organic papers
Acta Cryst.(2006). E62, o1291–o1292 doi:10.1107/S1600536806007823 Thorupet al. C
14H8N2
o1291
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
Acenaphtho[1,2-
b
]pyrazine
Niels Thorup,a* Jørgen Eskildsenb‡ and Jørn B. Christensenb
aDepartment of Chemistry, Technical University
of Denmark, Kemitorvet, DTU-207, DK-2800 Kgs. Lyngby, Denmark, andbDepartment of
Chemistry, University of Copenhagen, Universitetsparken 5, DK-2100 Copenhagen Ø, Denmark
‡ Present address: Acadia Pharmaceuticals AB PA, Hanssonsva¨g 35, S-20512 Malmo¨, Sweden.
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 120 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.043
wRfactor = 0.099
Data-to-parameter ratio = 11.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 2 March 2006 Accepted 3 March 2006
#2006 International Union of Crystallography All rights reserved
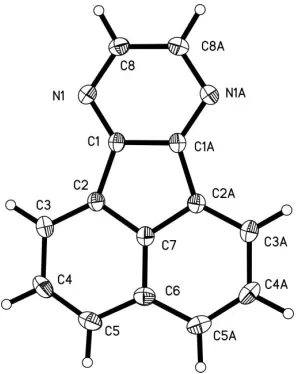
In the crystal structure of acenaphtho[1,2-b]pyrazine (or 7,10-diazafluoranthene), C14H8N2, the molecule has crystallo-graphicmsymmetry, but the observed symmetry is very close to mm2. The structure contains dimers of face-to-face antiparallel molecules.
Comment
Fluoranthene forms a series of 2:1 radical cation salts with anions such as PF6and AsF6(Enkelmannet al., 1982). 10c -Azoniafluoranthene forms a 1:1 radical cation salt with the anion PF6 (Boubekeuret al., 1989). The present molecule,
7,10-diazafluoranthene or acenaphtho[1,2-b]pyrazine, (I), is of interest as a modified fluoranthene with nitrogen substitution along the periphery.
The molecule has crystallographic m symmetry. As expected, the observed symmetry is very close tomm2 (C2v).
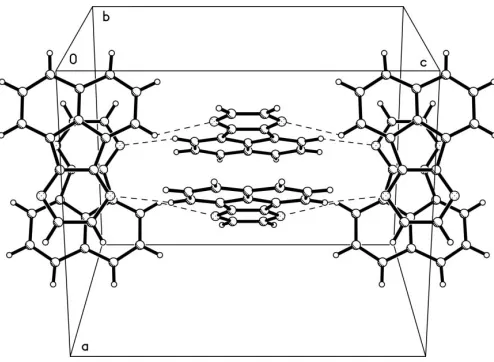
[image:1.610.253.401.515.702.2]The structure contains dimers of face-to-face antiparallel
Figure 1
molecules. The interplanar distance within a dimer is 3.417 (2) A˚ . The dimers probably interact via a weak C— H N hydrogen bond (see Table 2). The interacting dimers are stacked orthogonally to each other (see Fig. 2). The intermolecular attractions appear to be rather weak, which is in good accordance with the observed low melting point (ca 419 K).
Experimental
Acenaphtho[1,2-b]pyrazine was prepared as previously reported by Eskildsen & Christensen (2004). Crystals for X-ray analysis were obtained by slow evaporation of a solution in ethanol of a sublimed sample.
Crystal data
C14H8N2
Mr= 204.22 Tetragonal,P42=mbc
a= 11.9243 (2) A˚
c= 14.4040 (3) A˚
V= 2048.09 (6) A˚3
Z= 8
Dx= 1.325 Mg m 3
MoKradiation Cell parameters from 7188
reflections = 2.4–26.4 = 0.08 mm1
T= 120 (2) K Prism, yellow 0.300.090.06 mm
Data collection
Bruker SMART 1K CCD area-detector diffractometer !scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2003)
Tmin= 0.900,Tmax= 0.995
17778 measured reflections
1088 independent reflections 910 reflections withI> 2(I)
Rint= 0.052
max= 26.4
h=14!14
k=14!14
l=17!17
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.043
wR(F2) = 0.099
S= 1.14 1088 reflections 93 parameters
All H-atom parameters refined
w= 1/[2
(Fo2) + (0.041P)2
+ 0.8729P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.27 e A˚
3
min=0.20 e A˚
3
Extinction correction:SHELXTL
[image:2.610.47.294.69.250.2]Extinction coefficient: 0.0035 (6)
Table 1
Selected geometric parameters (A˚ ,).
N1—C1 1.3347 (17) N1—C8 1.3489 (18) C1—C1i
1.428 (3) C1—C2 1.4734 (19) C2—C3 1.376 (2) C2—C7 1.4179 (17)
C3—C4 1.424 (2) C4—C5 1.382 (2) C5—C6 1.4251 (18) C6—C7 1.406 (3) C8—C8i
1.392 (3)
C1—N1—C8 113.94 (12) N1—C1—C1i
122.76 (8) N1—C1—C2 128.87 (12) C1i
—C1—C2 108.36 (8) C3—C2—C7 119.34 (13) C3—C2—C1 135.20 (13) C7—C2—C1 105.45 (12) C2—C3—C4 117.84 (14)
C5—C4—C3 122.91 (14) C4—C5—C6 120.27 (15) C7—C6—C5 115.83 (10) C5—C6—C5i
128.3 (2) C6—C7—C2 123.81 (9) C2i—C7—C2 112.38 (17) N1—C8—C8i
123.29 (8)
Symmetry code: (i)x;y;z.
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C3—H3 N1ii 0.982 (17) 2.605 (16) 3.3663 (19) 134.5 (12) Symmetry code: (ii)y;x;zþ1
2.
Data collection:SMART(Siemens, 1995); cell refinement:SAINT
(Siemens, 1995); data reduction:SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2001); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL (Sheldrick, 2001) andPLATON(Spek, 2003); software used to prepare material for publication:SHELXTL.
References
Boubekeur, K., Fourmigue, M., Batail, P. & Bechgaard. K. (1989).Acta Cryst.
C45, 1636–1637.
Enkelmann, V. Morra, B. S., Kro¨hnke, C., Wegner, G. & Heinze, J. (1982).
Chem. Phys.66, 303–313.
Eskildsen, J. & Christensen, J. (2004).Molbank, 2004, M386.
Sheldrick, G. M. (2001).SHELXTL. Version 6.12. Bruker AXS Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (2003).SADABS. Version 2.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Siemens (1995). SMART and SAINT. Versions 4.05. Siemens AXS Inc., Madison, Wisconsin, USA.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Figure 2
[image:2.610.313.567.196.326.2]supporting information
sup-1
Acta Cryst. (2006). E62, o1291–o1292
supporting information
Acta Cryst. (2006). E62, o1291–o1292 [https://doi.org/10.1107/S1600536806007823]
Acenaphtho[1,2-
b
]pyrazine
Niels Thorup, J
ø
rgen Eskildsen and J
ø
rn B. Christensen
Acenaphtho[1,2-b]pyrazine
Crystal data
C14H8N2
Mr = 204.22
Tetragonal, P42/mbc
Hall symbol: -P 4c 2ab
a = 11.9243 (2) Å
c = 14.4040 (3) Å
V = 2048.09 (6) Å3
Z = 8
F(000) = 848
Dx = 1.325 Mg m−3
Melting point: 419 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 7188 reflections
θ = 2.4–26.4°
µ = 0.08 mm−1
T = 120 K Prism, yellow
0.30 × 0.09 × 0.06 mm
Data collection
Bruker SMART 1K CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scan, frame data integration Absorption correction: multi-scan
(SADABS; Sheldrick, 2003)
Tmin = 0.900, Tmax = 0.995
17778 measured reflections 1088 independent reflections 910 reflections with I > 2σ(I)
Rint = 0.052
θmax = 26.4°, θmin = 2.4°
h = −14→14
k = −14→14
l = −17→17
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.043
wR(F2) = 0.099
S = 1.14 1088 reflections 93 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
All H-atom parameters refined
w = 1/[σ2(F
o2) + (0.041P)2 + 0.8729P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.27 e Å−3
Δρmin = −0.20 e Å−3
Extinction correction: SHELXTL, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Least-squares planes (x,y,z in crystal coordinates) and deviations from them (* indicates atom used to define plane) 6.5712 (0.0024) x - 9.9501 (0.0016) y - 0.0666 (0.0102) z = 1.7028 (0.0010)
* -0.0046 (0.0012) C1 * -0.0123 (0.0013) C2 * -0.0064 (0.0012) C3 * 0.0020 (0.0012) C4 * 0.0105 (0.0014) C5 * 0.0056 (0.0014) C6 * -0.0084 (0.0015) C7 * 0.0063 (0.0010) C8 * 0.0074 (0.0011) N1
Rms deviation of fitted atoms = 0.0076
6.5712 (0.0024) x - 9.9501 (0.0016) y + 0.0666 (0.0102) z = 1.7028 (0.0010) Angle to previous plane (with approximate e.s.d.) = 0.53 (0.03)
* -0.0046 (0.0012) C1_$2 * -0.0123 (0.0013) C2_$2 * -0.0064 (0.0012) C3_$2 * 0.0020 (0.0012) C4_$2 * 0.0105 (0.0014) C5_$2 * 0.0056 (0.0014) C6_$2 * -0.0084 (0.0015) C7_$2 * 0.0063 (0.0010) C8_$2 * 0.0074 (0.0011) N1_$2 Rms deviation of fitted atoms = 0.0076
6.5674 (0.0023) x - 9.9528 (0.0015) y - 0.0000 (0.0000) z = 1.7086 (0.0005) Angle to previous plane (with approximate e.s.d.) = 0.27 (0.03)
* -0.0069 (0.0012) C1 * -0.0130 (0.0013) C2 * -0.0016 (0.0014) C3 * 0.0063 (0.0014) C4 * 0.0093 (0.0013) C5 * -0.0013 (0.0019) C6 * -0.0148 (0.0018) C7 * 0.0049 (0.0010) C8 * 0.0090 (0.0011) N1 * -0.0069 (0.0012) C1_$2 * -0.0130 (0.0013) C2_$2 * -0.0016 (0.0014) C3_$2 * 0.0063 (0.0014) C4_$2 * 0.0093 (0.0013) C5_$2 * 0.0049 (0.0010) C8_$2 * 0.0090 (0.0011) N1_$2
Rms deviation of fitted atoms = 0.0083
- 6.5674 (0.0023) x + 9.9528 (0.0015) y + 0.0000 (0.0000) z = 1.7086 (0.0005) Angle to previous plane (with approximate e.s.d.) = 0.00 (0.03)
* -0.0069 (0.0012) C1_$3 * -0.0130 (0.0013) C2_$3 * -0.0016 (0.0014) C3_$3 * 0.0063 (0.0014) C4_$3 * 0.0093 (0.0013) C5_$3 * -0.0013 (0.0019) C6_$3 * -0.0148 (0.0018) C7_$3 * 0.0049 (0.0010) C8_$3 * 0.0090 (0.0011) N1_$3 * -0.0069 (0.0012) C1_$4 * -0.0130 (0.0013) C2_$4 * -0.0016 (0.0014) C3_$4 * 0.0063 (0.0014) C4_$4 * 0.0093 (0.0013) C5_$4 * 0.0049 (0.0010) C8_$4 * 0.0090 (0.0011) N1_$4 - 3.4042 (0.0016) C2 - 3.4024 (0.0021) C7 Rms deviation of fitted atoms = 0.0083
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
N1 −0.03065 (10) −0.19279 (10) 0.09972 (8) 0.0204 (3) C1 0.04718 (11) −0.13985 (11) 0.04957 (9) 0.0174 (3) C2 0.14477 (11) −0.07484 (11) 0.08179 (9) 0.0183 (3) C3 0.18878 (13) −0.04694 (12) 0.16699 (10) 0.0236 (4) H3 0.1536 (13) −0.0705 (13) 0.2255 (12) 0.030 (5)* C4 0.28878 (13) 0.01825 (13) 0.16870 (11) 0.0290 (4) H4 0.3209 (15) 0.0372 (13) 0.2305 (12) 0.034 (5)* C5 0.34289 (13) 0.05366 (13) 0.08905 (11) 0.0285 (4) H5 0.4118 (16) 0.0982 (16) 0.0926 (11) 0.036 (5)*
C6 0.29894 (17) 0.02572 (17) 0.0000 0.0230 (5)
C7 0.19992 (17) −0.03826 (16) 0.0000 0.0185 (4)
supporting information
sup-3
Acta Cryst. (2006). E62, o1291–o1292 Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
N1 0.0210 (6) 0.0210 (6) 0.0193 (6) 0.0002 (5) 0.0027 (5) 0.0012 (5) C1 0.0198 (7) 0.0163 (7) 0.0160 (7) 0.0031 (5) 0.0005 (5) −0.0003 (5) C2 0.0201 (7) 0.0170 (7) 0.0179 (7) 0.0021 (5) 0.0002 (6) −0.0013 (5) C3 0.0268 (8) 0.0255 (8) 0.0183 (8) 0.0005 (6) −0.0010 (6) −0.0025 (6) C4 0.0304 (9) 0.0317 (9) 0.0248 (8) −0.0032 (7) −0.0077 (7) −0.0073 (7) C5 0.0254 (8) 0.0269 (8) 0.0333 (9) −0.0065 (6) −0.0040 (7) −0.0045 (7) C6 0.0229 (11) 0.0194 (10) 0.0265 (11) −0.0022 (8) 0.000 0.000 C7 0.0213 (10) 0.0165 (9) 0.0177 (10) 0.0022 (8) 0.000 0.000 C8 0.0202 (7) 0.0228 (7) 0.0250 (8) −0.0030 (6) 0.0022 (6) 0.0010 (6)
Geometric parameters (Å, º)
N1—C1 1.3347 (17) C4—C5 1.382 (2)
N1—C8 1.3489 (18) C4—H4 0.996 (18)
C1—C1i 1.428 (3) C5—C6 1.4251 (18)
C1—C2 1.4734 (19) C5—H5 0.98 (2)
C2—C3 1.376 (2) C6—C7 1.406 (3)
C2—C7 1.4179 (17) C8—C8i 1.392 (3)
C3—C4 1.424 (2) C8—H8 0.982 (17)
C3—H3 0.982 (17)
C1—N1—C8 113.94 (12) C3—C4—H4 117.5 (10)
N1—C1—C1i 122.76 (8) C4—C5—C6 120.27 (15)
N1—C1—C2 128.87 (12) C4—C5—H5 120.9 (10)
C1i—C1—C2 108.36 (8) C6i—C5—H5 118.9 (10)
C3—C2—C7 119.34 (13) C6—C5—H5 118.9 (10)
C3—C2—C1 135.20 (13) C7—C6—C5 115.83 (10)
C7—C2—C1 105.45 (12) C5—C6—C5i 128.3 (2)
C2—C3—C4 117.84 (14) C6—C7—C2 123.81 (9)
C2—C3—H3 122.2 (9) C2i—C7—C2 112.38 (17)
C4—C3—H3 119.9 (9) N1—C8—C8i 123.29 (8)
C5—C4—C3 122.91 (14) N1—C8—H8 116.4 (9)
C5—C4—H4 119.6 (10) C8i—C8—H8 120.3 (9)
C8—N1—C1—C1i −0.93 (14) C5—C6—C7—C7i 0.00 (4)
C8—N1—C1—C2 179.75 (13) C5i—C6—C7—C7i 0.00 (4)
N1—C1—C2—C3 0.2 (3) C6i—C6—C7—C2i 0.00 (6)
C1i—C1—C2—C3 −179.17 (15) C5—C6—C7—C2i −179.34 (17)
N1—C1—C2—C7i 179.33 (14) C5i—C6—C7—C2i 0.2 (3)
C1i—C1—C2—C7i −0.07 (12) C6i—C6—C7—C2 0.00 (6)
N1—C1—C2—C7 179.33 (14) C5—C6—C7—C2 −0.2 (3)
C1i—C1—C2—C7 −0.07 (12) C5i—C6—C7—C2 179.34 (17)
C7i—C2—C3—C4 0.1 (2) C3—C2—C7—C7i 0.00 (4)
C7—C2—C3—C4 0.1 (2) C1—C2—C7—C7i 0.00 (2)
C2—C3—C4—C5 −0.3 (2) C1—C2—C7—C6i −179.06 (17)
C3—C4—C5—C6i 0.3 (3) C3—C2—C7—C6 0.2 (3)
C3—C4—C5—C6 0.3 (3) C1—C2—C7—C6 −179.06 (17)
C4—C5—C6—C6i 0.00 (5) C3—C2—C7—C2i 179.39 (10)
C4—C5—C6—C7i 0.0 (3) C1—C2—C7—C2i 0.1 (2)
C4—C5—C6—C7 0.0 (3) C1—N1—C8—C8i 0.94 (14)
C4—C5—C6—C5i −179.51 (15)
Symmetry code: (i) x, y, −z.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C3—H3···N1ii 0.982 (17) 2.605 (16) 3.3663 (19) 134.5 (12)