1H Indole 3 carbaldehyde
Full text
Figure
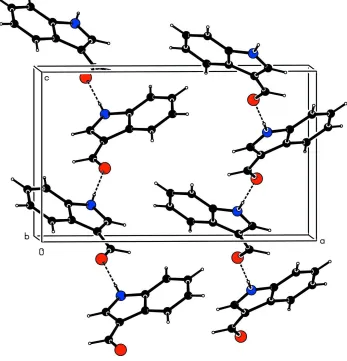
Related documents
In this study, we identified 9 protein markers for predicting time to recurrence using the protein expression data on 222 TCGA pri- marily high-grade serous ovarian cancers
For the purpose of analyzing the impurities in the water samples coming from different roofs, four building within the KCAET campus viz location 1(library -
To overcome the problems and weakness, this project need to do some research and studying to develop better technology. There are list of the objectives to be conduct
The above block diagram shows the SPV fed to Dc/Dc Converter for different dc applications, To analysis the performance of dc-dc converters(Buck, Boost,
22 subjects showing low or undetectable activities of BAT were randomly divided into 2 groups: one was exposed to cold at 17°C for 2 hours every day for 6 weeks (cold group; n
Foxo deletion on osteoblast differentiation in both bone marrow and calvaria cells suggests that the increases in ALP activity and mineralization observed in the bone
Histologically, the lesion is composed of fibrous connective tissue trabeculae (top quarter of image) and adipose connective tissue (bottom three quarters of image); within
• Data shows credit using and rationing of risk averts, risk neutrals and risk lovers respectively. As to risk averts, the credit is mainly used to pay children’s tuition, medical
