Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Identification of Medically Important Yeasts Using PCR-Based
Detection of DNA Sequence Polymorphisms in the
Internal Transcribed Spacer 2 Region of the rRNA Genes
Y. C. CHEN,1J. D. EISNER,1M. M. KATTAR,1S. L. RASSOULIAN-BARRETT,1K. LAFE,1S. L. YARFITZ,2A. P. LIMAYE,1,3ANDB. T. COOKSON1,4*
Departments of Laboratory Medicine,1Infectious Diseases,3and Microbiology,4and Health Sciences Library and Department of Medical Education, Division of Bioinformatics,2University of Washington, Seattle, Washington
Received 11 January 2000/Returned for modification 26 February 2000/Accepted 27 March 2000
Identification of medically relevant yeasts can be time-consuming and inaccurate with current methods. We evaluated PCR-based detection of sequence polymorphisms in the internal transcribed spacer 2 (ITS2) region of the rRNA genes as a means of fungal identification. Clinical isolates (401), reference strains (6), and type strains (27), representing 34 species of yeasts were examined. The length of PCR-amplified ITS2 region DNA was determined with single-base precision in less than 30 min by using automated capillary electrophoresis. Unique, species-specific PCR products ranging from 237 to 429 bp were obtained from 92% of the clinical isolates. The remaining 8%, divided into groups with ITS2 regions which differed by<2 bp in mean length, all contained species-specific DNA sequences easily distinguishable by restriction enzyme analysis. These data, and the specificity of length polymorphisms for identifying yeasts, were confirmed by DNA sequence analysis of the ITS2 region from 93 isolates. Phenotypic and ITS2-based identification was concordant for 427 of 434 yeast isolates examined using sequence identity of>99%. Seven clinical isolates contained ITS2 sequences that did not agree with their phenotypic identification, and ITS2-based phylogenetic analyses indicate the possi-bility of new or clinically unusual species in theRhodotorulaandCandidagenera. This work establishes an initial database, validated with over 400 clinical isolates, of ITS2 length and sequence polymorphisms for 34 species of yeasts. We conclude that size and restriction analysis of PCR-amplified ITS2 region DNA is a rapid and reliable method to identify clinically significant yeasts, including potentially new or emerging pathogenic species.
Opportunistic fungal infections have increased dramatically in recent years, often as a result of advanced medical treat-ments (22, 33). Aggressive chemotherapy compromises patient immunity against fungal infections, and broad application of antifungal agents has been associated with the emergence of resistant strains (7). Coupled with the risk of nosocomial fun-gal infection (32), the rapid and accurate identification of eti-ological agents and resistant strains is crucial in medical cen-ters caring for large groups of susceptible patients.
At least 150 fungal species have been identified as human pathogens and have been isolated from virtually all body sites (6). Identification of this increasing diversity of pathogens by conventional methods is often difficult and sometimes incon-clusive (25). Morphological features and reproductive struc-tures useful for identifying isolated fungi may take days to weeks to develop in culture, and evaluation of these charac-teristics requires expertise in mycology. Most fungal infections are caused by yeasts (21). Two commercial methods used to identify yeasts, the API and VITEK systems, require 2 to 3 days before biochemical reactions can be interpreted (5). In addition, their databases are limited (2, 24).
Molecular techniques utilizing amplification of target DNA provide alternative methods for diagnosis and identification (13). PCR-based detection of fungal DNA sequences can be rapid, sensitive, and specific (17). Coding regions of the 18S,
5.8S, and 28S nuclear rRNA genes evolve slowly, are relatively conserved among fungi, and provide a molecular basis of es-tablishing phylogenetic relationships (31). Between coding re-gions are the internal transcribed spacer 1 and 2 rere-gions (ITS1 and ITS2, respectively) which evolve more rapidly and may therefore vary among different species within a genus. Thus, PCR amplification may facilitate the identification of ITS re-gion DNA sequences with sufficient polymorphism to be useful for identifying fungal species.
In this study, ITS2 sequence polymorphisms are evaluated for their specificity in identifying 34 species of pathogenic yeasts. Using universal primers complementary to the coding regions of the fungal rRNA genes, we amplified the ITS2 region from 27 type strains and over 400 clinical isolates. When determined with single-base-pair precision, the PCR product length alone identified 92% of the clinical isolates. Sequence analyses of ITS2 DNA from 93 isolates confirmed the speci-ficity of this identification method, and phenotypic and ITS2-based identifications were concordant for⬎98% of the yeasts examined. Our data indicate ITS2 sequence polymorphisms are useful for identifying medically important yeasts and may facilitate taxonomic and phylogenetic classification of poten-tially new pathogenic species.
MATERIALS AND METHODS
Yeast isolates.Four hundred and one clinical isolates (Table 1), collected from
30 October 1998 to 17 February 1999, six reference strains (Table 2) from the mycology laboratory at the University of Washington Medical Center, 22 type strains from the American Type Culture Collection, and five type strains from the Centraalbureau voor Schimmelcultures (Table 2) were included in this study. Eleven isolates ofCandida dubliniensis were a gift from W. R. Kirkpatrick, University of Texas Health Science Center, San Antonio, Tex. (11). These 434
* Corresponding author. Mailing address: Departments of Labora-tory Medicine and Microbiology, University of Washington, 1959 NE Pacific St., NW 120, Box 357110, Seattle, WA 98195. Phone: (206) 598-6131. Fax: (206) 598-6189. E-mail: [email protected].
2302
on May 15, 2020 by guest
http://jcm.asm.org/
isolates represent 34 different species of pathogenic yeasts. Isolates were iden-tified by either API 20C AUX strip or VITEK automated systems and by formation of true hyphae, pseudohyphae, blastoconidia, or chlamydoconidia on cornmeal-Tween 80 agar (10). Morphological evaluation of pseudohyphae was also used in distinguishingCandidaspecies (14). The identities of 23 type strains were confirmed morphologically and biochemically (exceptEndomyces fibuliger,
Pichia farinosa,Trichosporon cutaneum, andTrichosporon jiroveciiwhich are not
in either the API 20C or VITEK databases).
Yeasts were subcultured onto Sabouraud dextrose agar plates (BBL-Emmon’s Mod, Cockeysville, Md.) and were incubated at 30°C for 2 days, and DNA was extracted by using a modified heat extraction method (17). Briefly, 2-day-old yeast colonies in 100l of lysis buffer (100 mM Tris, 30 mM EDTA, 0.5% [wt/vol] sodium dodecyl sulfate, pH 7.5) were vortexed briefly and incubated at 100°C for 15 min. One hundred microliters of 2.5 M potassium acetate was added, and the suspension was incubated on ice for 1 h and centrifuged at 14,000⫻gfor 5 min. The supernatant was transferred to a new tube, an equal volume of isopropanol was added, and the suspension was centrifuged for 5 min. The supernatant was then decanted, 500 l of 100% ethanol was added, and the suspension was centrifuged for 20 min. The supernatant was decanted, and the extracted DNA was dried in a Speed Vac, resuspended in 100l of sterile pharmacy water (Sterile Water for Irrigation; USP-Baxter, Deerfield, Ill.), and stored at⫺20°C.
PCR and DNA sequencing.ITS2 region DNA was PCR amplified from a 1:50
dilution of template DNA in 1⫻PCR buffer containing 3 mM MgCl2, 0.04 U of
AmpliTaq DNA Polymerase (Perkin-Elmer Corporation, Foster City, Calif.) per
l, 200M of each deoxynucleoside triphosphate (Pharmacia Biotech), 900M primer ITS3 (5⬘-GCATCGATGAAGAACGCAGC-3⬘) (GIBCO BRL, Grand Island, N.Y.), and 300M primer ITS4 (5⬘-TCCTCCGCTTATTGATATGC-3⬘) (16) in sterile pharmacy water. The ITS4 primer was labeled with fluorescent dye, NED (Perkin Elmer), FAM, or HEX (SYNTHEGEN, Houston, Tex.). The DNA Thermal Cycler (model 9700; PE Applied Biosystems) was set to the following parameters: 95°C for 6 min, followed by 25 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 30 s, followed by one extension at 72°C for 10 min.
To determine the size of fluorescently labelled PCR products, two parts PCR product and one part of GS-500 ROX size standard (Perkin-Elmer Applied Biosystems, Warrington, England) were added to deionized formamide (AMRESCO, Solon, Ohio), denatured at 95°C for 3 min, placed on ice for at least 2 min, and injected into a 47-cm by 50-m capillary column containing the high-performance polymer 4 in the ABI 310 genetic analyzer, an automated fluorescence capillary electrophoresis system (PE Applied Biosystems) utilizing denaturing conditions. Electrophoresis parameters were set on the instrument at 5-s injection time, 15 kV injection voltage, 15 kV electrophoresis voltage, and a constant temperature of 60°C. The average electrophoresis time was 30 min to ensure detection of product sizes below 500 bp, and PCR product lengths were determined by using the ABI310 GeneScan software (PE Applied Biosystems). To sequence ITS2 region PCR products, unlabeled ITS3 and ITS4 primers were used to sequence both the forward and reverse strands, respectively, and each sequence (listed in Table 2) was repeated at least in duplicate. PCR products were filtered by using a Microcon column (YM-100; Amicon, Inc., Beverly, Mass.) and were resuspended in approximately 85l of sterile pharmacy water. Cycle sequencing was performed with the Ready-Reaction mix (ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction Kit; PE Applied Biosystems) according to manufacturer’s instructions on a PE thermocycler, Model 9700, by using the preprogrammed BigDye cycling parameters on the instrument. Each sequencing product was concentrated to dryness using a Speed Vac after removing excess DyeDeoxyR terminators with CENTRI-SEP columns (Princeton Separations, Adelphia, N.J.). Each sequencing product was resus-pended in 20l of the template suppression reagent (PE Applied Biosystems) and was incubated at room temperature for approximately 10 min, heated at 95°C for 2 to 5 min, and placed on ice for 3 min. Using a 47-cm by 50-m capillary containing the high-performance polymer 6, each sample could be sequenced in 1 h on the ABI PRISM 310 Genetic Analyzer.
Sequence similarity and phylogenetic analyses.Sequences were assembled
and edited with the Sequencher program (version 3.1). Multiple sequence align-ment was performed by using CLUSTAL_X (28) with a gap opening penalty value of 20 and the default gap extension penalty value of 6.66. The multiple alignment output from CLUSTAL_X was imported and manually edited with the Java alignment editor, Jalview (version 1.3b) (M. Clamp, European Bioinfor-matics Institute [http://circinus.ebi.ac.uk:6543/jalview/]). Pairwise sequence com-parisons were expressed as the percentage of the total number of nucleotide differences divided by the total number of positions. Phylogenetic analysis was performed by using the phylogeny inference package, PHYLIP (version 3.573) (J. Felsenstein, Department of Genetics, University of Washington, Seattle [http://evolution.genetics.washington.edu/phylip.html]), with three treeing algo-rithms: neighbor-joining, Fitch-Margoliash, and maximum likelihood (18).
Pneu-mocystis cariniiwas used as the outgroup. The branching orders of the
Fitch-Margoliash and the neighbor-joining dendrograms were evaluated with 1,000 bootstrap analyses using the SEQBOOT program in PHYLIP.
RESULTS
Approximately 128 bp of the 5.8S rRNA gene, the entire ITS2 region, and approximately 59 bp of the 28S rRNA gene can be amplified from fungal template DNA by using one primer pair (Fig. 1) (15, 16). The PCR products amplified from 17 yeast species (including nine from the same genus) ap-peared as single bands from 200 to 500 bp on a 1% agarose gel (data not shown). This suggested that related species could be distinguished by length polymorphisms in their ITS2 region DNA. To test this hypothesis, we utilized fluorescence capillary electrophoresis to more precisely measure the length of ITS2 region PCR products amplified from a large number of yeasts: 401 clinical isolates, 6 reference strains, and 27 type strains representing 34 species of yeasts.
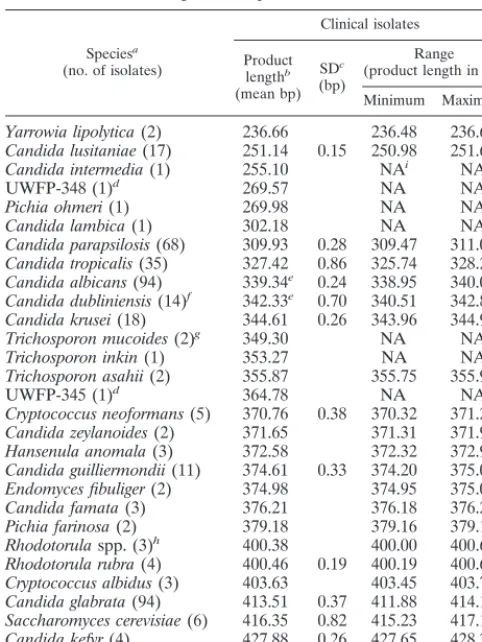
ITS2 length polymorphisms. ITS2 region PCR products from clinical isolates ranged in size from 237 bp (Yarrowia lipolytica) to 429 bp (Cryptococcus uniguttulatus, Table 1). Product lengths were measured for 434 yeasts and for multiple isolates in at least two separate PCRs:Candida albicans(n⫽ 46), Candida glabrata(n ⫽ 36),Candida tropicalis (n ⫽ 27),
Candida parapsilosis(n⫽23),Candida krusei(n⫽15), Can-TABLE 1. ITS2 region PCR products from clinical isolates
Speciesa
(no. of isolates)
Clinical isolates Product lengthb (mean bp) SDc (bp) Range (product length in bp) Minimum Maximum
Yarrowia lipolytica(2) 236.66 236.48 236.62
Candida lusitaniae(17) 251.14 0.15 250.98 251.68
Candida intermedia(1) 255.10 NAi NA
UWFP-348 (1)d 269.57 NA NA
Pichia ohmeri(1) 269.98 NA NA
Candida lambica(1) 302.18 NA NA
Candida parapsilosis(68) 309.93 0.28 309.47 311.01
Candida tropicalis(35) 327.42 0.86 325.74 328.25
Candida albicans(94) 339.34e 0.24 338.95 340.00
Candida dubliniensis(14)f 342.33e 0.70 340.51 342.87
Candida krusei(18) 344.61 0.26 343.96 344.98
Trichosporon mucoides(2)g 349.30 NA NA
Trichosporon inkin(1) 353.27 NA NA
Trichosporon asahii(2) 355.87 355.75 355.98
UWFP-345 (1)d 364.78 NA NA
Cryptococcus neoformans(5) 370.76 0.38 370.32 371.2
Candida zeylanoides(2) 371.65 371.31 371.99
Hansenula anomala(3) 372.58 372.32 372.91
Candida guilliermondii(11) 374.61 0.33 374.20 375.09
Endomyces fibuliger(2) 374.98 374.95 375.00
Candida famata(3) 376.21 376.18 376.25
Pichia farinosa(2) 379.18 379.16 379.19
Rhodotorulaspp. (3)h 400.38 400.00 400.64
Rhodotorula rubra(4) 400.46 0.19 400.19 400.63
Cryptococcus albidus(3) 403.63 403.45 403.75
Candida glabrata(94) 413.51 0.37 411.88 414.19
Saccharomyces cerevisiae(6) 416.35 0.82 415.23 417.17
Candida kefyr(4) 427.88 0.26 427.65 428.23
Cryptococcus uniguttulatus(1) 428.73 NA NA
aIdentification of species by biochemical and morphological assessment.
Number of isolates collected is indicated in parenthesis.
bPCR product sizes determined by capillary electrophoresis as described in
Materials and Methods.
cStandard deviation is calculated for species with four or more strains. dAlso see Table 4.
ePCR product sizes ofC. albicansandC. dubliniensiswere statistically
differ-ent (P⬍0.001 byttest).
fTen strains obtained from the University of Texas Health Science Center at
San Antonio.
gUWFP-366 and -367, also see Table 4. hUWFP-370, -373, and -380, also see Table 4. iNA, nonapplicable.
on May 15, 2020 by guest
http://jcm.asm.org/
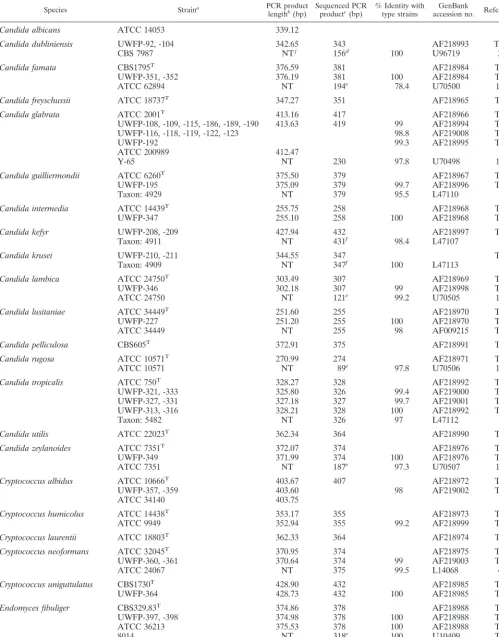
[image:2.612.53.294.83.404.2]TABLE 2. Length and sequence polymorphisms of ITS2 region DNA from clinical strains and type strains
Species Straina PCR product
lengthb(bp) Sequenced PCRproductc(bp) % Identity withtype strains accession no.GenBank Reference
Candida albicans ATCC 14053 339.12
Candida dubliniensis UWFP-92, -104 342.65 343 AF218993 TSi
CBS 7987 NTj 156d 100 U96719 3
Candida famata CBS1795T 376.59 381 AF218984 TS
UWFP-351, -352 376.19 381 100 AF218984 TS
ATCC 62894 NT 194e 78.4 U70500 15
Candida freyschussii ATCC 18737T 347.27 351 AF218965 TS
Candida glabrata ATCC 2001T 413.16 417 AF218966 TS
UWFP-108, -109, -115, -186, -189, -190 413.63 419 99 AF218994 TS
UWFP-116, -118, -119, -122, -123 98.8 AF219008 TS
UWFP-192 99.3 AF218995 TS
ATCC 200989 412.47
Y-65 NT 230 97.8 U70498 15
Candida guilliermondii ATCC 6260T 375.50 379 AF218967 TS
UWFP-195 375.09 379 99.7 AF218996 TS
Taxon: 4929 NT 379 95.5 L47110
Candida intermedia ATCC 14439T 255.75 258 AF218968 TS
UWFP-347 255.10 258 100 AF218968 TS
Candida kefyr UWFP-208, -209 427.94 432 AF218997 TS
Taxon: 4911 NT 431f 98.4 L47107
Candida krusei UWFP-210, -211 344.55 347 TS
Taxon: 4909 NT 347f 100 L47113
Candida lambica ATCC 24750T 303.49 307 AF218969 TS
UWFP-346 302.18 307 99 AF218998 TS
ATCC 24750 NT 121e 99.2 U70505 15
Candida lusitaniae ATCC 34449T 251.60 255 AF218970 TS
UWFP-227 251.20 255 100 AF218970 TS
ATCC 34449 NT 255 98 AF009215 TS
Candida pelliculosa CBS605T 372.91 375 AF218991 TS
Candida rugosa ATCC 10571T 270.99 274 AF218971 TS
ATCC 10571 NT 89e 97.8 U70506 15
Candida tropicalis ATCC 750T 328.27 328 AF218992 TS
UWFP-321, -333 325.80 326 99.4 AF219000 TS
UWFP-327, -331 327.18 327 99.7 AF219001 TS
UWFP-313, -316 328.21 328 100 AF218992 TS
Taxon: 5482 NT 326 97 L47112
Candida utilis ATCC 22023T 362.34 364 AF218990 TS
Candida zeylanoides ATCC 7351T 372.07 374 AF218976 TS
UWFP-349 371.99 374 100 AF218976 TS
ATCC 7351 NT 187e 97.3 U70507 15
Cryptococcus albidus ATCC 10666T 403.67 407 AF218972 TS
UWFP-357, -359 403.60 98 AF219002 TS
ATCC 34140 403.75
Cryptococcus humicolus ATCC 14438T 353.17 355 AF218973 TS
ATCC 9949 352.94 355 99.2 AF218999 TS
Cryptococcus laurentii ATCC 18803T 362.33 364 AF218974 TS
Cryptococcus neoformans ATCC 32045T 370.95 374 AF218975 TS
UWFP-360, -361 370.64 374 99 AF219003 TS
ATCC 24067 NT 375 99.5 L14068 4
Cryptococcus uniguttulatus CBS1730T 428.90 432 AF218985 TS
UWFP-364 428.73 432 100 AF218985 TS
Endomyces fibuliger CBS329.83T 374.86 378 AF218988 TS
UWFP-397, -398 374.98 378 100 AF218988 TS
ATCC 36213 375.53 378 100 AF218988 TS
8014 NT 318e 100 U10409 TS
Hansenula anomala UWFP-396 372.52 375 AF218991 TS
ATCC 8168 NT 188d 100 U96720 3
Continued on following page
on May 15, 2020 by guest
http://jcm.asm.org/
dida lusitaniae(n ⫽ 15), C. dubliniensis (n ⫽ 13), Candida famata (n ⫽ 3), Saccharomyces cerevisiae (n ⫽ 3), Candida zeylanoides(n⫽2),Rhodotorula rubra(n⫽2), and one isolate each ofC. lambica,Candida intermedia,Cryptococcus neofor-mans,Cryptococcus humicolus,C. uniguttulatus,Pichia ohmeri,
Pichia farinosa, Trichosporon asahi, Y. lipolytica, E. fibuliger, and Trichosporon inkin. Between-run standard deviations of the mean PCR product length from 14 isolates ofC. albicans
were 0.07, 0.17, and 0.31 bp in a series of separate PCRs. Standard deviations for 11 species with ⱖ4 isolates ranged from 0.15 to 0.38 bp (Table 1). Interstrain variation (see below) contributed to higher standard deviations observed for
prod-ucts fromC. dubliniensis(0.70 bp),C. tropicalis(0.86 bp), and
S. cerevisiae(0.82 bp). Thus, the length of PCR products can be determined with single-base precision by using capillary elec-trophoresis.
Ninety-two percent of the clinical isolates (368 of 401 strains), comprising 17 species, produced distinct and species-specific ITS2 PCR products which differed in mean length by
ⱖ2 bases (Table 1). Eleven isolates were correctly reidentified because their ITS2 PCR product length did not agree with their initial designation: new identities were confirmed by bio-chemical and morphological phenotypes. Four of these misi-dentifications failed to distinguishC. dubliniensisfromC. albi-cans(data not shown); these closely related organisms were reliably identified by ITS2 region PCR product length alone (Table 1). Confirming these data, the PCR product lengths from clinical isolates correlated well with those from their respective type strains (Table 2) and differed by only 0.05 to 0.85 bp, except forCandida lambica (1.31 bp) andP. ohmeri
(1.14 bp).
[image:4.612.52.554.83.386.2]Seventeen species and one unknown clinical isolate, UWFP-348, could be divided into eight groups withⱕ2-base difference in the mean length of their ITS2 region PCR products (Tables
[image:4.612.72.276.647.698.2]FIG. 1. ITSs are noncoding regions flanked by the structural rRNA genes. Approximate binding sites of the ITS3 and ITS4 PCR primers are shown by arrows.
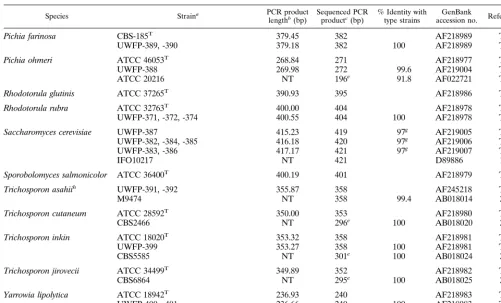
TABLE 2—Continued
Species Straina PCR product
lengthb(bp) Sequenced PCRproductc(bp) % Identity withtype strains accession no.GenBank Reference
Pichia farinosa CBS-185T 379.45 382 AF218989 TS
UWFP-389, -390 379.18 382 100 AF218989 TS
Pichia ohmeri ATCC 46053T 268.84 271 AF218977 TS
UWFP-388 269.98 272 99.6 AF219004 TS
ATCC 20216 NT 196e 91.8 AF022721 TS
Rhodotorula glutinis ATCC 37265T 390.93 395 AF218986 TS Rhodotorula rubra ATCC 32763T 400.00 404 AF218978 TS
UWFP-371, -372, -374 400.55 404 100 AF218978 TS
Saccharomyces cerevisiae UWFP-387 415.23 419 97g AF219005 TS
UWFP-382, -384, -385 416.18 420 97g AF219006 TS
UWFP-383, -386 417.17 421 97g AF219007 TS
IFO10217 NT 421 D89886 19
Sporobolomyces salmonicolor ATCC 36400T 400.19 401 AF218979 TS Trichosporon asahiih UWFP-391, -392 355.87 358 AF245218 TS
M9474 NT 358 99.4 AB018014 26
Trichosporon cutaneum ATCC 28592T 350.00 353 AF218980 TS
CBS2466 NT 296e 100 AB018020 27
Trichosporon inkin ATCC 18020T 353.32 358 AF218981 TS
UWFP-399 353.27 358 100 AF218981 TS
CBS5585 NT 301e 100 AB018024 27
Trichosporon jirovecii ATCC 34499T 349.89 352 AF218982 TS
CBS6864 NT 295e 100 AB018025 27
Yarrowia lipolytica ATCC 18942T 236.93 240 AF218983 TS
UWFP-400, -401 236.66 240 100 AF218983 TS
ATCC 9773 236.88 240 100 AF218983 TS
aUWFP, University of Washington Fungal Project; ATCC, American Type Culture Collection; CBS, Centraalbureau voor Schimmelcultures; IFO, Institute of Fermentation; ATCC type strains are labeled with superscript T.
bPCR product length determined with capillary electrophoresis as described in Materials and Methods. cExact number of nucleotides determined by direct sequencing of the PCR products.
dPartial sequence compared to ITS2 region DNA sequences from clinical strains in this study. ePartial sequence compared to ITS2 region DNA sequences from type strains in this study. fSequence compared to ITS2 region DNA sequences from clinical strains in this study. gITS2 region DNA sequence compared with D89886.
hT. asahiiidentified as such by the API 20C and asT. beigeliiby the VITEK system. iTS, this study.
jNT, not tested.
on May 15, 2020 by guest
http://jcm.asm.org/
1 and 2): T. cutaneum and T. jirovecii; C. famata, Candida guilliermondii, andE. fibuliger;T. inkinand C. humicolus;R. rubraand Sporobolomyces salmonicolor; Hansenula anomala,
C. zeylanoides, andC. neoformans;Candida utilisand Crypto-coccus laurentii; C. uniguttulatus and Candida kefyr; and P. ohmeriand UWFP-348.
ITS2 sequence polymorphisms. The DNA sequences of ITS2 region PCR products from 66 clinical strains and 27 type strains were analyzed to confirm the specificity of length poly-morphisms for identifying yeasts and to resolve our other find-ings: three species displayed mean product lengths with stan-dard deviations ⱖ0.5 bases, and eight groups of species produced products differing in mean length byⱕ2 bases. To
reduce the possibility of errors, sequence was obtained directly from both strands of the PCR products, and each strain was either sequenced twice or confirmed by two or more strains with the same sequence (Table 2). The length of the PCR product determined by capillary electrophoresis showed excel-lent correlation (R2⫽0.9992) with the actual number of
nu-cleotides enumerated by direct sequencing (Fig. 2). The single-base precision of this method (see above) allowed us to determine that the PCR fragment sizes determined by Gene-Scan were slightly shorter than the actual sizes determined by sequencing, with a small proportional bias (Fig. 2, inset). Al-though similar discrepancies have been observed by others using capillary electrophoresis (9), our data demonstrate both the precision and accuracy of this technique for determining length polymorphisms among PCR products.
ITS2 region DNA from clinical strains of C. famata, C. intermedia, C. lusitaniae, C. zeylanoides, C. uniguttulatus, E. fibuliger, P. farinosa, R. rubra, T. inkin, andY. lipolytica had 100% sequence similarity compared with their type strains. Similarity between the type and clinical strains of C. guillier-mondii,P. ohmeri,C. tropicalis,C. humicolus,C. lambica, and
C. neoformansexceeded 99% (Table 2).H. anomalaand Can-dida pelliculosa had identical sequences since they are the teleomorphic (sexual) and anamorphic (asexual) forms of the same organism (30). Thus, ITS2 region sequence similarity exceeded 99% among members of a single species (Table 2), except forC. glabrata(98.8 to 99.3%) andCryptococcus albidus
(98.0%).
[image:5.612.73.271.72.229.2]The exception amongC. glabrataresulted from a 417-base product from the type strain which differed from clinical strains at up to five nucleotide positions. In contrast, the three se-quences found among 12 clinical strains were 419 bp (Table 2) and 99.5 to 99.8% similar to each other (data not shown). Six clinical isolates ofS. cerevisiaedisplayed products of 421, 420, or 419 bp with 99.3 to 99.8% similarity to each other, versus 97% similarity to a published ITS2 sequence fromS. cerevisiae
FIG. 2. ITS2 region PCR product length as determined by capillary electro-phoresis versus actual PCR product length as determined by direct sequencing. Eighty-nine independent length determinations plotted against actual length reveals a small underestimate of actual PCR product size by capillary electro-phoresis, with a small proportional bias (inset, actual length minus length deter-mined by electrophoresis versus actual sequence length).
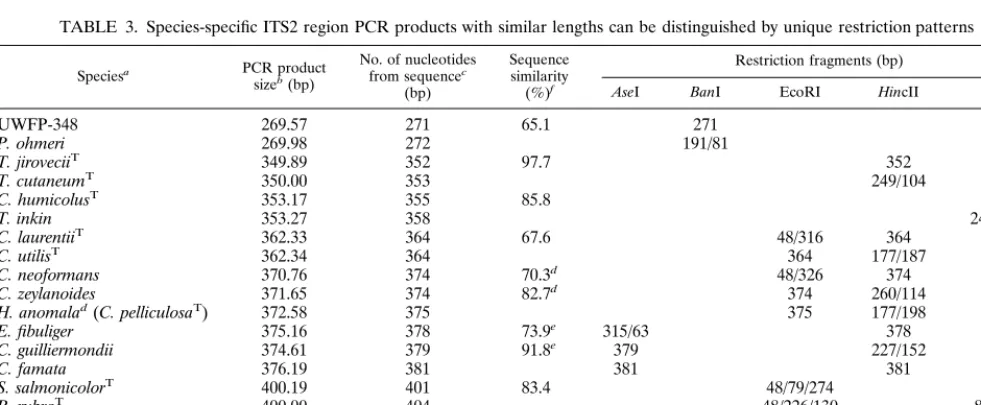
TABLE 3. Species-specific ITS2 region PCR products with similar lengths can be distinguished by unique restriction patterns
Speciesa PCR product sizeb(bp)
No. of nucleotides from sequencec
(bp)
Sequence similarity (%)f
Restriction fragments (bp)
AseI BanI EcoRI HincII StyI
UWFP-348 269.57 271 65.1 271
P. ohmeri 269.98 272 191/81
T. jiroveciiT 349.89 352 97.7 352
T. cutaneumT 350.00 353 249/104
C. humicolusT 353.17 355 85.8 355
T. inkin 353.27 358 249/109
C. laurentiiT 362.33 364 67.6 48/316 364
C. utilisT 362.34 364 364 177/187
C. neoformans 370.76 374 70.3d 48/326 374 C. zeylanoides 371.65 374 82.7d 374 260/114 H. anomalad(C. pelliculosaT) 372.58 375 375 177/198 E. fibuliger 375.16 378 73.9e 315/63 378 C. guilliermondii 374.61 379 91.8e 379 227/152
C. famata 376.19 381 381 381
S. salmonicolorT 400.19 401 83.4 48/79/274 401
R. rubraT 400.00 404 48/226/130 89/315
C. kefyr 427.88 432 66.4 432 176/256 432
C. uniguttulatus 428.73 432 48/384 432 89/207/136 aSuperscript T indicates type strains.
bFrom Tables 1 and 2. cFrom Table 2.
dSequence similarity betweenC. neoformansandC. zeylanoideswas 70.3% and was 82.7% betweenC. zeylanoidesandH. anomala. H. anomalaandC. pelliculosa are the teleomorphic and anamorphic forms of the same organism, respectively.
eSequence similarity betweenE. fibuligerandC. guilliermondiiwas 73.9% and was 91.8% betweenC. guilliermondiiandC. famata. fSequence similarity with the species immediately following with similarly sized PCR products is indicated.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:5.612.57.548.470.673.2](Table 2). Similarly, two clinical isolates of C. albidus were identical but 98% similar to their type strain (Table 2). To-gether with length variation observed among six clinical strains ofC. tropicalis(Table 2), these data indicateⱖ99% similarity in ITS2 region DNA exists among recent clinical isolates of the same species and that intraspecific polymorphism occurs in this region for some species.
Seventeen species and one isolate, UWFP-348, could be divided into eight groups with ITS2 region PCR productsⱕ2 bases apart in mean length (Table 3). Products in each group contained species-specific ITS2 region DNA: sequence simi-larities ranged from 65.1 to 97.7%, and products were easily distinguished from each other by unique restriction patterns after digestion withAseI,BanI,EcoRI,HincII, orStyI (Table 3). In total, 434 isolates were analyzed in this study, and 427 (98.4%) had concordant identification by phenotypic, bio-chemical, and ITS2-based methods. Forty-three species-spe-cific ITS2 region DNA sequences were submitted to GenBank (Table 2).
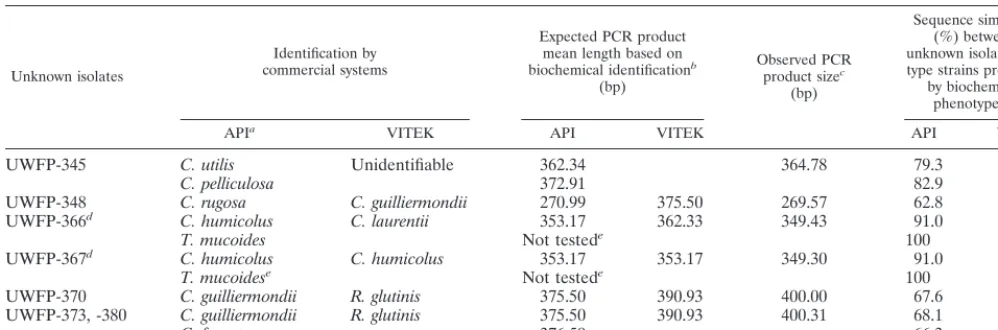
Unusual isolates identified by ITS2 sequence polymor-phisms. Seven clinical isolates provided discrepant data for phenotypic and ITS2-based identification: the observed ITS2 region PCR product length was not the expected length pre-dicted by the biochemical phenotype of each isolate (Table 4). Observed ITS2 region sequences also differed from expected, and their similarity ranged from 57.5 to 100%.
Isolates UWFP-366 and -367 biochemically resembled C. laurentii,C. humicolus, andTrichosporon mucoides(Table 4). Type strains of these yeasts displayed ITS2 region similarities of 79.4, 91, and 100%, respectively, when compared to UWFP-366 and -367. Our observations (Table 2) and the work of others (26) indicate conspecific strains generally have fewer than 1% nucleotide substitutions in ITS2 region DNA, and we conclude that UWFP-366 and -367 are probably isolates ofT. mucoides. Also of interest, two clinical strains identified as
Trichosporon beigeliiby the VITEK system (Trichosporon asa-hii, Table 1) had identical ITS2 region sequences which were ⬎99% similar to sequences ofT. asahii,Trichosporon ovoides, andT. inkin(26). These isolates were identified by the API 20C system asT. asahii. However, note thatT. ovoidesis not part of the API 20C database.
Isolates UWFP-370, -373, and -380 biochemically resembled
C. guillermondii,C. famata, andRhodotorula glutinis(Table 4), and type strains of these yeasts displayed 66.3 to 91.1% se-quence similarity with UWFP-370, -373, and -380. Although identity of these strains will require additional studies, their ITS2 region sequences most closely resembled those of R. rubra(99.5% similarity for UWFP-370 and 99.3% similarity for UWFP-373, and -380).
The remaining isolates, UWFP-345 and -348, displayed
ⱕ83% ITS2 region sequence similarity with the type strains predicted from their respective biochemical phenotypes (Table 4). Previous observations indicate ITS2 similarities less than 95% correspond to nuclear DNA complementarity of less than 20% (26). Nuclear DNA complementarity among different species is less than 40%, varieties or subspecies display 40 to 80%, and members of a biological species generally exceed 80% (23). These isolates are therefore very likely to be species absent from the current ITS2 region databases (GenBank, EMBL, DDBJ, and Table 2).
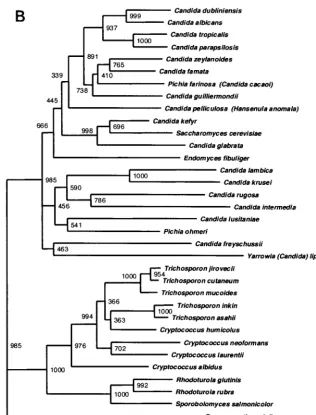
[image:6.612.53.552.85.250.2]These data are supported by the phylogenetic trees con-structed with ITS2 region sequences (Fig. 3A) by using differ-ent treeing algorithms. The global tree topology among the different treeing methods was very similar (data not shown) with species from each genus generally segregating into a dis-tinct cluster; however, the local topology within the major clusters varied slightly. Moreover, the phylogenetic positions of the clinical strains which remained phenotypically unidentified could be inferred. UWFP-345 was most closely related to C. glabrata, C. kefyr, andS. cerevisiae with 73.6, 72.5, and 66% ITS2 region DNA similarity, respectively. UWFP-348 also clus-tered with otherCandidaspp. but displayed only 63.1% ITS2 region DNA similarity to Y. lipolytica. (Candida lipolytica). ITS2-based trees also supported the clustering of UWFP-370, -373, and -380 withRhodotorulaspp. (Fig. 3A). Similar phylo-genetic relationships were observed in trees constructed with 26S rDNA sequences (Fig. 3B) and with the limited number of 18S rDNA sequences available for the species we examined (data not shown). However, ITS2 sequences provided better resolution of the generaTrichosporonandCryptococcus.
TABLE 4. Unusual isolates identified by ITS2 region DNA sequence polymorphisms
Unknown isolates
Identification by commercial systems
Expected PCR product mean length based on biochemical identificationb
(bp)
Observed PCR product sizec
(bp)
Sequence similarity (%) between unknown isolates and type strains predicted
by biochemical phenotype(s)
APIa VITEK API VITEK API VITEK
UWFP-345 C. utilis Unidentifiable 362.34 364.78 79.3
C. pelliculosa 372.91 82.9
UWFP-348 C. rugosa C. guilliermondii 270.99 375.50 269.57 62.8 57.5
UWFP-366d C. humicolus C. laurentii 353.17 362.33 349.43 91.0 79.4
T. mucoides Not testede 100
UWFP-367d C. humicolus C. humicolus 353.17 353.17 349.30 91.0 91.0
T. mucoidese Not testede 100
UWFP-370 C. guilliermondii R. glutinis 375.50 390.93 400.00 67.6 91.1
UWFP-373, -380 C. guilliermondii R. glutinis 375.50 390.93 400.31 68.1 90.8
C. famata 376.59 66.3
aMore than one possible identification is sometimes given by the API system. bFrom Tables 1 and 2.
cFrom Table 1.
dUWFP-366 and -367 have identical ITS2 region sequences. eSequence ofT. mucoides(AB018031) (26) from GenBank.
on May 15, 2020 by guest
http://jcm.asm.org/
DISCUSSION
By examining 401 clinical isolates, 6 reference strains, and 27 type strains, we demonstrated that ITS2 region sequence poly-morphisms can be used to identify 34 species of pathogenic yeasts. ITS2 region PCR product lengths were rapidly deter-mined with single-base precision by using capillary electro-phoresis and were sufficient to identify 92% of the clinical isolates in this study. The remaining isolates produced ITS2 region PCR products with similar mean lengths (ⱕ2 bases apart) and were distinguished by restriction enzyme analysis. These species-specific ITS2 region polymorphisms were con-firmed by sequence analysis of 93 isolates. Thus, of 434 isolates
examined, 427 (⬎98%) had concordant identification by phe-notypic and ITS2-based methods.
Our results confirm and extend the work of Turenne et al. (29), demonstrating ITS2 length polymorphisms among a col-lection of molds and yeasts. Their study examined 26 clinical isolates and a large number of reference strains, but they did not observe intraspecies variability when more than one strain was examined. In contrast, we found intraspecies variability and clinical isolates that were more similar to each other than they were to their respective type strains (Table 2). We also identified eight groups of species whose PCR product lengths were sufficiently similar that restriction analysis was required for specific identification (Table 3). These observations
indi-FIG. 3. (A) ITS2 sequence-based phylogenetic tree of clinical yeast isolates. Neighbor-joining dendrogram with 1,000 bootstraps was based on 463 aligned positions of complete ITS2 sequences and adjacent partial sequences of 5.8S and 28S rRNA genes from 38 yeast isolates, including 26 type strains. TheP. cariniiITS2 sequence retrieved from GenBank (accession no. U07226) was used as the outgroup, and sequences ofC. albicansandC. parapsilosiswere previously published (GenBank accession no. L28817 and U10988, respectively). UWFP-373 and UWFP-380 have 100% identical sequence to UWFP-370. UWFP-373 and -380 were therefore not included in the tree. (B) 26S sequence-based phylogenetic tree showing the relationships of 34 yeast taxa. Neighbor-joining dendrogram was based on 557 aligned positions of the 5⬘end of 26S rRNA gene. Sequences with the following accession numbers were retrieved from GenBank:C. albicans, no. U45776;C. dubliniensis, no. AB031020;C. famata, no. U94927;Candida freyschussii, no. AF017242;C. glabrata, no. U44808;C. guilliermondii, no. U45709;C. intermedia, no. U44809;C. kefyr, no. U94924;C. krusei, no. U76347;C. lambica, no. U75726;C. lusitania, no. U44817;C. parapsilosis, no. U45754;Candida rugosa, no. U45727;C. tropicalis, no. U45749; C. zelanoides, no. U45832;C. albidus, no. AF137605;C. humicolus, no. AF189854;C. laurentii, no. AF075469;C. neoformans, no. AF189845;E. fibuliger, no. U40089; H. anomala, no. U74592;P. farinosa, no. U45739;P. ohmeri, no. U45702;R. glutinis, no. AF070430;R. rubra, no. AF189961;S. cerevisiae, no. U44806;S. salmonicolor, no. AF189979;T. asahii, no. AF105393;T. cutaneum, no. AF075483;T. inkin, no. AF105396;T. jirovecii, no. AF105398;T. mucoides, no. AF075515;Y. lipolytica, no. U40080; andP. carinii, no. M86760 used for outgroup. Lower bars indicate the genetic distance. Numbers at each node indicate percent bootstrap values.
on May 15, 2020 by guest
http://jcm.asm.org/
cate that strain variability is an important consideration for applying this method in the clinical laboratory and underscores the importance of including clinical isolates when developing new diagnostic methods. For example, by examining a large number of strains, we confirmed that ITS2 region PCR product length alone can reliably distinguishC. albicans and C. dub-liniensis (Table 1). Because these organisms produce germ tubes and share biochemical characteristics (1),C. dubliniensis
is easily misidentified asC. albicans(20). Xylose assimilation distinguishesC. albicans(88 to 90% positive) andC. dublini-ensis(0% positive), but is not 100% reliable (I. F. Salkin, W. R. Pruitt, A. A. Padhye, D. Sullivan, D. Coleman, and D. H. Pincus, Letter, J. Clin. Microbiol.36:1467, 1998) and takes 2 to 3 days (5). SinceC. dubliniensisis more likely thanC. albicans
to develop resistance to fluconazole, misidentification can im-pact the outcome of antifungal treatment (27).
Our results also agree with the examination of the ITS1 and ITS2 regions ofTrichosporonspp. by Sugita et al. (26). They also found intraspecies or strain-specific sequence variability in the ITS2 region and, with rare exceptions, documented that different species contain ITS2 region DNA with less than 99%
sequence similarity. Our results extend these observations to a broader population of clinically relevant yeasts, including members of the basidiomycetes (Cryptococcus, Rhodotorula,
Sporobolomyces, andTrichosporonspp.) and ascomycetes (Ta-ble 2 and Fig. 3A).
The difficulty commercial systems have with identifying some yeast species (2, 5) was evident for seven isolates in our study (Table 4) and demonstrates the potential usefulness of sequence-based identifications. Two isolates likely to be T. mucoideswere correctly suggested as one of two possible iden-tifications by the API system and were incorrectly designated asCryptococcusspp. by the VITEK; the genus of three prob-ableRhodotorulaspp., closely related toR. rubra, was correctly suggested by the VITEK system but designated asCandida
[image:8.612.143.459.69.484.2]spp. by the API; and two isolates were either unidentifiable (VITEK) or incorrectly designated asCandidaspp. (API and VITEK) (Table 4). Sequence analysis of these isolates con-firmed that the ITS2 region length polymorphisms correctly resolved discrepant results between phenotypic and genotypic methods (Table 4), and phylogenetic trees constructed with the
FIG. 3—Continued.
on May 15, 2020 by guest
http://jcm.asm.org/
available ITS2 sequences provided additional support for the genotypic data (Fig. 3A).
In general, the topology of a phylogenetic tree depends on the characteristics of DNA sequences and tree construction algorithms utilized (18). The global topology of trees con-structed with ITS2, 26S, and 18S rDNA sequences displayed striking similarities (Fig. 3A and B). Our ITS2-based trees displayed generally good bootstrap support at terminal branches, distinguishing different species, and provided better distinction of the generaCryptococcus and Trichosporon. In-terestingly, we found thatC. humicolusclustered with the Tri-chosporonclade in trees constructed with both ITS2 and 26S rDNA sequences as noted previously by others (8). However, the bootstrap support at the basal branches was stronger with 26S and 18S rDNA trees when compared to ITS2. The branch-ing pattern of the more basal lineages within each cluster differed among ITS2, 26S, and 18S rRNA gene-based phylo-genetic trees. These results were expected, as the bootstrap values at the basal branches within the major clusters were low with all three markers. This observation has also been made by others (26), and the most accurate phylogenetic relationships may be provided by the less rapidly evolving structural rRNA genes (12, 18). However, the ability to evaluate short ITS2 region amplicons with sufficient polymorphism to distinguish yeast species provides advantages as a diagnostic method. Analysis is rapid and amenable to automated methods as dem-onstrated here and by others (29), and the polymorphisms are sufficient that array-based hybridization schemes will be useful analytical tools in the future. Our data establish an initial database of ITS2 length and sequence polymorphisms for 34 species of yeasts that is validated with over 400 clinical isolates, and we anticipate that this will facilitate the rapid diagnosis of fungal infections directly from patient specimens.
REFERENCES
1.Bikandi, J., R. S. Millan, M. D. Moragues, G. Cebas, M. Clarke, D. C.
Coleman, D. J. Sullivan, G. Quindos, and J. Ponton.1998. Rapid
identifi-cation ofCandida dubliniensisby indirect immunofluorescence based on differential localization of antigens onC. dubliniensisblastospores and
Can-dida albicansgerm tubes. J. Clin. Microbiol.36:2428–2433.
2.Dooley, D. P., M. L. Beckius, and B. S. Jeffrey.1994. Misidentification of
clinical yeast isolates by using the updated Vitek Yeast Biochemical Card. J. Clin. Microbiol.32:2889–2892.
3.Elie, C. M., T. J. Lott, E. Reiss, and C. J. Morrison.1998. Rapid
identifi-cation ofCandidaspecies with species-specific DNA probes. J. Clin. Micro-biol.36:3260–3265.
4.Fan, M., B. P. Currie, B. P. Guttell, M. A. Ragan, and A. Casadevall.1994.
The 16S-like, 5.8S, and 23S-like rRNA’s of the two varieties of Cryptococcus neoformans: sequence, secondary structure, phylogenetic analysis, and re-striction fragment polymorphisms. J. Med. Vet. Mycol.32:163–180.
5.Fenn, J. P., H. Segal, B. Barland, D. Denton, J. Whisenant, H. Chun, K.
Christofferson, L. Hamilton, and K. Carroll.1994. Comparison of updated
Vitek Yeast Biochemical Card and API 20C yeast identification systems. J. Clin. Microbiol.32:1184–1187.
6.Fromtling, R. A.1995. Mycology, p. 697–855.InP. R. Murray, E. J. Baron,
M. A. Pfaller, F. C. Tenover, and R. H. Yolken (ed.), Manual of clinical microbiology, 6th ed. ASM Press, Washington, D.C.
7.Gleason, T. G., A. K. May, D. Caparelli, B. M. Farr, and R. G. Sawyer.1997.
Emerging evidence of selection of fluconazole-tolerant fungi in surgical intensive care units. Arch. Surg.132:1197–1201. (Discussion, 1202.)
8.Gueho, E., L. Improvisi, R. Christen, and G. S. de Hoog.1993. Phylogenetic
relationships of Cryptococcus neoformans and some related basidiomycet-ous yeasts determined from partial large subunit rRNA sequences. Antonie Leeuwenhoek.63:175–189.
9.Hernandez, S. M., G. P. Morlock, W. R. Butler, J. T. Crawford, and R. C.
Cooksey.1999. Identification of Mycobacterium species by PCR-restriction
fragment length polymorphism analyses using fluorescence capillary electro-phoresis. J. Clin. Microbiol.37:3688–3692.
10.Kern, M. E., and K. S. Blevins.1997. Medical mycology: a self-instructional
text, 2nd ed., p. 146–152. Jean-Franc¸ois Vilain, Philadelphia, Pa.
11.Kirkpatrick, W. R., S. G. Revankar, R. K. McAtee, J. L. Lopez-Ribot, A. W.
Fothergill, D. I. McCarthy, S. E. Sanche, R. A. Cantu, M. G. Rinaldi, and
T. F. Patterson.1998. Detection ofCandida dubliniensisin oropharyngeal
samples from human immunodeficiency virus-infected patients in North America by primary CHROMagar candida screening and susceptibility test-ing of isolates. J. Clin. Microbiol.36:3007–3012.
12.Kurtzman, C. P., and C. J. Robnett.1998. Identification and phylogeny of
ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences. Antonie Leeuwenhoek.73:331–371.
13.Kurtzman, C. P., and C. J. Robnett.1997. Identification of clinically
impor-tant Ascomycetous yeasts based on nucleotide divergence in the 5⬘end of the large-subunit (26S) ribosomal DNA gene. J. Clin. Microbiol.35:1216–1223.
14.Lodder, J. (ed.).1970. The yeasts: a taxonomic study, 2nd ed., p. 917–1079.
North-Holland publishing company, Amsterdam, The Netherlands.
15. Lott, T. J., B. M. Burns, R. Zancope-Oliveira, C. M. Elie, and E. Reiss.1998.
Sequence analysis of the internal transcribed spacer 2 (ITS2) from yeast species within the genus Candida. Curr. Microbiol.36:63–69.
16. Lott, T. J., R. J. Kuykendall, and E. Reiss.1993. Nucleotide sequence
analysis of the 5.8S rDNA and adjacent ITS2 region of Candida albicans and related species. Yeast9:1199–1206.
17. Makimura, K., S. Y. Murayama, and H. Yamaguchi.1994. Detection of a
wide range of medically important fungi by the polymerase chain reaction. J. Med. Microbiol.40:358–364.
18. Morrison, D. A.1996. Phylogenetic tree-building. Int. J. Parasitol.26:589–
617.
19. Oda, Y., M. Yabuki, K. Tonomura, and M. Fukunaga.1997. A phylogenetic
analysis of Saccharomyces species by the sequence of 18S-28S rRNA spacer regions. Yeast13:1243–1250.
20. Odds, F. C., L. Van Nuffel, and G. Dams.1998. Prevalence ofCandida
dubliniensisisolates in a yeast stock collection. J. Clin. Microbiol.36:2869–
2873.
21. Pfaller, M., and R. Wenzel.1992. Impact of the changing epidemiology of
fungal infections in the 1990s. Eur. J. Clin. Microbiol. Infect. Dis.11:287– 291.
22. Pfaller, M. A., S. A. Messer, A. Houston, M. S. Rangel-Frausto, T. Wiblin,
H. M. Blumberg, J. E. Edwards, W. Jarvis, M. A. Martin, H. C. Neu, L. Saiman, J. E. Patterson, J. C. Dibb, C. M. Roldan, M. G. Rinaldi, and R. P.
Wenzel.1998. National epidemiology of mycoses survey: a multicenter study
of strain variation and antifungal susceptibility among isolates of Candida species. Diagn. Microbiol. Infect. Dis.31:289–296.
23. Price, C. W., G. B. Fuson, and H. J. Phaff.1978. Genome comparison in yeast
systematics: delimitation of species within the generaSchwanniomyces,
Sac-charomyces,Debaryomyces, andPichia. Microbiol. Rev.42:161–193.
24. Ramani, R., S. Gromadzki, D. H. Pincus, I. F. Salkin, and V. Chaturvedi.
1998. Efficacy of API 20C and ID 32C systems for identification of common and rare clinical yeast isolates. J. Clin. Microbiol.36:3396–3398.
25. Reiss, E., K. Tanaka, G. Bruker, V. Chazalet, D. Coleman, J. P. Debeaupuis,
R. Hanazawa, J. P. Latge, J. Lortholary, K. Makimura, C. J. Morrison, S. Y. Murayama, S. Naoe, S. Paris, J. Sarfati, K. Shibuya, D. Sullivan, K. Uchida,
and H. Yamaguchi.1998. Molecular diagnosis and epidemiology of fungal
infections. Med. Mycol.36(Suppl. 1):249–257.
26. Sugita, T., A. Nishikawa, R. Ikeda, and T. Shinoda.1999. Identification of
medically relevantTrichosporonspecies based on sequences of internal tran-scribed spacer regions and construction of a database forTrichosporon iden-tification. J. Clin. Microbiol.37:1985–1993.
27. Sullivan, D., and D. Coleman.1998.Candida dubliniensis: characteristics and
identification. J. Clin. Microbiol.36:329–334.
28. Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G.
Higgins.1997. The CLUSTAL_X windows interface: flexible strategies for
multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res.25:4876–4882.
29. Turenne, C. Y., S. E. Sanche, D. J. Hoban, J. A. Karlowsky, and A. M.
Kabani.1999. Rapid identification of fungi by using the ITS2 genetic region
and an automated fluorescent capillary electrophoresis system. J. Clin. Mi-crobiol.37:1846–1851.
30. Warren, N. G., and K. C. Hazen.1999. Candida, Cryptococcus, and other
yeasts of medical importance, p. 1191–1197.InP. R. Murray, E. J. Baron, M. A. Pfaller, F. C. Tenover, and R. H. Yolken (ed.), Manual of clinical microbiology, 7th ed. American Society for Microbiology, Washington, D.C.
31. White, T. J., T. Bruns, S. Lee, and J. Taylor.1990. Amplification and direct
sequencing of fungal ribosomal RNA genes for phylogenetics, p. 315–322.In
M. A. Innis, D. H. Gefland, J. J. Sninsky, and T. J. White (ed.), PCR protocols: a guide to methods and applications. Academic Press, Inc., New York, N.Y.
32. W right, W. L., and R. P. Wenzel.1997. Nosocomial Candida. Epidemiology,
transmission, and prevention. Infect. Dis. Clin. N. Am.11:411–425.