive pSVLori (Fig. 1B).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.71.555.71.253.2](ii)pSVoriA21 and pSVLoriA21. pHS102 (3) was digested with BamHI, repaired with Klenow fragment DNA pol I, recut with EcoRI, and the 233-nt fragment of SV40 DNA extending from nt 113 (the uniqueBamHI site) to nt 346 (the
EcoRI site at the pBR322-SV40 junction as in pSV1) was
isolated. pSVori was cut with NcoI (SV40 nt 38), repaired
with Klenow fragment DNA pol I, and recut with EcoRI
(SV40nt 346). The large pSVorifragment wasligated to the
233-ntfragment isolated from pHS102. The resultant plasmid
(pSVoriA21)has a deletion from nt 38 to 113 which removes
the 21-bp repeats (see Fig. 7A). The SV40 late region was inserted into pSVoriA21 by using the sameconstruction as described for pSVLori, to givepSVLoriA21 (Fig. 1B).
(iii) pSVoriA72 and pSVLoriA72. pSVori was cleaved with PvuI and PvuII, and the704-ntfragmentextendingfrom nt 3735 (pBR322) to nt 270(SV40) was isolated. Separately, a 296-nt fragment extending from nt 128 (SV40) to nt 471 (pBR322) was isolated by cleavage of pSVori with SphI, Klenow fragment DNA pol I repair, and cleavage with
BanII. These fragments were ligated to a 3,250-ntfragment
isolated from pBR322 viaPvuI-BanII cleavage. This recom-binant plasmid (pSVoriA72) has a deletion from nt 128 to 272 in SV40which removes the major part ofthe 72-bp repeats (see Fig. 7A). The SV40 late region was inserted into
pSVoriA72 as before, to give pSVLoriA72 (Fig. 1B).
pOLO series. The pOLO series includes pOLOandpOLA3.
(i) pOLO. pA0, which contains only one copyof the72-bp
repeats (nt107 to 178 aredeleted;42) wasdigestedwithKpnI
and Stul, and the 275-nt fragmentfrom SV40nt 294 to 5190
was isolated. Theplasmid pL113b, containingthe SV40late
region from nt 113 to 2770 (30), wasdigestedwith KpnIand
NruI, and the large fragment obtained was ligated to the
275-nt fragment described above. The resultant plasmid,
pOLO, contains the SV40lategenes and anSV40 regulatory
region deleted for one copy of the 72-bp repeats. It has a
unique BamHI site between the 72-bp sequence and the
21-bp repeats generated in theconstruction ofpAO(Fig. 1C;
see Fig. 7B).
(ii) pOLA3. pOLO was digested with BamHI, repaired with Klenow fragment DNA pol I, and recut with ClaI
(pBR322 nt 23). The 3,568-nt fragment obtained wasligated
to a2,921-nt fragment obtained by SphI digestion, Klenow
fragmentDNA pol I repair, and ClaI (pBR322 nt 23)
diges-tion ofpOLO (Fig. 1C).pOLA3 istherefore identicaltopOLO
except fora deletionfrom nt 179 to 204(between the novel
BamHI site and the SphI site in the 72-bp sequence),
removing the early end of the single 72-bp repeat (see Fig.
7B).
Construction of the internalmarkerpSVL7. AnXhoI linker
[d(CCTCGAGG), New England BioLabs] was ligated into
pSVL at theEcoRIsite (nt 1782 in SV40),aftercleavageand KlenowfragmentDNA pol I repair of this site. Theaddition
of an XhoI linker preserved the proper reading frame and
permittedthe translation ofprotein.
Quantitative Si nuclease analysis. HeLa, CV-1P, and
COS-7 cells were seeded at 106 cells per 10-cm-diameter petri dish. At 24 h after seeding, the cells were transfected with 20 ,ug of the recombinant plasmid and 10
jig
of the marker plasmid pSVL7 by the calcium phosphatecoprecipitation techniqueaspreviouslydescribed(12). At 48
hposttransfection,total RNA wasextractedwith hot phenol
(33) and quantitativeSi nuclease analysis wascarriedout as
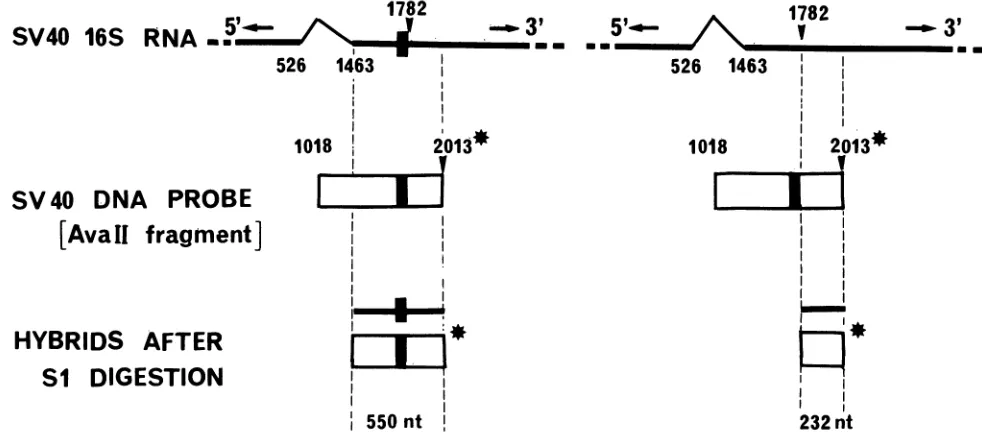
follows. The single-stranded probe used was the coding strand of the SV40 AvaIl fragment (nt 2013 to 1018) of pSVL7, 5'-endlabeledwith
32P
atnt 2013 (see Fig. 2). 5'- endlabelingandstrand separation were carried out by standard
techniques (26). Total RNA (25 ,ugfrom HeLa cells, 50 ,ug
from CV-1P cells, or2.5
,ug
fromCOS-7 cells) washybrid-ized, for 15 h at
42°C,
to an excess of 5'-end labeledsingle-stranded probe in the presence of0.4 M NaCl-50%
formamide-40 mM piperazine-N-N'-bis(2-ethanesulfonic
acid) (PIPES),pH 6.4. Trial hybridization experimentswere
performed to ensure the presence of excess probe (data not
shown). Si nuclease digestion was performed by ninefold
dilution of the hybridization mixture in0.23 M NaCl, 40 mM
ZnCl2, 30 mM sodium acetate (pH 4.5), and 250 U ofSi
nuclease (Bethesda Research Laboratories, Inc.,
Gaithersburg, Md.), followed by 2h of incubation at 25°C.
The digestion products were ethanol precipitated and
sepa-rated on 5% polyacrylamide-7 M urea gels. Theamount of
RNAtranscribed from the SV40 recombinant plasmids was
estimated by scanning the autoradiograms. Each gel was
exposed to film, without an intensifying screen, for various
lengths of time to avoid saturation of the film. Levels of
transcription for each plasmid were normalized relative to
transcription of the internal standard (pSVL7). The final
figures presented in Fig.7represent the averageresultsofat
least five independent experiments with different plasmid
preparations.
Analysis of DNA replication. COS-7 cells were seeded at
106 cells per 10-cm-diameter petri dish. At24hafter seeding,
the cells were transfected with0.5 p.g of recombinant
plas-mid and 5 ,ug of marker plasmid pBR327 by the calcium
phosphate coprecipitation technique aspreviously described
(12). At 48 h posttransfection, the low-molecular-weight DNA was extracted bytheprocedure ofHirt(21). This DNA
(in the Hirt supernatant) was deproteinized by two
extrac-tions with phenol saturated in 10mMTrishydrochloride (pH
8)-i
mM EDTA and was then precipitated with ethanol.After linearization of the DNA by Clal (pSVL series) or
EcoRI (pSVLori series), each DNAsample wasfractionated
by electrophoresis through a 1% agarose gel in 40 mMTris
hydrochloride (pH
7.8)-50
mM sodium acetate-4 mMEDTA.The gels wereimmersedin 0.5 MNaOH-1.5 MNaCl
for 45 min, and then neutralized in 1 M Tris hydrochloride
(pH
7.8)-3
mM NaCl, for 45 min before transfer to anitrocellulose filter (Schleicher & Schuell, Inc.,
Keene,
N.H.; BA 85) by the method ofSouthern (35). Transferred DNA was detected by hybridization to a pBR327 probe labeled with32P by nick translation (31). The relative copy number of each plasmid at 48 h posttransfection was esti-mated by quantitative scanning of the autoradiograms ob-tained. Eachfilterwasexposed tofilm for various periods of time, without intensifying screen, to avoidsaturation ofthe film. The levels ofeach plasmid present were normalized relative to the amount of the internal marker pBR327 to controlforvariations in theefficiency of transfection and for extraction of DNA. The relative amount of each plasmid present at 48 h posttransfection was obtained by averaging the results of at least four independent experiments with different plasmid preparations.
RESULTS
Linearrelationship between thequantityof transfectedgene and the level of transcription. Initially, to ensure that no experiments were carried out under conditions of excess input DNA, we determined the dose-response relationships for the wild-type plasmid pSVL in all three cell lines used. The pSVLplasmid contains the complete SV40 late region and an intact origin of replication. For these experiments, HeLa, CV-1P, or COS-7 cells were cotransfected with
on November 10, 2019 by guest
http://jvi.asm.org/
TRANSFECTING
DNA
INTERNAL
MARKER
PLASMIDS
TO
BE TESTED
1'
1782
SV40
16S
RNA
N
A
_.
526
1463
1018
2013SV40
DNA
PROBE
liiZI[AvaIl
fragment]
HYBRIDS AFTER
Si
DIGESTION
I
526
1463
1018
II
:5
I
550
nt!
1782
v
I.^
2013
201
I*
rI
232 nt
FIG. 2. Illustration of the method used forquantitativeSinucleaseanalysis. The 16SmRNAobtained from the marker andtestplasmids areshown.Spliced-out sequencesareindicatedby the thin black line, and the nucleotide numbersatthesplice junctionsaregiven.The XhoI linker atnt1782in the markerplasmid isrepresentedby theblack box. Under eachRNA, the open rectangle represents thesingle-stranded DNAprobelabeledatits 5' end(*). This probe isprepared from the AvaIl fragmentofpSVL7(nt 2013to1018) which contains the XhoI linker. At thebottomof thefigure, the hybrids obtained afterS1 nucleasedigestionareshown. When theprobeDNAwashybridizedto16SmRNA transcribed from the markerplasmid,DNAsequencehomology continued until the acceptorsplice junctionat nt1463 anda550-nthybrid fragmentwasprotected from S1 digestion. However, when the probeDNA hybridized with 16SmRNAtranscribed fromthe testplasmid, the sequence homology stoppedattheXhoIlinkeratnt1782anda232-ntfragmentwasprotected.
aconstant amountof the internal markerpSVL7 (10 ,ug).At 48 h after transfection, total RNA was extracted and the
amount of late transcription directed by pSVL was deter-mined by quantitative Si nuclease analysis as described in
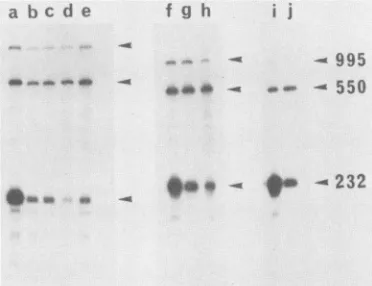
Materials and Methods. A schematic illustration of this methodology is shownin Fig. 2. These resultsarepresented inFig. 3.Aspredicted in Fig. 2,bandsof 232and550ntwere observed (panel A). The 232-nt protected fragment corre-sponds to 16S and 19S mRNA transcribed from the pSVL plasmid, whereas 16S mRNA transcribedfrom the internal marker protects a fragment of550 nt. 19S mRNA synthe-sizedby the internal markerprotectsthe entireprobeandso yields a band at 995 nt. As the majority of late mRNA species are 16S molecules (41), this band is much weaker than the 550-nt band. It can be seen in Fig. 3A that the
intensity of the 232-nt band increases with input DNA concentration in all three cell types. These results were quantified by scanning suitably exposed autoradiograms from fiveindependent experiments similartothoseshownin Fig.3A. Theaveragelevelsofexpression ofpSVL,
normal-izedrelative totranscriptionof the internal markerpSVL7,
are plotted versus the amount of DNA transfected in Fig. 3B. Inall threecell linesthecotransfection ofupto30 ,ug of pSVLand10,ugofpSVL7didnotresult in saturation ofthe system.
Forallsubsequentexperiments,wecotransfected 20jigof
thetestplasmidand10 ,ug oftheinternal markersince these levels arewithinthe linearresponse rangeofthesystem.
Effectof deletions within theG-C-richand72-bp repeatson
late geneexpressionprecedingviralreplication. Thedeletion mutants used, which contain the complete SV40 late
region
andanintact
origin
ofreplication
but havevarying deletionswithin the G-C-rich sequence and the 72-bp repeats, are
depicted in Fig. 1 and7. The SV40early genes aredeleted
from allthese
plasmids,
arid
consequently replication fromthe SV40origin is blocked. The
efficiency
of latetranscrip-tion of each ofthese mutants wasmeasured after
transfec-tion into semipermissive HeLa cells or permissive CV-1P
cells. At 48 h after transfection, total RNA was extracted and theamountof
late-specific
RNAinitiatedby
thevarious mutants wasdeterminedbyquantitative
S1 nucleaseanaly-sis.Theresults ofonesuch
experiment
areillustratedinFig.
4. The efficiency of late transcription directed by each
plasmidwasestimatedby densitometric scanningofsuitably
exposed autoradiograms (see Materials and
Methods)
andnormalizationrelativetotranscription directed bythe
inter-nal markerpSVL7.Theresultswerethenexpressedrelative tototaltranscriptionfrom thewild-typerecombinants
pSVL
or pSVLori
(taken
as100%).
pSVL comprises SV40se-quencesfromnt 4739 to2770in pBR328, whereas
pSVLori
includes sequences fromnt5170to2770inpBR322.Thedatapresented in Fig. 7A represent the average resultsfrom at
leastfiveindependent quantification experiments.
Theeffectof deletionswithinthe21-bprepeatsonlategene
expression.
It canbeseeninFig.4and 7A thatdeletionofthe21-bp repeats (pSVLoriA21) resulted in a 3.5- to 4-fold
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.60.551.139.355.2]A
ab
cd
e
_. .
f gh i
j
..._
_ _ --__
6
5
4
3
1 2 34
if.4 .
AP *
12 3 4
- 995
-- -550
B
*a00 -a232
*a
.* e--aa b cd e
*-.-a
2 f2
5 1 0 20 30
DNIA pg added per Petri dish
FIG. 3. Dose-response data for pSVL transcription in HeLa, CV-1P, and COS-7 cells.(A)HeLa, CV-1P,andCOS-7 cellswere
cotransfected by calcium phosphate precipitation with increasing
concentrations ofpSVL DNAandaconstantconcentration of the internal markerpSVL7 (10
jig).
RNAextraction, hybridization, S1 digestion, and electrophoresis were performed as described inMaterials and Methods.Atotalof5(lane 1), 10 (lane 2), 20(lane3),
or30,ug (lane 4)ofpSVLDNAwereaddedper10-cm-diameterpetri
dish. (B)TherelativeexpressionofpSVLwasevaluatedby
densi-tometric scanning of suitably exposed autoradiograms similar to those shown inpanelA.ThetranscriptionofpSVLwasnormalized
relativetotranscriptionfrom theinternalmarkerpSVL7.Eachpoint
in thisfigure is the average of five independent experiments with different DNApreparations.Symbols: (-),HeLacells;(A),CV-1P
cells; (0),COS-7 cells.
O0-.232
f g h i j
-- - MO- 955
- 00-*,so0 550
a -'0 232
FIG. 4. Quantitative S1 nuclease analysis of late transcription directed by the pSVL, pSVLori, and pOLO series of deletion mutantsbeforereplication. Total RNA was extracted from HeLa or CV-1P cells cotransfected with 20 ,ug of each of the deletionmutants
and 10 ,ugof theinternal marker pSVL7. Total RNAfrom HeLa cells(25 ,ug) (A) or50pugof totalRNA from CV-1P cells (B)was
hybridizedto an excessof5'-end-labeled, single-stranded probe (see Fig. 2). AfterSi nucleasetreatmentandseparation by electropho-resis through 5% polyacrylamide-7 M urea gels, protected
frag-ments232 and 550 ntlong were obtained, corresponding to RNA
transcribed from thetestandmarkerplasmids, respectively. Lanes
areidentical inpanels AandB. Lanes: a, pSVL; b, pSVL101; c, pSVL208;d,pSVLBK3;e,pSVLBS6; f,pSVLori;g,pSVLoriA21; h, pSVLoriA72; i,pOLO;j, pOLA3.
expression.ItcanbeseeninFig.4and 7Athat deletionofthe
21-bp repeats (pSVLoriA21) resulted in a 3.5- to 4-fold
increase in latetranscriptionboth in HeLaand CV-1Pcells.
We have recently reported a similar phenomenon with
chimeric transposition plasmids in which T antigen was
expressed under the control ofputative late promoter
frag-ments (30). These resultsgive further support tothe hypo-thesis thatbefore viral DNAreplicationthe presenceofthe
21-bprepeatsbetween theenhancer and theearly startsites
favors early transcription. When the 21-bp repeats are
ab-sent,
transcription
isnolonger directedpreferentially intheearly
direction,
andtranscription
in the late direction isconsequently increased.
This increase in late transcription is not seen with the
plasmid pSVLBS6.Thedeletioninthisplasmidextends into
the21-bp repeatsasfaras nt66, thusremovingfour of the
sixG-C-rich motifs.However,aportionof the72-bprepeats
is
alsoremoved,
and it may be that an increase in theefficiencyof late transcriptionduetothe removalof four of
the six G-C repeats is offset by a decrease in promoter
strength due to the deletion ofimportant sequences within
the enhancerregion.
Theeffect of deletions within the72-bp repeatsonlategene expression. Theplasmid pSVL208,which hasadeletionof 95 bpwithin the72-bprepeats,showed
only
amoderate reduc-AHeLa Cv-tP COS-7
1 2 3 3 4
_... e.
-- 995
B
10
9
8
7
c 0
(n
V.
CL x
._
'a% . -- --m -. - 55 0
._ MO^^ _
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.324.541.79.342.2] [image:5.612.57.293.102.578.2]a bc d e f g h ,!Pql~ lq~ 9 4'.. '.4
4 4 4~'4 A 44
e coC e9e coe e e
Q q Q q Q q Q Q q
i
i
*.4 - 995
go,A* -- w
-_
550*
*i,s
-
232
9467
1418 do.L #*.deN
em.4
4362 1W 3273 4P
FIG. 5. Analysis of the replication of the pSVL series plasmids. Low-molecular-weight DNA was e
COS-7 cells 48 h aftercotransfection with 0.5F±goftU and 5
jig
of the internal control pBR327 and ana Southern blotting technique (35) as described in Methods. The relative amount of eachtestplasmidw by scanning of suitably exposed autoradiograms. Tlinear pBR327 is indicated (arrowontheright), and ti themolecular-weightmarkersareshownontheleft.
7A). However, thedeletion in pSVL101, which
265 in the late direction, brought about a 2
reduction in late promoter strength (33 to 40'0
Similarly, pSVLoriA72 from which sequenc
from nt 128 to 272 are missing, directed late
with an efficiency of 46% of pSVLori in
Surprisingly, this plasmid appeared to directla
tion moreefficiently in HeLacells (75% ofpS
unsureof the significance of this observation.
suggested the existence ofa region important
scription before viral replication between nt,
Extension of the deletiontont300
(pSVLBK3;
did not further affect the efficiency oftranscr
pared with pSVL101) suggesting that, in the,
largepartof the enhancer(nt130 to272),the
rn
TABLE 1. Relativeamountsofreplicationdirecte
mutantsinCOS-7 cells
Mutanta ofR(
pSVL (wild type) pSVL101 pSVL208 pSVLBK3 pSVLBS6
pSVLori (wild type) pSVLoriA21 pSVLoriA72
aThe deletionmutantsusedaredescribed in the legends
bThe values from four independent experiments with
preparations areexpressed as apercentageoftheactivit) undeleted mutants pSVLor pSVLori and are normalizes
amountoftheinternal markerpBR327asdescribedinMateri
FIG. 6. Quantitative S1 nuclease analysis of late transcription directed by the pSVL, pSVLori, and pOLO series of deletion
mutants after replication. Total RNA was extracted from COS-7 pBR 327 cells cotransfected with 20,ugof each of the plasmids and 10jigof the internalmarkerpSVL7. Thiswas analyzedasdescribed in the legendtoFig.4exceptthat 2.5 jigoftotalRNAwashybridizedto the 5' end-labeled probe. Lanes: a, pSVL; b, pSVL101; c, pSVL208; d, pSVLBK3;e,pSVLBS6; f, pSVLori;g,
pSVLoriA21;
and pSVLori h,pSVLoriA72; i,pOLO;j, pOLA3.xtracted from
le testplasmid
Llyzed
via the 265 to 300 is not critical for latetranscription
before repli-Materials and cation.'as
determined Effect of deletions within the regulatory sequencesonlate heposition
of gene transcription after the onset of viral replication. The hepositions
of effectof deletionswithin theSV40regulatory
sequenceson late genetranscription
afterDNAreplication
wasexamined.To allow
replication
of theseplasmids,
which contain anextendstont intact viral
origin
ofreplication, they
were transfected into'.5- to 3-fold COS-7 cells which
constitutively
expressfunctional Tanti-3'o
ofpSVL).
gen. Since it waspossible
that the differentplasmids
repli-es
extending
cate to different extents, we first determined the relativetranscription
amount of eachplasmid
present when thetranscriptional
CV-1P cells. analyses were carried out. COS-7 cells were
cotransfected
ate
transcrip-
with 0.5 ,ug of each testplasmid
and 5pug
ofpBR327
asVL). We are
nonreplicative
internal marker. Theplasmid
pSVLAS,
These results which is unable to
replicate
due to a deletion at the SV40for latetran-
origin
ofreplication,
was includedas anegative
control.At232 and 265. 48 h
posttransfection, low-molecular-weight
DNA wasiso-see
Fig. 7A)
lated andanalyzed
as described in Materials and Methods.ription (com- The
autoradiogram
presented
inFig.
5represents theresultsabsence of a ofone such
experiment.
Itcanbe seenthat,by
comparison
egion
fromnt with thenonreplicative
controlpSVLAS,
all theplasmids
replicated
after transfection into COS-7 cells(pSVLAS
wasd bydeletion
faintly
visible on overexposure of theautoradiogram;
datanot
shown).
The relative amount of eachplasmid
presentwasthen estimated
by
scanning suitably exposed
autoradio-elativeefficiency grams (see Materials and Methods) and normalizing relative f replication (%)b to the amount ofthe internal marker pBR327. The results 100 (Table
1)
of at least four suchexperiments
were then 90averaged
toprovide
an estimation of the relative copy 104 number of eachplasmid48 hposttransfection ofCOS-7 cells. 150 The relative levels of transcription were determined in a 165 series of quantitativeS1
nuclease analysis experiments by 100 using the same internal marker and probe as in the studies 60 carried out before replication. The results of one such 153 experiment are presented in Fig. 6. The variations in the amountof late RNAsynthesized by
eachtestplasmid
were toFig.1and 7. quantified by densitometric scanning of suitably exposed different DNA autoradiogramsasbefore. A summary ofthe results of this y determinedford relative to the sertes of experiments is presented in Fig. 7A. All results are ialsandMethods.
again
expressed
as apercentage oftheappropriate
wild-type
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.60.302.81.303.2] [image:6.612.343.529.82.225.2] [image:6.612.62.302.590.699.2]RELATIVE EFFICIENCY OF TRANSCRIPTION
(%)
A
PLASM ID TRANSFECTED pSVL or pSVLori
HeLa CV-IP COS-7
Major Late m RNA5'ends
HpaIl Kp
346 29
I 11
pn1
94
SphI 200
I 11 TATTTAT Major Early
I
m1Nrn
Ie
- dsSphI Bgl
128 5235
100
a b
100 100 100
265 162
33 40 16 18
232 137
*
70 69 22 22
130
29 30 10 7
105 96 14 9
350 404 35 58
75 46 21 14
272 128
100 100 ND
[image:7.612.56.555.65.510.2]72 79 18 ND
FIG. 7. Summaryoflevels of latetranscription directed bythepSVL,pSVLori,and pOLO seriesof deletionmutantsin HeLa,CV-1P,
andCOS-7 cells. Aftertransfection of cells with each of the deletionmutantsand the internal markerpSVL7,theamountof RNA transcribed
was measured by quantitative S1 nuclease analysis(see Fig.4and 6), followed by scanning of suitably exposedgels. The resultswere
normalizedrelative' transcriptionofthe internal markerpSVL7andareexpressedas apercentageoftranscription of the undeleted plasmid pSVL,pSVLori, andpOLO. Theyarecorrected(column b)ornot(column a)to accountfor variationsin thecopynumber of the different plasmidsinCOS-7 cells.The valuespresentedaretheaveragesof fiveindependent experiments performedwith different DNApreparations. (A)Levels oflatetranscription directed bythepSVLandpSVLoriseries. Aschematicrepresentation of SV40 regulatorysequencesand the
locations of the deletions inmutantplasmidsarepresented. Thetopof thefigureshows, fromrighttoleft:the5'ends ofearly mRNA sites
(arrowshowsthe direction ofearly transcription);the BgIl site(theminimaloriginofreplicationis locatedaround theBgllsite fromnt5208 to30); theTATTTAT sequence, (theTATAbox of theearlypromoter);theG-C-richregion(one 22-bp degenerate repeatandtwo21-bp directedrepeats)inwhich the six5'-CCGCCC-3' repeatedsequencesareindicated bysix blackblocks;thetwo72-bpdirectrepeats(open
boxes) withthepositionsof domains I and II indicated above;andthemajorlatetranscriptioninitiation site(arrow showsthe direction of late transcription). The deletionmutantsusedare depicted underneath. The nucleotidesatthe boundaries of each deletion (inclusive)are given. (B)Levels of latetranscription directedbythepOLO series.Aschematic representationof theSV40 regulatorysequencesincluded
inpOLO is presented. The symbols usedareasinpanelA.Thenucleotidesatthejunctionof the 21-bprepeatsand thesingle 72-bpsequence areindicated, asarethenucleotidesatthe boundaries of thepOLA3 deletion. ND, Notdetermined.
plasmid (pSVLorpSVLori), both ofwhichcan replicate in COS-7 cells. The variations in the relativeamount of
repli-cation ofthe differentplasmidsdid notsignificantlyaffect the
efficiencyof late transcription. Ingeneral, thevarious dele-tions hadamorepronouncedeffectonthe relativeefficiency
of late transcription afterreplication. pSVL101, pSVL208, pSVLBK3, and pSVLoriA72 all showed relative promoter
strengthstwo- tothreefold less than beforereplication.After correction for activity of replication, the plasmid pSVLoriA21showed anefficiencyoftranscription58% that
of pSVLori. This is in contrast to the situation before replication, in which the removal of the 21-bp repeats
resulted in a stimulation of late transcription. The fact that thiseffectwasnolongerseensuggests that,afterreplication, pSVI. 101
pSVL 208 pSVLBK3
pSVL BS6
300
pSVLori A21
204 66
113 38
pSVLori A72
B
pOLO
179 101
I
ISphI BamHI 200 101
I
Hpa34
346
Kpnt 294
pOLA3
BII
853
5235
100
204 179
_-- b
I,I
on November 10, 2019 by guest
http://jvi.asm.org/
the
21-bp
repeats favorearly
transcription
to amuchlesser extent.Themost
interesting result, however,
isthatobtained withthe
plasmid
pSVLBS6. Beforereplication,
this plasmidshowed an
unimpaired efficiency
of latetranscription,
whereas afterwarditshowedlevelsoflate
transcription
only9% those of
pSVL. Thus,
thedeletioninpSVLBS6
removedsequences critical for late
transcription
afterreplication,
andtranscription prereplication
remained unaffected.Significance
of the late distalmoietyof the72-bprepeats forlategene expression. The results described above suggested that sequences
important
for latetranscription
both beforeand after
replication
are found between nt 130 and 300.Furthermore,
the results obtained withpSVLBS6
show thatthedeletion in this
plasmid clearly
affectslatetranscription
more
severely
afterreplication.
Wenoticedthatthisplasmid
istheonlyoneinwhichthesequences downstreamfromthe
SphI
siteof both72-bp
repeatsweredeleted(Fig. 7A).
Theseresults suggestedthat sequencesrequired onlyafter
replica-tion arelocated in theseregions.
However,
since the deletion of four ofthe six G-C-richmotifs in
pSVLBS6
can have astimulatory
effect on latetranscription,
it ispossible
that the deletion in the 72-bprepeatsdoes indeed removelatepromotersequencesactive
before
replication.
Toclarify
thispoint,
weconstructed theplasmid pOLA3
in which the sequences downstream fromthe
SphI
site(nt
179to204)
aredeletedfrom thesingle 72-bp
sequence of the
plasmid
pOLO.
Thisplasmid
retained theG-C-rich motifs.
pOLA3
orpOLO
were cotransfected intoHeLa,
CV-1P,
or COS-7 cells with the internal markerpSVL7.
Relative levels oftranscription
weredeterminedinaseries of
quantitative Si
nucleaseanalysis experiments
asdescribedabove. The resultsoftheseanalysesarepresented
in
Fig.
4 and 6 and summarized inFig.
7B. We found that,whereas beforereplication
pOLA3
directedlatetranscriptionwith an
efficiency slightly
lessthan wild type(70 to80%
ofpOLO),
afterreplication
itwas moreinhibited,
with a pro-moterstrength only 18%thatofpOLO.
Thisresultthereforeconfirms that the late distal
moiety
of each72-bp
repeat iscritically important
afterreplication
andoflesserimportance
before
replication.
DISCUSSION
Inthis
study,
weanalyzed
andcompared
the structureoftheSV40 late promoterbefore and after the onset of DNA
replication.
Tothisend,
we constructedanextensiveseriesofmutants deleted within the late promoter
region.
These mutants all contain the altered promoter in its normalposition
relative to theSV40
late genes. The deletions encompass both the G-C-rich repeats and the enhancerregion sothat the contribution of both theseregions can be
assessed. An
important
observation that can be madecon-cerning
theoperation
of the late promoter beforereplicationis that this promoterapparently functionsinasimilar manner in HeLa and CV-1P cells. This confirms thesignificance of
previous
resultsdealing
with thefunctioning
ofthis promoterin HeLacells(12, 30).
The analysis of the plasmid
pSVLoriA21
indicates that,whereas before replication removal of the G-C-rich region
stimulates late transcription approximately fourfold, after
replication
the removal of these sequences results in amoderatedrop in late promoterstrength (Fig.7A). The fact that the removal of the G-C-rich region did not entail an
increase in late transcription postreplication suggests that
the G-C-rich repeats do not favor
early
transcription tothesame extent afterreplication asbefore. Theslight decrease inthe late promoterstrengthofpSVLoriA21after
replication
may be due to a reduction in transcription from the late initiation sites closest to the G-C-rich repeats in the72-bp
repeats. These sitesarepreferentially
used invitro(28,
32,
38)
orin vivo after viral DNAreplication (11,15).
Thestudy ofmutantswithin the enhancerelement allowed
us to identify two distinct domains in this region which
appeared to be particularly important for late promoter
activity.Oneimportant domain,whichwecalldomain I,was
showntobebetweennt232and265(compare pSVL101and
pSVL208, Fig. 7A) comprisingthe late proximal endof the
72-bprepeatsand 15bptothelate sideof theserepeats.Our
results show that domain I is activebeforeviral
replication.
Itis interesting that this domain includes sequences which
other workers have shown are important for early gene
transcription. Weiher et al. (40) used a series of
multiple-point mutations within the 72-bp repeats to define a core
sequence from nt 239 to 246 (5'-TGGAAAGT-3') which is essential for the enhancer activity of this region in CV-1 cells. Shawetal. (34) have subsequentlyfound that certain of the mutants studied by Weiher et al. (40) also display reduced late transcription in vivo beforereplication. More
recently, Zenke et al. (42)have used an extensive series of
pointanddeletion mutations to examine the relative
impor-tance of all sequences within the enhancer region.
By
analysis of an SV40 early promoter with only one
72-bp
sequence, they identified two broad domains, A and
B,
corresponding to theearly and late moieties, respectively, of the enhancer element used. Our results nowshow that some 6fthe sequences within domain B (nt 226 to 278) are also required for latetranscription. Notice,however,that wefind that deletion of this domian is less deleterious to late transcription than to early transcription. Moreau et al. (29) reported that the deletion of TB1l1causes adropof greater than 100-fold in the activity of the early promoter, whereas we find here that the same deletion reduces late promoteractivity three- to fivefold. It might be that the differential
effect of this deletion reflects the possible ability of the21-bp repeats to function as a substitute late promoter in the absence of the enhancer sequences, as suggested by Fromm
and Berg(14).
The second domain ofimportance (domain II, nt 179 to 204) functionedchiefly after the onset ofreplication. Thisis clearly demonstratedby the results obtained with the plas-midpOLA3 (Fig. 7B). Again we notice that the domain we identify here includes sequences that Zenke et al. (42) have shown are important for the early enhancer effect. The deletion in pOLA3 removes sequences belonging to the domain A describedbythese workers.
The finding that the enhancer element is required for late transcription both before and after replication would appear to be in disagreement with the results of Shaw et al. (34), who were unable to demonstrate that enhancer mutations have a deleterious effect on late transcription postreplica-tion. We do not know the basisforthis apparent contradic-tion.However,wenote thatthe resultsof Hartzell et al. (20) alsoindicate that sequences in the 72-bprepeats are impor-tantfor late geneexpression in vivopostreplication. Indeed, these authors found that the late distal 22-bpportion of the
72-bp repeat, corresponding inpositiontodomain II
identi-fiedhere,is sufficient for induction of late promoteractivity
when the minimal origin of replication is present. Other authors have alsoprovided evidence supportingthe impor-tance of the domainsidentified. Both Brady and Khoury (5) and Keller and Alwine (23) found that sequences in the
on November 10, 2019 by guest
http://jvi.asm.org/
enhancerregion are required for late geneexpression in the presence of T antigen. The latter authors further subdivided the sequences of importance into two domains, extending from nt 200 to 270 and nt 168 to 200, which are in good agreement with the domains describedhere. However, our results show that the presence of Tantigen is not required for the activation of domain I (nt 232 to 265).
Theexistence of elements of the latepromoter which are morecriticalafter replication can account for the factthat all the enhancer deletion mutants examined aremore deficient postreplication,sinceall these mutants lack at least one copy of this domain. Conversely, in none of the mutants has late transcription beencompletelyabolished. Inthisrespect,it is important to note that allmutants retain at least onecopy of eitherdomain. It is possible that each copy of each domain can function, to a certainextent, independentlyof the other important sequences (asZenke et al. [42]have demonstrated for the two domains of the earlyenhancer).Note that if this is so, then the results obtained with pSVLBS6 and pOLA3 suggest thatthedomain I (in the presenceof the +7 to -53 element) continues to function after replication. These re-sults are confirmed by a study of point mutations in this domain (manuscript in preparation).
The increasedimportance of the domain II after replica-tion suggests that there is a change in the operation ofthe latepromoterconcomitant withreplication. Wehave previ-ously observed thatthe 5' termini of late mRNAs are more heterogeneous after viralreplicationthan before (11), which
also suggests the occurrence of an alteration in the late
promoter. Itseems highly likely that this change reflects the
differential interaction ofatranscriptional factor(s) with the
promoter region. Indeed the domain II (nt 179 to 204)
virtually corresponds to the sequence identified by Keller
and Alwine (23) from nt 160 to 200, which appeared to be
responsible forT-antigen-mediated trans activation.
A striking observation made in this study was of the
potential identity ofelements within the enhancerrequired
for thestimulationofearly and latetranscription. Inlight of our results concerning the role of the G-C-rich region, we notice that this raises an interesting possibility concerning the change in strength of the later promoter pre- and
postreplication. We have already shown that, before
repli-cation, the location of the G-C-rich repeats between the enhancer and the early start sites causes transcription to proceed moreefficiently in the early direction (30). Results
presented here suggest that the G-C-rich region does not
favor earlytranscriptionto the same extent afterreplication.
Simultaneously, domainII(whichcorresponds in positionto
domain A of Zenke et al.[42]) becomes more criticalforlate
transcription. Takahashi et al. (37) have provided evidence
which strongly suggests thatprotein-protein contacts exist between factorsboundtotheG-C-rich regionand todomain A of theenhancer. Thus, it is possible that, whereas before
replication the G-C-rich region directs transcription chiefly
inthe earlydirection, afterreplication this effectis reduced anddomain II is now free to stimulate transcription in the late direction.
ACKNOWLEDGMENTS
We thank P. May forhelpful discussions.We thank C. Benoist, P. Moreau, M. Zenke, and P. Chambon for the generous gifts of the plasmids pSV1, HS102,TB1l1, TB208, andpAO. Wealso thank J. Borde and C.Breugnotfor their experttechnical assistance.Weare grateful to M. Maillot for thepreparationof this manuscript.
This work was supported by grant ATP003001 from the Centre National de laRecherche Scientifique.
LITERATURE CITED
1. Acheson,H. N.1981.Lytic cycleofSV40 andpolyoma virus, p. 125-204. In J. Tooze(ed.), DNA tumor viruses. Cold Spring HarborLaboratory, ColdSpring Harbor, N.Y.
2. Banerji, J.,S.Rusconi,and W. Schaffner. 1981. Expression of a
P-globin
gene is enhanced by remote SV40 DNA sequences. Cell 27:299-308.3. Benoist, C., and P. Chambon. 1981. In vivo sequence
require-ments of the SV40 early promoter region. Nature (London) 290:304-310.
4. Brady, J., B. J. Bolen, M. Radonovich, N. Salzman, and G. Khoury. 1984. Stimulation of simian virus 40 late gene expres-sionby simian virus 40tumorantigen. Proc. Natl. Acad. Sci. USA 81:2040-2044.
5. Brady,J.,andG.Khoury. 1985. transactivation of the simian virus 40 late transcription unit by T-antigen. Mol. Cell. Biol. 5:1391-1399.
6. Brady, J., M. R. Loeken, and G. Khoury. 1985. Interaction between two transcriptional control sequences required for tumor-antigen-mediated simian virus 40 late gene expression.
Proc. Natl. Acad.Sci. USA 82:7299-7303.
7. Brady, J., M. Radonovich, M. Vodkin, V. Natarajan, M. Thoren, G. Das, J. Janik,and N. P.Salzman. 1982. Site-specific base substitution and deletion mutations that enhance or sup-press transcription of the SV40 major late RNA. Cell 31:624-633.
8. Breathnach, R., and P. Chambon. 1981. Organization and
ex-pression of eucaryotic split genes coding for proteins. Annu. Rev. Biochem. 50:349-383.
9. Buchman, A. R., L. Burnett, and P. Berg. 1981. The SV40 nucleotide sequence, p. 799-829. In J. Tooze (ed.), DNAtumor
viruses. Cold Spring Harbor Laboratory,Cold Spring Harbor,
N.Y.
10. Dynan, W. S., and R.Tjian. 1983. The promoter-specific
tran-scription factorSpl binds toupstream sequences in the SV40 early promoter. Cell 35:79-87.
11. Ernoult-Lange, M.,and E.May. 1983.Evidence oftranscription from the late region of the integrated simian virus 40 genome in transformed cells: location of the 5' ends of late transcripts in cells abortively infected and in cells transformed by simian virus
40.J. Virol. 46:756-767.
12. Ernoult-Lange, M., P. May, P. Moreau, and E. May. 1984. Simian virus 40 late promoterregion abletoinitiate simian virus
40early gene transcription in the absence of the simian virus 40 origin sequence. J. Virol. 50:163-173.
13. Everett, R. D., D. Baty, and P. Chambon. 1983. The repeated G-C motif upstream from the TATA boxareimportantelements of the SV40 early promoter. Nucleic Acids Res.
11:2447-2464.
14. Fromm, M., and P. Berg. 1982. Deletion mapping of DNA regions required for SV40 early region promoter function in vivo. J. Mol. Appl. Genet. 1:457-481.
15. Ghosh,P.K., V. B.Reddy, J.Swinscoe, P.Lebowitz,andS. W. Weissman. 1978.Heterogeneity and 5' terminalstructuresof the lateRNAs of simian virus 40. J. Mol. Biol. 126:813-846. 16. Gluzman, Y. 1981. SV40-transformed simian cells support the
replication of early SV40 mutants. Cell 23:175-182.
17. Gruss, P., R. Dhar, and G. Khoury. 1981. Simian virus 40 tandemrepeated sequences as an element of the early promoter. Proc. Natl. Acad. Sci. USA 78:943-947.
18. Haegeman, G., H. VanHeuverswyn,D. Gheysen, and W. Fiers.
1979.Heterogeneity of the5'terminusof late mRNA inducedby
aviable simian virus 40 deletionmutant. J. Virol. 31:484-493. 19. Hansen, U., and P. A. Sharp. 1983. Sequences controlling in
vitro transcription of SV40 promoters. EMBO J. 2:2293-2303. 20. Hartzell, S. W., B. J. Byrne, andK. N. Subramanian. 1984.The
simian virus 40 minimal origin and the72 base pair repeat are
required simultaneously for efficient induction of late gene expression with large tumor antigen. Proc. Natl. Acad. Sci. USA 81:6335-6339.
21. Hirt, B. 1967. Selective extraction of polyoma from infected mouse cell cultures. J. Mol. Biol. 26:365-369.
on November 10, 2019 by guest
http://jvi.asm.org/
22. Keller,J. M., and J. C. Alwine. 1984. Activation of the SV40 latepromoter: direct effects of T antigen in the absence of viral DNAreplication. Cell 36:381-389.
23. Keller,J. M., and J. C. Alwine. 1985.Analysis ofanactivatable promoter: sequences in the simian virus 40 late promoter required for T-antigen-mediated trans activation. Mol. Cell. Biol. 5:1859-1869.
24. Lai, C., R. Dhar,and G. Khoury. 1978. Mapping of spliced and unspliced late lytic SV40 RNAs. Cell 14:971-982.
25. Lange, M., E. May, and P. May. 1981.Ability of nonpermissive mouse cells to express a simian virus 40 late function(s). J. Virol. 38:940-951.
26. Maxam, A. M., and W. Gilbert. 1980. Sequencing end-labeled DNAwith base-specific chemical cleavages. Methods Enzymol. 65:499-560.
27. Messing, J., R. Crea, and P H. Seeburg. 1981. A system for shotgunDNAsequencing. Nucleic Acids Res.9:309-321.
28. Mishoe, H., J. N. Brady, M. Radonovich, and N. P. Salzman. 1984. Simianvirus 40guanine-cytosine-rich sequences function
as independent transcriptional control elements in vitro. Mol. Cell. Biol. 4:2911-2920.
29. Moreau, P., R. Hen, B. Wasylyk, R. Everett, M. P. Gaub, and P. Chambon. 1981. The SV40 72 base pair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res. 9:6047-6067.
30. Omilli, F., M. Ernoult-Lange, J. Borde, and E. May. 1986. Sequences involved in the initiation of simian virus 40 late transcription in the absence of T antigen. Mol. Cell. Biol. 6:1875-1885.
31. Rigby, P.W. J., M.Dieckmann, C. Rhodes,and P.Berg. 1977. Labelling deoxyribonucleic acidtohighspecific activity in vitro by nick translation with DNA polymerase. J. Mol. Biol. 113:237-251.
32. Rio, D. C., and R. Tjian. 1984. Multiple control elements involved in the initiation of SV40 late transcription. J. Mol. Appl. Genet. 2:423-435.
33. Scherrer, K. 1969. Isolation and sucrose gradient analysis of RNA, p. 413-432. In K. Habel and N. P. Salzman (ed.), Fundamental techniques in virology. Academic Press, Inc., New York.
34. Shaw, P. E., D. Bohmann, and A. Sergeant. 1985. The SV40 enhancerinfluences viral late transcription in vitro and in vivo butnot onreplicating templates. EMBO J. 4:3247-3252. 35. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated by gel electrophoresis. J. Mol. Biol. 98:503-518.
36. Tack, L. C., andP.Beard. 1985. Bothtrans-acting factors and chromatinstructure areinvolvedin the regulation of transcrip-tion from the early and late promoters in simian virus 40
chromosomes. J. Virol. 54:207-217.
37. Takahashi, K., M. Vigneron, H. Matthes, A. Wildeman, M.
Zenke,and P. Chambon. 1986. Requirement ofstereospecific alignments for initiation from the simian virus 40 early
pro-moter.Nature(London) 319:121-126.
38. Vigneron, M., H. A. Barrera-Saldana, D. Baty, R. E. Everett, and P. Chambon. 1984. Effect of the 21-bp repeat upstream element on in vitrotranscription from the early and late SV40 promoters.EMBO J. 3:2373-2382.
39. Wasylyk, B., C.Wasylyk, P. Augereau,andP. Chambon. 1983. The SV40 72 bp repeat preferentially potentiates transcription starting from proximal naturalorsubstitute promoter elements. Cell 32:503-514.
40. Weiher, H., M. Konig, and P. Gruss. 1983. Multiple point mutations affecting the simian virus 40 enhancer. Science 219:626-631.
41. Weinberg,R.A.,Z.Ben-Ishai,andJ. E.Newbold. 1974.Simian virus40transcription in productivelyinfected and transformed cells. J. Virol. 13:1263-1273.
42. Zenke, M., T. Grundstrom, H. Matthes, M. Wintzerith, C. Schatz, A. Wildeman, and P. Chambon. 1986.Multiple sequence motifs are involved in SV40 enhancer function. EMBO J.
5:387-397.