Janina Szeląg
1, Anna Sadakierska-Chudy
2, Jan Łyczek
3, Anna Paradowska
1Molecular Background of Ectodermal Dysplasia
Molekularne podłoże dysplazji ektodermalnej
1 Division of Facial Anomalies, Department of Dentofacial Orthopedics and Orthodontics, Wroclaw Medical University, Poland
2 Molecular Techniques Unit, Department of Forensic Medicine, Wroclaw Medical University, Poland 3 Student’s Scientific Association of Facial Anomalies, Wroclaw Medical University, Poland
Abstract
Ectodermal dysplasia is a disease of genetic background in which the main symptoms are anomalies of the teeth, hair, nails, and sweat glands. It occurs in 7/10,000 births and may be divided into 170 different syndromes. The main symptoms are weak thin hair, hypodontia and other dental anomalies, a small number of sweat glands, and malfor-mation and weakening of nails. The most common type of the disease is hypohidrotic ectodermal dysplasia related to chromosome X, which is Christ-Siemens-Touraine syndrome (Adv Clin Exp Med 2010, 19, 2, 257–269).
Key words: ectodermal dysplasia, gene, mutation, transcription factors, X-linked disease.
Streszczenie
Dysplazja ektodermalna jest chorobą o etiologii genetycznej, której kluczowymi objawami są nieprawidłowości zębów, włosów, paznokci i gruczołów potowych. Do schorzeń typu dysplazji zalicza się ponad 170 jednostek cho-robowych wspólnie występujących z częstością 7/10 000 urodzeń. Objawami dysplazji ektodermalnej są osłabione i cienkie włosy, hipodoncja i zniekształcenia zębów, zmniejszenie liczby gruczołów potowych oraz zniekształcenie i osłabienie paznokci. Najczęściej występującym typem schorzenia jest hipohydrotyczna dysplazja ektodermalna sprzężona z chromosomem X, czyli zespół Christ-Siemens-Touraine (Adv Clin Exp Med 2010, 19, 2, 257–269). Słowa kluczowe: dysplazja ektodermalna, gen, mutacja, czynniki transkrypcyjne, choroba sprzężona z chromo-somem X.
Adv Clin Exp Med 2010, 19, 2, 257–269 ISSN 1230-025X
rEvIEWS
© Copyright by Wroclaw Medical University
Ectodermal dysplasia is a hereditary disorder whose major manifestations are abnormalities of the teeth, hair, nails, and sweat glands. There are more than 170 different disorders regarded as ectodermal dysplasia, occurring with a fre-quency of 7 in 10,000 births (Tab. 1) [1, 2]. The crucial symptoms are hypodontia, malformed teeth, decreased number of sweat glands, sparse and fine hair, and deformed nails [3]. However, the particular disorders have different manifes-tations which can also involve other structures, such as the nervous system or thyroid glands. Several classifications of ectodermal dysplasia have been introduced. The first, proposed by Pinheiro and Freire-Maia (1994), was based on clinical manifestations. Ectodermal dysplasia was a disease with abnormalities of at least two ecto-dermal appendages. Gradual elucidation of the
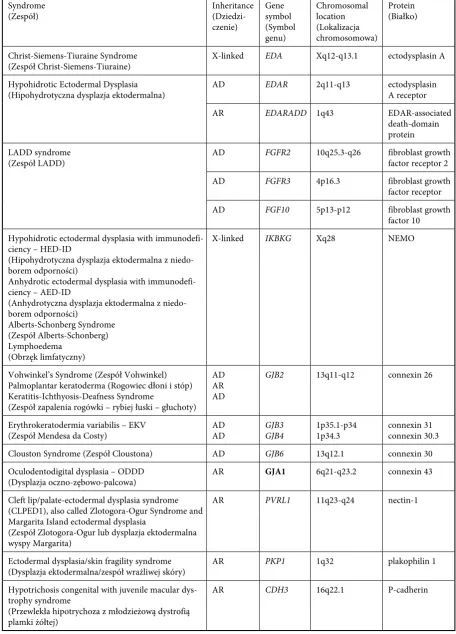
caus-Table 1. Genes and coded molecules in molecular dysplasias
Tabela 1. Geny i kodowane molekuły w dysplazjach molekularnych Syndrome
(Zespół) Inheritance (Dziedzi-czenie)
Gene symbol (Symbol genu)
Chromosomal location (Lokalizacja chromosomowa)
Protein (Białko)
Christ-Siemens-Tiuraine Syndrome
(Zespół Christ-Siemens-Tiuraine) X-linked EDA Xq12-q13.1 ectodysplasin A Hypohidrotic Ectodermal Dysplasia
(Hipohydrotyczna dysplazja ektodermalna) AD EDAR 2q11-q13 ectodysplasin A receptor
Ar EDARADD 1q43 EDAr-associated
death-domain protein LADD syndrome
(Zespół LADD) AD FGFR2 10q25.3-q26 fibroblast growth factor receptor 2
AD FGFR3 4p16.3 fibroblast growth
factor receptor
AD FGF10 5p13-p12 fibroblast growth
factor 10 Hypohidrotic ectodermal dysplasia with
immunodefi-ciency – HED-ID
(Hipohydrotyczna dysplazja ektodermalna z niedo-borem odporności)
Anhydrotic ectodermal dysplasia with immunodefi-ciency – AED-ID
(Anhydrotyczna dysplazja ektodermalna z niedo-borem odporności)
Alberts-Schonberg Syndrome (Zespół Alberts-Schonberg) Lymphoedema
(Obrzęk limfatyczny)
X-linked IKBKG Xq28 NEMO
vohwinkel’s Syndrome (Zespół vohwinkel) Palmoplantar keratoderma (rogowiec dłoni i stóp) Keratitis-Ichthyosis-Deafness Syndrome
(Zespół zapalenia rogówki – rybiej łuski – głuchoty) AD Ar AD
GJB2 13q11-q12 connexin 26
Erythrokeratodermia variabilis – EKv
(Zespół Mendesa da Costy) AD AD GJB3 GJB4 1p35.1-p34 1p34.3 connexin 31 connexin 30.3 Clouston Syndrome (Zespół Cloustona) AD GJB6 13q12.1 connexin 30 Oculodentodigital dysplasia – ODDD
(Dysplazja oczno-zębowo-palcowa) Ar GJA1 6q21-q23.2 connexin 43 Cleft lip/palate-ectodermal dysplasia syndrome
(CLPED1), also called Zlotogora-Ogur Syndrome and Margarita Island ectodermal dysplasia
(Zespół Zlotogora-Ogur lub dysplazja ektodermalna wyspy Margarita)
Ar PVRL1 11q23-q24 nectin-1
Ectodermal dysplasia/skin fragility syndrome
(Dysplazja ektodermalna/zespół wrażliwej skóry) Ar PKP1 1q32 plakophilin 1 Hypotrichosis congenital with juvenile macular
dys-trophy syndrome
(Przewlekła hipotrychoza z młodzieżową dystrofią plamki żółtej)
Ar CDH3 16q22.1 P-cadherin
ing particular diseases, less than 30 types of dys-plasia have a fully elucidated background.
The aim of this study was to present the molec-ular and genetic causes of ectodermal dysplasias. The paper supports Lamartin’s classification as it directly corresponds to the topic of the study. Table 1 presents information on genes and coded molecules in molecular dysplasias and Table 2 pres-ents basic information on genetic terminology.
Signalization
Teeth, hair, nails, and sweat glands are ecto-dermal appendages and their development strictly depends on communication between the ecto-derm and mesoecto-derm. Despite obvious differenc-es in final morphology, the early stagdifferenc-es of their development are regulated by common mediators. The consequence is that deficiency of one media-tor caused by mutation in one gene disables the proper development of all ectodermal appendages except nails.
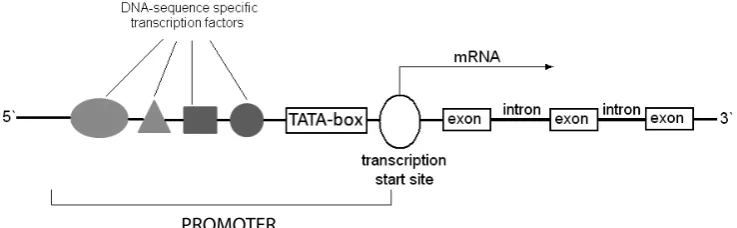
The most frequent form of the disorder is X-linked hypohidrotic ectodermal dysplasia, also known as Christ-Siemens-Touraine syndrome [6]. It occurs with a frequency of 1 in 100,000 births [7]. The major symptoms are individually consistent and comprise fine and sparse hair, hypodontia, microdontia, malformed teeth, and decreased number of sweat glands. Moreover, a prominent forehead, hypoplastic maxilla, and low nose base may be present. Hypohidrotic dysplasia is caused by mutations in the EDA gene located on chromo-some X at locus q12-q13.1 [3]. EDA codes ectodys-plasin A, a transmembrane protein that belongs to the tumor necrosis factor ligand family [8]. One of the most important functions of ectodysplasin is the activation of nuclear factor-κB (NF-κB tran-scription factor). A trantran-scription factor is a pro-tein that binds to a specific sequence of DNA, such as the promoter, enhancer, or silencer. All transcription factors contain one or more DNA
binding domains and an activator domain. The transcription factor together with other proteins as a complex can stimulate or repress the tran-scription of specific genes. Without trantran-scription factors, copying rNA from DNA cannot occur. These proteins can be activated or inhibited by physiological, therapeutic, environmental, and pathological stimuli (i.e. mutations) (Fig. 1).
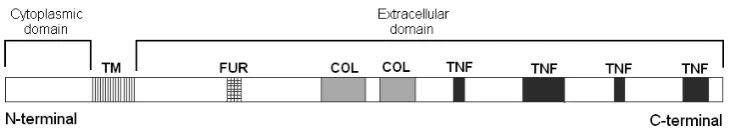
NF-κB is responsible for regulating the tran-scription of many genes important for bone morphogenesis, proliferation and differentiation of keratinocytes, the formation of ectodermal appendages, and stimulation of inflammation and an immunological response [9–11]. Several splice isoforms of EDA gene have been identified. The most common and longest ectodysplasin isoforms, EDA-A1 (391 amino acids) and EDA-A2 (389 amino acids), differ from each other only by the presence of Gln 308 and val 309 in the receptor binding region of EDA-A1 (Fig. 2). This two-ami-no-acid (aa) difference confers receptor specificity [12,13]. EDA-A1 is essential for the development of ectodermal appendages and binds to ectodys-plasin receptor (EDAr). EDA-A2 is responsible for the synchronization and finalization of this process and binds to X-linked EDA-A2 receptor (XEDAr) [14]. Ectodysplasin contains a small N-terminal cytoplasmic domain and a large C-terminal extracellular domain. An extracellu-lar domain contains TNF-homologous regions, a (Gly-X-Y)19 collagen repeat, and a sequence of
seven amino acids that is recognized by furinase. The proper structure of every protein motif is crucial for the activity of ectodysplasin. The TNF-homologous region is responsible for pro-tein folding and receptor binding. The function of the collagen-like region is the multimerization of ectodysplasin homotrimers. The 7-amino-acid sequence is recognized by furin protease; cleav-age by this enzyme releases the soluble C-terminal fragment with TNF motifs. Mutations are located in the region of EDA that encodes a sequence of TNF regions that cause disability of EDA to bind
Fig. 1. Structure of the promoter and coding region of the gene Ryc. 1. Struktura promotora i regionu kodującego genu
to its receptor. Improprieties in the structure of the collagen-like repeat disallow multimerization of EDA homotrimers, which decreases its biologi-cal activity. Changes in the region of 7-amino-acid sequence cause furinase to cleave EDA at an inap-propriate position, i.e. Arg 159. This decreases the solidity of EDA and inhibits its function. Mutations in all motifs can trigger hypohidrotic ectoder-mal dysplasia, depending on the level of inhibi-tion of EDA function. The disorder is inherited as X-linked, with low penetration [15]. The conse-quence is that manifestations differ in severity in men and women. Men, having only one X chro-mosome, always develop the entire spectrum of symptoms. In addition to ectodermal append-ages there are also abnormalities of hypoplastic maxilla, prominent forehead, low and broad base of the nose, and wrinkles around the eyes [16]. Women, in contrast, having two X chromosomes, may be homo- and heterozygous mutation carri-ers. Heterozygous patients have milder symptoms with microdontia, hypodontia, and increased body temperature caused by the lower number of sweat glands and improprieties in peripheral blood cir-culation [17, 18]. If a person is a homozygous car-rier, the syndrome is much more severe. A case of twins was described in whom the proper chromo-some X was inactivated and the second one had a mutation in the EDA gene. One of the girls died at the age of 2.5 years because of airway obstruc-tion by epithelial debris. Autopsy revealed an absolute lack of sweat, mucous, and ceruminous glands [19].
Autosomal hypohidrotic ectodermal dysplasia can also be caused by mutations in two other genes:
EDAR and EDARADD. Nevertheless, mutations in
EDA are the most frequent reason triggering the X-linked disorder; they evoke 63% of cases in the whole patient population and 95% of cases among the males [20]. It has been shown that the clinical images of the disorder caused by mutations in all three genes are indistinguishable [21].
The EDAR gene is located on chromosome 2 at locus q11-q13 and encodes a protein called
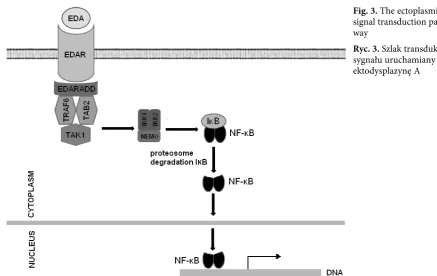
EDAr, which belongs to the TNF receptor super-family [22]. EDAr transcript consists of a sig-naling sequence, an extracellular cysteine-rich domain responsible for EDA binding, a trans-membrane fragment, and an intracellular domain which is a putative death domain. The function of the death domain is transduction of a sig-nal to the EDArADD-EDAr-associated death domain which eventually activates NF-κB [22–24]. Mutations inhibiting the function of EDAr are mainly located in the ligand-binding domain and death domain. Mutations in the binding domain decrease to EDA’s affinity to bind to its receptor, which inhibits signal transduction [14]. Missense and nonsense mutations cause premature termi-nation of translation or incorrect splicing position, which leads to improper structure of the death domain or its complete lack [24]. This results in inability of the death domain to form homotri-mers, which makes the interaction between EDAr and EDArADD receptor impossible and, finally, NF-κB is not activated [21, 25, 27].
Whereas mutations of EDA are X-linked, mutations of EDAR are both autosomal recessive and autosomal dominant. Mutations in regions encoding the extracellular fragment are mainly recessive, but mutations occurring in the intracel-lular region are dominant [22]. Patients with auto-somal dominant mutations are more heat tolerant and have milder hair abnormalities than carriers of autosomal recessive mutations [28]. Mutations
of EDAR account for 25% of all cases that are not
caused by mutations of EDA [22, 29].
Hypohidrotic ectodermal dysplasia may also be caused by mutations in EDARADD gene, located on chromosome 1 at locus q42.2-q43 [30].
EDARADD encodes an EDAr-associated death
domain. It is essential for the intracellular trans-duction of signal to NF-κB [25]. receptors (e.g. EDAr) belonging to the TNFr superfamily con-tain a death domain which is used to form an intracellular complex that activates the next ele-ment of the signaling path, NF-κB in this case [31–33]. EDArADD is essential for the formation Fig. 2. Schematic drawing of ectodysplasin A protein showing domains and predicted protein motifs. TM – trans-membrane domain, FUr – putative furin cleavage sequence, COL – collagen-like region, TNF – regions homologous to tumor necrosis factor
of this complex. Contrary to other TNF receptors, EDAr does not transduce a signal for apoptosis, but for the development and differentiation of cells. Mutations in the death domain-encoding region of EDARADD result in improper struc-ture or lack of the death domain, which makes the formation of a transducing complex impos-sible [27]. EDArADD also contains the sequence Pro-X-Gln-X-Thr, which is responsible for bind-ing Traf, which are needed for signal transduction. However, mutations in the region encoding this sequence do not significantly decrease NF-κB acti-vation (Fig. 3). Mutations in EDARADD gene can be both autosomal dominant and recessive [30].
Lacrimo-auriculo-dento-digital (LADD) syn-drome is characterized by hypoplasia or aplasia of the lacrimal puncta and obstruction of naso-lacri-mal ducts, naso-lacri-malformation of the auricula and hear-ing impairment, microdontia of the upper lateral incisors, enamel dysplasia, syndactyly, and clinod-actyly (fifth finger curved like an arch towards the other fingers) [34]. The disorder is inherited as an autosomal dominant trait. Mutations causing the disease occur in genes encoding fibroblast growth factor (FGF) receptors (FGFR2 and FGFR3) and the gene encoding FGF-10 (FGF10) [35]. FGFs play a crucial role in the transduction of signals for migration, proliferation, and differentiation dur-ing the development of the embryo. Signalization with FGFs also takes place between the ectoderm and mesoderm, which is essential for ectodermal appendage development [36]. FGF receptors con-sist of two or three extracellular
immunoglobulin-like domains that bind heparin, the transmem-brane region, and intracellular tyrosine kinase [37]. Mutations causing LADD syndrome occur in the regions of FGFR2 (10q26) and FGFR3 (4p16.3) that encode tyrosine kinase and result in a decrease in its activity. This inhibits the transduction of sig-nals and eventually triggers the disorder. LADD syndrome can also be caused by mutations in the
FGF10 gene [35].
IKBKG gene is located on chromosome X at
locus 28and encodes IKK-γ (inhibitor of kappa-B kinase gamma), also known as NEMO (NF-κB essential modulator) [38]. NEMO is a crucial regu-lator of activation of NF-κB [39]. It is a part of the IKK complex, which consists of IKK-α, IKK-β, and NEMO. The role of this complex is phosphoryla-tion of IκB, inhibitors of NF-κB. Phosphorylaphosphoryla-tion of IkB is then followed by their ubiquitination and degradation in proteasomes. This releases NF-κB, which is then transported to the nucleus and regu-lates the transcription of particular genes. NEMO consists of four domains: an N-terminal spiral domain, a C-terminal spiral domain, a zinc fin-ger domain, and a leucine zipper. The N-terminal spiral domain is responsible for binding to IKKs [40, 41]. The C-terminal domains play a role in the homotrimerization of NEMO [41–43]. It is supposed that the leucine zipper is needed for the ubiquitination of NEMO [44–46]. Several NEMO mutations are located in the C-terminal encoding region. Changes in the structure of the C-terminal spiral domain and leucine zipper may affect homotrimerization. Aberrations in the
leu-Fig. 3. The ectoplasmin signal transduction path-way
cine zipper disable full activation of the IKK com-plex. NEMO-dependent inhibition of NF-κB leads to skin and immunological disorders. Deletion of exons 4-10 causes incontinentia pigment type 2, with such manifestations as skin hyperpigmenta-tion and abnormalities of the eyes, teeth, bones, and other organs [47]. Changes in the structure of NEMO cause hypohidrotic ectodermal dyspla-sia with immunodeficiency (HED-ID), which is characterized by low immunoglobulin level and recurrent infections. Premature termination of transcription causes a more severe type, i.e. anhi-drotic ectodermal dysplasia with immunodeficien-cy (AED-ID), Albers-Schonberg syndrome (osteo-sclerosis, fragile bones, teeth malformations), and lymphoedema [48].
Some of the proteins essential for intercellular signaling are connexins which form nexus junc-tions. They are used for the exchange of molecules such as ions, metabolites, and second messengers smaller than 1000 Da [49]. In the skin, nexus junc-tions are important for the development and dif-ferentiation of keratinocytes, whereas in cochlear epithelium they convey potassium ions. In every cell, six connexins form a connexon (Fig. 4). Connexons of two adjacent cells connect and the nexus junction is created. Connexons can consist of one of several kinds of connexins. The result is that they are homo- or heteromers and they differ in conductance and selectivity [50, 51]. Mutations of genes encoding connexins result in hearing impairment and improper development of skin and its appendages.
The gene GJB2 is located on chromosome 13 at locus q11-12 [52]. It encodes connexin 26 and consists of two exons, one of which is not trans-lated [53]. Mutations in GJB2 cause impaired association of connexins and the connection of connexons as well as changes in the conductance of particular molecules [54]. This can result in several disorders such as vohwinkel`s syndrome (hyperkeratosis, hearing impairment), palmo-plantar keratoderma (hyperkeratosis of the palms and soles), and Keratisis-Ichthyosis-Deafness (KID) syndrome [49]. Mutations of the GJB3 and
GJB4 genes evoke erythrokeratoderma variabilis [55, 56]. Mutations of the GJB6 gene encoding con-nexin 30 cause hidrotic ectodermal dysplasia, also known as Clouston syndrome (presence of sweat and ceruminous glands, complete lack of hair, nail dystrophy, skin hyperpigmentation, lack of dental abnormalities) [49, 57]. Mutations of the GJA1
gene causeoculodentodigital dysplasia character-ized by microphthalmia, small nose, sparse hair, dental deformations, and syndactyly [58]. All dis-orders caused by mutations in connexin-encoding genes are inherited as autosomal dominant traits.
Adhesion Molecules
Intercellular junctions ensure the consistence and durability of the epithelium. resistance to mechanical factors depends on tight junctions, adherens junctions, and desmosomes. The junc-tions consist of proteins called adhesion molecules. These are cadherins in tight and adherens junc-tions and desmogleins and desmokolins in desmo-somes. These two kinds of junctions connect not only cell the membrane, but also the cytoskeleton, which ensures high consistence and mechani-cal resistance. Structural alternations of adhesion proteins will decrease the mechanical durability of epithelium.
PVRL1 gene is located on chromosome 11 at
locus 23 and encodes nectin-1 [59, 60]. Nectins are a family of five transmembrane immunoglobulin-associated proteins. They consist of five domains: an extracellular immunoglobulin-like v-shaped domain and two immunoglobulin-like C-shaped domains, a transmembrane sequence, and an intra-cellular C-terminal domain [61]. The extraintra-cellular fragments of nectins form dimers which connect with identical dimers located on an adjacent cell. The intracellular terminus of nectins is connect-ed through afadin with actin filaments [61–63]. Nectin-1 is also a basic receptor for the internal-ization of herpes simplex virus [64]. Mutations
in PVRL1 result in hidrotic ectodermal dysplasia,
also known as Margarita Island or Zlotogora-Ogur
Fig. 4. Schematic model illustrating the assembly of connexins into gap junctions. Six connexins oligomerize into connexons, which combine into gap junction channels
syndrome [65]. The disorder has an autosomal recessive mode of inheritance [66]. Manifestations of the syndrome are cleft palate and lip, fine and sparse hair, deformed auricula, syndactyly, insuf-ficient eyebrows, and severe teeth abnormalities, particularly frequent hypodontia of the upper lat-eral incisors [65–69].
PKP1 gene encodes plakophilin (PKP)-1, a protein essential for the proper formation of desmosomes (Fig. 5) [70–73]. PKP1 is located on chromosome 1 at locus 32 [74]. There are at least two different isoforms: PKP-1a (126 amino acids) and PKP-1b (747 aa). Isoform 1b is generated by alternative splicing, during which exon 7 encoding 21 aa is excised. PKP-1b protein occurs exclusively in the nucleus and plays a role in signal transduc-tion [75]. PKP-1a has been found in the nucleus
and desmosomal plaque. PKP-1a is responsible for the organization and stabilization of proteins of the desmosomal plaque [76]. Mutations of PKP-1
gene cause improper protein structure, which results in decreased the number and size of des-mosomes (Fig. 6) [77]. This leads to extension of the intercellular space [78]. Ectodermal dysplasia with skin fragility is evoked with other manifesta-tions, such as nail dystrophy, hyperkeratinization of the palms and soles, decreased sweating, and sparse hair [72, 79].
CDH3 gene is located on chromosome 16 at
locus q22.1 [80]. Itencodes P-cadherin, a protein essential for the formation of tight junction and punctum adherens. It is expressed intensively in the retinal epithelium and hair follicles. P-cadherin is a transmembrane protein consisting of an extra-cellular domain, a transmembrane fragment, and an intracellular tail. The extracellular domain con-tains five sequences characteristic of all cadherins which is called the cadherin motif/EC domain. They play a crucial role in Ca2+-dependent
inter-cellular junctions [81, 82]. A parallel connection is made between the two extracelluar fragments of P-cadherin. Such a dimer connects to an identical one on the adjacent cell [83]. The intracellular tails
Fig. 5. Cellular location of desmosome and gap junc-tion
Ryc. 5. Komórkowa lokalizacja desmosomów i połączeń szczelinowych
Fig. 6. The desmosomal plaque component. Dsg – desmoglein, Dsc – desmocollin, DP – desmoplakin, IF – cytoskeleton components, PKP – pakophilin, Pg – plakoglobin
Ryc. 6. Elementy płytki desmosomalnej. Dsg – desmo-gleina, Dsc – desmokolina, DP – desmoplakina, IF – elementy cytoszkieletu, PKP – pakofilina, Pg – plakoglobina
Fig. 7. Schematic model of P-cadherin structure and cellular transduction pathway
bind to β-catenin and γ-catenin [84]. β-catenin connects with α-catenin, which binds directly to actin filaments (Fig. 7) [85]. Two kinds of muta-tions of CDH3 gene (missense and frameshift) result in hypotrichosis with juvenile macular dys-trophy (HJMD) syndrome. The disorder is char-acterized by fine, twisted hair, early hair loss, and progressive retinal atrophy leading to vision loss between 20 and 30 years of age [86]. The disease is inherited as an autosomal recessive trait. The
frameshift mutation causes a premature termina-tion of transcriptermina-tion. The truncated protein, lack-ing the intracellular tail and three EC domains, cannot interact with β-catenin, which is essential for hair follicle development [87–89]. This muta-tion type has been identified in 11 pedigrees of 4 consanguineous families of Muslim origin [87].
A missense mutation in the eleventh exon of
CDH391 affects the structure of the Ca2+-binding
domain [80, 82]. It reduces the ability to bind
Table 2. Basic definitions of molecular biology
Tabela 2. Podstawowe definicje biologii molekularnej Adhesion molecules
(Cząsteczki adhezyjne) extracellular and cell-surface proteins mediating in cell-cell and cell-extracellular matrix adhesion Alternative splicing
(Splicing alternatywny) mechanism in which introns are removed from the primary gene transcript, resulting in various protein isoforms Codon (Kodon) a sequence of three nucleotides that encode one amino acid
Enhancer (Enhancer) a short region of DNAbinding activator proteins and increasing the transcription level Exon (Egzon) coding sequence of DNA present in mature messenger rNA; it encodes the amino-acid
product or some types of rNA Frameshift mutation
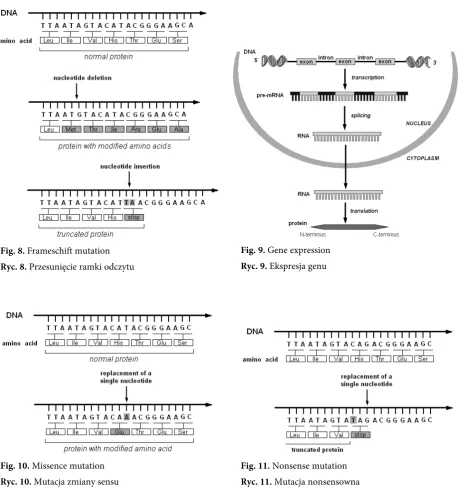
(Zmiana ramki odczytu) a nucleotide insertion or deletion can change the reading frame, resulting in a different amino-acid sequence or a truncated protein (Fig. 8) Gene expression
(Ekspresja genu) the process by which a nucleotide sequence of a gene is translated into rNA (Fig. 9) Heterodimer protein
(Białko heterodimerowe) two different polypetides forming a protein subunit Homodimer protein
(Białko homodimerowe) two identical polypetides forming a protein subunit
Intron (Intron) non-coding sequence of DNA spliced out of the pre-mature messenger rNA prior to translation
Messenger rNA (mrNA) rNA that serves as a template for protein synthesis Missense mutation
(Mutacja zmiany sensu) mutation in DNA molecule leading to amino-acid changes (Fig. 10) Nonsense mutation
(Mutacja nonsensowna) substitution of single base pair which introduces a stop codon instead of an amino-acid codon (Fig. 11) Promoter (Promotor) the specific region of a gene that acts as a binding site for transcription factors and rNA
polymerase. The process of transcription is initiated at the promoter Signal transduction
(Przewodzenie sygnału) the intercellular or intracellular transfer of information through a signaling pathway. It can come from a biologically active molecule (hormone, neurotransmitter) and can trans-mit an activation/inhibition signal via a specific receptor
Silencer
(Sekwencja wyciszająca) control elements binding repressors and decreasing or fully suppressing rNA synthesis Start or initiation codon
(Kodon „start”) codon ATG (or AUG in rNA) encoding the amino-acid methionine (Met) Stop or termination codon
(Kodon „stop”) a triplet nucleotide within mrNA (UAG, UAA, or UGA) which signals the end of a poly-peptide chain Transcription (Transkrypcja) copy a DNA sequence to rNA; this is the first step in gene expression
Ca2+ ions and alters the proper conformation of
the EC4 domain, finally decreasing the capabil-ity of P-cadherin to form intercellular junctions and impairing its function in cell differentiation. This mutation type has been identified in a large Arabic-Israeli family; four out of six children whose parents were first degree cousins developed the syndrome [90].
Missense mutations in the eighth exon of
CDH3 can also result in ectodermal dysplasia
with ectrodactyly (lack of one or more fingers or toes with cleft palm or foot) and retinal dys-trophy. In this case, binding of Ca2+ ions is also
impaired. In an affected family, the parents also
were first degree cousins and heterozygous muta-tion carriers. Both chromosomes inherited by the patient had mutated CDH3 [91]. Ectodermal dys-plasia with ectrodactyly and macular dystrophy (EEM syndrome) can be triggered by a frameshift mutation in the seventh exon of CDH3 gene. It produces truncated P-cadherin lacking both the intracellular and transmembrane domains as well as its extracellular domains 3–5. In an affect-ed Brazilian family the parents, a brother, and a son of the probant were heterozygous carriers [91, 92]. The brother had syndactyly of fingers 1–4 and microdontia, but no retinal dystrophy or sparse hair.
Fig. 8. Frameschift mutation Ryc. 8. Przesunięcie ramki odczytu
Fig. 9. Gene expression Ryc. 9. Ekspresja genu
Fig. 10. Missence mutation Ryc. 10. Mutacja zmiany sensu
References
Pinheiro M, Freire-Maia N:
[1] Christ-Siemens-Touraine syndrome – a clinical and genetic analysis of a large Brazilian kindred. I. Affected females. II. Affected males. III. Carrier detection. Am J Med Genet 1979,4, 113–134.
McKusick V:
[2] Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. Baltimore, MD: Johns, Hopkins University Press 1994.
Kere J, Srivastava AK, Montonen O, Zonana J, Thomas N, Ferguson, B, Munoz F, Morgan D, Clarke A, [3]
Baybayan P, Chen EY, Ezer S, Saarialho-Kere U, de la Chapelle A, Schlessinger D: X-linked anhidrotic (hypo-hidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein. Nat Genet 1996, 13(4), 409–416.
Priolo M, Lagana C:
[4] Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet 2001, 38, 579–585. Lamartine J:
[5] Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol 2003, 28, 351–355. Reed WB, Lopez DA, Landing B:
[6] Clinical spectrum of anhidrotic ectodermal dysplasia. Arch Dermatol 1970,
102, 2, 134–143.
Stevenson AC, Kerr CB:
[7] On the distribution of frequencies of mutation to genes determining harmful traits in man. Mutat res 1967, 4, 3, 339–352.
Schneider P, Street SL, Gaide O, Hertig S, Tardivel A, Tschopp J, Runkel L, Alevizopoulos K, Ferguson BM, [8]
Zonana J: Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J Biol Chem 2001, 276, 22, 18819–18827.
Schmidt-Ullrich R, Aebischer T, Hülsken J, Birchmeier W, Klemm U, Scheidereit C:
[9] requirement of
NF-kappaB/rel for the development of hair follicles and other epidermal appendices. Development 2001, 128, 19, 3843–3853.
Hayden MS, Ghosh S:
[10] Signaling to NF-kappaB. Genes Development 2004, 18, 2195–2224. Karin M, Ben-Neriah Y:
[11] Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu rev Immunol 2000, 18, 621–663.
Hymowitz D, Compaan M, Yan H, Wallweber V, Dixit M, Starovasnik A, de Vos:
[12] The Crystal Structures of
EDA-A1 and EDA-A2 Splice variants with Distinct receptor Specificity. Structur 2003, 11, 12, 1513–1520 S. Zhang XJ, Chen JJ, Song YX, Yang S, Xiong XY, Zhang AP, He PP, Gao M, Li YB, Lin D, Huang W:
[13] Mutation
analysis of the ED1 gene in two Chinese Han families with X-linked hypohidrotic ectodermal dysplasia. Arch Dermatol res 2003, 295, 38–42.
Lind K, Stecksén-Blicks Ch, Lejon K, Schmitt-Egenolf M:
[14] EDAr mutation in autosomal dominant hypohidrotic
ectodermal dysplasia in two Swedish families. BMC Med Genet 2006, 7, 80–??
Zonana J, Clarke A, Sarfarazi M, Thomas NST, Roberts K, Marymee K, Harper PS:
[15] X-linked hypohidrotic
ectodermal dysplasia: localization within the region Xq11-21.1 by linkage analysis and implications for carrier detection and prenatal diagnosis. Am J Hum Genet 1998, 43, 75–85.
Pinheiro M, Freire-Maia N:
[16] Christ-Siemens-Touraine syndrome – a clinical and genetic analysis of a large Brazilian kindred. I. Affected females. II. Affected males. III. Carrier detection. Am J Med Genet 1979, 4, 113–134. Nakata M, Koshiba H, Eto K, Nance WE:
[17] A genetic study of anodontia in X-linked hypohidrotic ectodermal dysplasia. Am J Hum Genet1980, 32, 908–919.
Clark RP, Goff MR, MacDermot KD:
[18] Identification of functioning sweat pores and visualization of skin tem-perature patterns in X-linked hypohidrotic ectodermal dysplasia by whole body thermography. Hum Genet 1990, 86, 7–13.
Zankl A, Addor MC, Cousin P, Gaide AC, Gudinchet F, Schorderet DF:
[19] Fatal outcome in a female monozygotic
twin with X-linked hypohydrotic (sic) ectodermal dysplasia (XLHED) due to a de novo t(X;9) translocation with probable disruption of the EDA gene. Eur J Pediat 2001, 160, 296–299.
Schneider P, Street SL, Gaide O, Hertig S, Tardivel A, Tschopp J, Runkel L, Alevizopoulos K, Ferguson BM, [20]
Zonana J: Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A. J Biol Chem 2001, 276, 22, 188, 19–27.
Munoz F, Lestringant G, Sybert V, Frydman M, Alswaini A, Frossard PM, Jorgenson R, Zonana J:
[21] Definitive
evidence for an autosomal recessive form of hypohidrotic ectodermal dysplasia clinically indistinguishable from the more common X-linked disorder. Am J Hum Genet 1997, 61, 94–100.
Monreal AW, Ferguson BM, Headon DJ, Street SL, Overbeek PA, Zonana J:
[22] Mutations in the human
homo-logue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia. Nat Genet 1999, 22, 4, 366–369.
Headon DJ, Overbeek PA:
[23] Involvement of a novel Tnf receptor homologue in hair follicle induction. Nat Genet 1999, 22, 4, 370–374.
Shimomura Y, Sato N, Miyashita A, Hashimoto T, Ito M, Kuwano R:
[24] A rare Case of Hypohidrotic Ectodermal
Dysplasia Caused by Compound Heterozygous Mutations in the EDAr Gene. Nat Genet 1999, 22, 4, 370–374. Döffinger R, Smahi A, Bessia C, Geissmann F, Feinberg J, Durandy A, Bodemer C, Kenwrick S, Dupuis- [25]
-Girod S, Blanche S, Wood P, Rabia SH, Headon DJ, Overbeek PA, Le Deist F, Holland SM, Belani K, Kumararatne DS, Fischer A, Shapiro R, Conley ME, Reimund E, Kalhoff H, Abinun M, Munnich A, Israël A, Courtois G, Casanova JL: X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat Genet 2001, 27, 3, 277–285.
van der Hout A, Oudesluijs G, Venema A, Verheij JBGM, Mol BGJ, Rump P, Brunner HG, Vos Y, van Essen AJ: [26]
Headon DJ, Emmal SA, Ferguson BM, Tucker AS, Justice MJ, Sharpe PT, Zonana J, Overbeek PA:
[27] Gene defect
in ectodermal dysplasia implicates a death domain adapter in development. Nature 2001, 41, 913–916. Jorgenson RJ, Dowben JS, Dowben SL:
[28] Autosomal dominant ectodermal dysplasia. J Craniofac Genet Dev Biol 1987, 7, 403–412.
Chassaing N, Bourthoumieu S, Cossee M, Calvas P, Vincent MC:
[29] Mutations in EDAr account for one-quarter
of non-ED1-related hypohidrotic ectodermal dysplasia. Hum Mutat 2006, 27, 3, 255–259.
Bal E, Baala L, Cluzeau C, El Kerch F, Ouldim K, Hadj-Rabia S, Bodemer C, Munnich A, Courtois G, Sefiani A, [30]
Smahi A: Autosomal dominant anhidrotic ectodermal dysplasias at the EDArADD locus. Hum Mutat 2007, 28, 703–709.
Hofmann K:
[31] The modular nature of apoptotic signaling proteins. Cell Molec Life Sci 1999, 55, 1113–1128. Ashkenazi A, Dixit VM:
[32] Death receptors: signaling and modulation. Science 1998, 281, 1305–1308. Locksley RM, Killeen N, Lenardo MJ:
[33] The TNF and TNF receptor superfamilies: Integrating mammalian biology. Cell 2001, 104, 487–501.
Hollister DW, Klein SH, Dejager HJ, Lachman RS, Rimoin DL:
[34] The lacrimo-auriculo-dento-digital syndrome.
J Pediat 1973,83, 438–444.
Rohmann E, Brunner HG, Kayserili H, Uyguner O, Nurnberg G, Lew ED, Dobbie A, Eswarakumar VP, [35]
Uzumcu A, Ulubil-Emeroglu M, Leroy JG, Li Y: Mutations in different components of FGF signaling in LADD syndrome. Nat Genet 2006,38, 414–417.
Hogan BL:
[36] Morphogenesis. review article highlighting the roles of multigene families, such as Fgfs, Bmps,
Hedgehogs, Wnts and Egfs in morphogenesis. Cell 1999, 96, 225–233.
Johnson DE, Lee PL, Lu J, Williams LT:
[37] Diverse forms of a receptor for acidic and basic fibroblast growth factors. Mol Cell Biol 1990, 10, 4728–4736.
Jin DY, Jeang KT:
[38] Isolation of full-length cDNA and chromosomal localization of human NF-kappaB modulator NEMO to Xq28. J Biomed Sci1999, 6, 115–120.
Vinolo E, Sebban H, Chaffotte A, Israel A, Courtois G, Ve´ron M, Agou F:
[39] A Point Mutation in NEMO
Associated with Anhidrotic Ectodermal Dysplasia with Immunodeficiency Pathologyn results in Destabilization of the Oligomer and reduces Lipopolysaccharide- and Tumor Necrosis Factor-mediated NF-B Activation. J Biol Chem 2006, 281, 6334–6348.
May MJ, D’Acquisto F, Madge LA, Glöckner J, Pober JS, Ghosh S:
[40] Selective Inhibition of NF-kappa B Activation
by a Peptide that Blocks the Interaction of NEMO with the Ikappa B Kinase Complex. Science 2000, 289, 1550– 1554.
Rothwarf DM, Zandi E, Natoli G, Karin M:
[41] IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 1998, 395 (6699), 297–300.
Huang GJ, Zhang ZQ, Jin DY:
[42] Stimulation of IKK-γ oligomerization by the T-cell leukemia virus oncoprotein Tax. FEBS Letters 2002, 531, 494–498.
Agou F, Ye F, Goffinont S, Courtois G, Yamaoka S, Israel A, Veron M:
[43] NEMO Trimerizes through Its
Coiled-coil C-terminal Domain. J Biol Chem 2002, 277, 17464–17475. Tang ED, Wang CY, Xiong Y, Guan KL:
[44] A role for NF-kappaB essential modifier/IkappaB kinase-gamma (NEMO/IKKgamma) ubiquitination in the activation of the IkappaB kinase complex by tumor necrosis factor-alpha. J Biol Chem 2003, 278, 37297–37305.
Zhou H, Wertz I, O’Rourke K, Ultsch M, Seshagiri S, Eby M, Xiao W, Dixit VM:
[45] Bcl10 activates the NF-kappaB
pathway through ubiquitination of NEMO. Nature 2004, 427, 167–171.
Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G:
[46] CYLD is a deubiquitinating
enzyme that negatively regulates NF-kB activation by TNFr family members. Nature 2003, 424, 793–796. Smahi A, Courtois G, Rabia SH, Döffinger R, Bodemer C, Munnich A, Casanova JL, Israël A:
[47] The NF-kappaB
signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-defi-ciency syndromes. Hum Mol Genet 2002, 11, 2371–2375.
Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, Shapira SK, Farndon PA, Wara DW, [48]
Emmal SA, Ferguson BM: A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dys-plasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000, 67, 1555–1562.
Kellermayer R, Keller M, Ratajczak P, Richardson E, Harangi F, Mérei E, Melegh B, Kosztolányi G, Richard G: [49]
Bigenic connexin mutations in a patient with hidrotic ectodermal dysplasia. Eur J Dermatol 2005, 15, 2, 75–79. Kumar NM, Gilula NB:
[50] The Gap Junction Communication Channel. Cell 1996, 84, 3, 381–388. Bruzzone R, White TW, Paul DL:
[51] Connections with Connexins: The Molecular Basis of Direct Intercellular Signaling. Eur J Biochem 1996, 238, 1, 1–27.
Mignon C, Fromaget C, Mattei MG, Gros D, Yamasaki H, Mesnil M:
[52] Assignment of connexin 26 (GJB2) and
46 (GJA3) genes to human chromosome 13q11-q12 and mouse chromosome 14D1-E1 by in situ hybridization. Cytogenet Cell Genet 1996, 72, 185–186.
Kiang DT, Jin N, Tu Z-J, Lin HH:
[53] Upstream genomic sequence of the human connexin26 gene. Gene 1997, 199, 165–171.
Maestrini E, Korge BP, Ocańa-Sierra J, Calzolari E, Cambiaghi S, Scudder PM, Hovnanian A, Monaco AP, [54]
Richard G, Smith LE, Bailey RA, Itin P, Hohl D, Epstein EH Jr, DiGiovanna JJ, Compton JG, Bale SJ:
[55] The
Molecular Basis of Direct Intercellular Signaling. Nat Genet 1998, 20, 4, 366–369.
Macari F, Landau M, Cousin P, Mevorah B, Brenner S, Panizzon R, Schorderet DF, Hohl D, Huber M: [56]
Mutation in the gene for connexin 30.3 in a family with erythrokeratodermia variabilis. Am J Hum Genet 2000, 67, 1296–1301.
Lamartine J, Essenfelder GM, Kibar Z, Lanneluc I, Callouet E, Laoudj D, Lemaître G, Hand C, Hayflick SJ, [57]
Zonana J, Antonarakis S, Radhakrishna U, Kelsell DP, Christianson AL, Pitaval A, Der Kaloustian V, Fraser C, Blanchet-Bardon C, Rouleau GA, Waksman G: Mutations in GJB6 cause hidrotic ectodermal dysplasia. Nat Genet 2000, 26, 142–144.
Paznekas WA, Boyadjiev SA, Shapiro RE, Daniels O, Wollnik B, Keegan CE, Innis JW, Dinulos MB, Christian C, [58]
Hannibal MC, Jabs EW: Connexin 43 (GJA1) Mutations Cause the Pleiotropic Phenotype of Oculodentodigital Dysplasia. Am J Hum Genet 2003, 72, 2, 408–418.
Lopez M, Eberle F, Mattei MG, Gabert J, Birg F, Bardin F, Maroc C, Dubreuil P:
[59] Complementary DNA
charac-terization and chromosomal localization of a human gene related to the poliovirus receptor-encoding gene. Gene 1995, 155, 261–265.
Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, [60]
Mizoguchi A, Takai Y: Nectin/Prr: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J Cell Biol 1999, 139, 517–549.
Cocchi F, Lopezt M, Menotti L, Aoubalat M, Dubreuilt P, Campadelli-Fiume G:
[61] The v domain of herpesvirus
Ig-like receptor (HIgr) contains a major functional region in herpes simplex virus-1 entry into cells and interacts physically with the viral glycoprotein D. Proc Natl Acad Sci USA, 1998, 95, 15700–15705.
Mandai K, Nakanishi H, Satoh A, Obaishi H, Wada M, Nishioka H, Itoh M, Mizoguchi A, Aoki T, Fujimoto T, [62]
Matsuda Y, Tsukita S, Takai Y: Afadin: a novel actin filament-binding protein with one PDZ domain localized at cadherin-based cell-to-cell adherens junction. J Cell Biol 1997, 139, 517–528.
Mandai K, Nakanishi H, Satoh A, Takahashi K,
[63] Satoh K, Nishioka H, Mizoguchi A, Takai Y: Ponsin/SH3P12: an l-afadin- and vinculin-binding protein localized at cell-cell and cell-matrix adherens junctions. J Cell Biol 1999, 144, 1001–1017.
Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG:
[64] Entry of alphaherpesviruses mediated by
poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms JA, Spitz RA:
[65] Mutations of PvrL1, encoding
a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palate ectodermal dysplasia. Nat Genet 2000, 25, 8, 427–430.
Zlotogora J, Zilberman Y, Tenenbaum A, Wexler MR:
[66] Cleft lip and palate, pili torti, malformed ears, partial syndactyly of fingers and toes and mental retardation. A new syndrome? J Med Genet 1987, 24, 291–293. Zlotogora J:
[67] Syndactyly, ectodermal dysplasia, and cleft lip/palate. J Med Genet 1994, 31, 957–959. Ogur G, Yüksel M:
[68] Association of syndactyly, ectodermal dysplasia, and cleft lip and palate: report of two sibs from Turkey. J Med Genet 1998, 25, 37–40.
Zlotogora J, Ogur G:
[69] Syndactyly, ectodermal dysplasia, and cleft lip and palate. J Med Genet 1988, 25, 503. McGrath JA, Hoeger PH, Christiano AM, McMillan JR, Mellerio JE, Ashton GHS,
[70] Dopping-Hepenstal PJC,
Lake BD, Leigh IM, Harper JI, Eadyr AJ: Skin fragility and hypohidrotic ectodermal dysplasia resulting from ablation of plakophilin 1. Br J Dermatol 1999, 140, 297–307.
McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RAJ:
[71] Mutations
in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 1997, 17, 240–244. Whittock NV, Haftek M, Angoulvant N, Wolf F, Perrot H, Eady RAJ, McGrath JA:
[72] Genomic amplification of
the human plakophilin 1 gene and detection of a new mutation in ectodermal dysplasia/skin fragility syndrome. J Invest Dermatol 2000, 115, 368–374.
Hamada T, South AP, Mitsuhashi Y,
[73] Kinebuchi T,Bleck O, Ashton GHS, Hozumi Y, Suzuki T, Hashimoto T, Eady RAJ, McGrath JA: Genotype-phenotype correlation in skin fragility – ectodermal dysplasia syndrome result-ing from mutations in plakophilin 1. Exp Dermatol 2002, 11, 107–114.
Bonne S, van Hengel J, van Roy F:
[74] Chromosomal mapping of human armadillo genes belonging to the p120(ctn)/ plakophilin subfamily. Genomics 1998, 51, 452–454.
Schmidt A, Langbein L, Rode M, Prätzel S, Zimbelmann R, Franke WW:
[75] Plakophilins 1a and 1b: Widespread
nuclear proteins recruited in specific epithelial cells as desmosomal plaque components. Cell Tissue res 1997, 290, 481–499.
Bornslaeger EA, Godsel LM, Corcoran CM, Park JK, Hatzfeld M, Kowalczyk AP, Green KJ:
[76] Plakophilin 1
inter-feres with plakoglobin binding to desmoplakin, yet together with plakoglobin promotes clustering of desmosomal plaque complexes at cell-cell borders. J Cell Sci 2001, 114, 727–738.
Hatzfeld M, Haffner C, Schulze K, Vinzens U:
[77] The function of plakophilin 1 in desmosome assembly and actin filament organisation. J Cell Biol 2000, 149, 209-222.
McMillan JR, Haftek M, Akiyama M, South AP, Perrot H, McGrath JA, Eady RAD, Shimizu H:
[78] Alterations in
Sprecher E, Molho-Pessach V, Ingber A, Sagi E, Indelman M, Bergman R:
[79] Homozygous Splice Site Mutations
in PKP1 result in Loss of Epidermal Plakophilin 1 Expression and Underlie Ectodermal Dysplasia/Skin Fragility Syndrome in Two Consanguineous Families. J Invest Dermatol 2004, 122, 647–651.
Kremmidiotis G, Baker E, Crawford J, Eyre HJ, Nahmias J, Callen DF:
[80] Localization of human cadherin genes to
chromosome regions exhibiting cancer-related loss of heterozygosity. Genomics 1998, 49, 467–471. Takeichi M:
[81] Cadherins: a molecular family important in selective cell-cell adhesion. Annu rev Biochem 1990, 59, 237–252.
Ivanov DB, Philippova MO, Tkachuk VA:
[82] Structure and function of classical cadherins. Biochemistry 2001, 10, 1174–1186.
Troyanovsky SM:
[83] Mechanisms of cell-cell adhesion complex assembly. Curr Opin Cell Biol 1999, 11, 561–566. Tsukita S, Tsukita S, Nagafuchi A, Yonemura S:
[84] Molecular linkage between cadherins and actin filaments in cell-cell adherens junctions. Curr Opin Cell Biol 1992, 4, 834–839.
Rimm DL, Koslov ER, Kebriaei P, Cianci CD, Morrow JS:
[85] α1(E)-Catenin is an actin-binding and -bundling
protein mediating the attachment of F-actin to the membrane adhesion complex. Proc Natl Acad Sci USA 1995, 92, 8813–8817.
Souied E, Amalric P, Chauvet ML, Chevallier C, Le Hoang P, Munnich A, Kaplam J:
[86] Unusual association of
juvenile macular dystrophy with congenital hypotrichosis: occurrence in two siblings suggesting autosomal reces-sive inheritance. Ophthal General 1995, 16, 11–15.
Sprecher E, Bergman R, Richard G, Lurie R, Shalev S, Petronius D, Shalata A, Anbinder Y, Leibu R, Perlman I, [87]
Cohen N, Szargel R: Hypotrichosis with juvenile macular dystrophy is caused by a mutation in CDH3 encoding P-cadherin. Nat Genet 2001, 29, 134–136.
Huelsken J, Vogel R, Erdmann B, Cotsarelis G, Birchmeier W:
[88] Beta-catenin controls hair follicle morphogenesis
and stem cell differentiation in the skin. Cell 2001, 105, 533–545. Gat U, DasGupta R, Degenstein L, Fuchs E:
[89] De novo hair follicle morphogenesis and hair tumors in mice express-ing a truncated beta-catenin in skin. Cell 1998, 95, 605–614.
Indelman M, Bergman R, Lurie R, Richard G, Miller B, Petronius D, Ciubutaro D, Leibu R, Sprecher E:
[90] A
mis-sense mutation in CDH3, encoding P-cadherin, causes hypotrichosis with juvenile macular dystrophy. J Invest Dermatol 2002, 119, 1210–1213.
Kjaer KW, Hansen L, Schwabe GC, Marques-de-Faria AP, Eiberg H, Mundlos S, Tommerup N, Rosenberg T: [91]
Distinct CDH3 mutations cause ectodermal dysplasia, ectrodactyly, macular dystrophy (EEM syndrome). J Med Genet 2005, 42, 4, 292–298.
Balarin Silva V, Simőes AM, Marques-de-Faria AP:
[92] EEM syndrome: report of a family and results of a ten-year follow-up. Ophthalmic Genet 1999, 20, 2, 95–99.
Yang A, Kaghad M, Wang Y, Gillett E, Fleming M, Dötsch V, Andrews N, Caput D, McDeon F:
[93] p63, a p53
Homolog at 3q27–29, Encodes Multiple Products with Transactivating, Death-Inducing, and Dominant-Negative Activities. Mol Cell 1998, 2, 3, 305–316.
Address for correspondence:
Anna Paradowska
Department of Maxillofacial Orthopedics and Orthodontics Wroclaw Medical University
Krakowska 26 50-425 Wrocław Poland
Tel.: +48 71 784 02 99 Email: [email protected]