ABSTRACT
BASSETT, HARRIS. Peptide Modified Microfluidic Devices for the Affinity Isolation of Circulating Tumor Cells. (Under the direction of Dr. Steve Soper.)
Metastasis from the primary tumor and invasion of distant organs is the predominate cause of mortality in cancer patients. Metastasis is initiated by cancer cells circulating in the blood, called Circulating Tumor Cells (CTCs), that are released from the primary tumor and potentially metastatic sites as well. Microfluidic devices have shown
great potential for the detection of tumor metastasis via enumeration of CTCs, which permits for more effective treatments and consequently, potentially increased survival rates. Herein we describe the design and operation of a microfluidic polymeric device with channel surfaces decorated with anti-EpCAM peptides for the isolation of CTCs of
epithelial origin. Low-cost fabrication of the microfluidic device was accomplished via hot embossing into a polymeric substrate, Cyclic Olefin Copolymer (COC) in particular. A parallel array of sinusoidal channels associated with the device permitted high-throughput sample processing rates (>1.5 mL/h), high recovery and also, high purity of the affinity selected CTCs. Isolation of CTCs as represented by the SKBR3 model cell line
Peptide Modified Microfluidic Devices for the Affinity Isolation of Circulating Tumor Cells
by Harris Bassett
A thesis submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Master of Science
Biomedical Engineering
Raleigh, North Carolina 2017
APPROVED BY:
Glenn Walker Michael Gamcsik
DEDICATION
BIOGRAPHY
A native of Raleigh, I graduated from Enloe High School by Summer of 2008. I entered North Carolina State University in the Fall of that same year, to pursue
undergraduate studies for Biochemistry. I took a break from science during the year of 2010-2011, to study sociology and economy in Grenoble France. In doing so, I finished the requirements for a Minor in French. A wonderful two years of core biochemistry coursework was to follow, culminating in my graduation from the program by the
Summer of 2013. My classmate Garrett and I, then embarked on a self-supported summer-long cycling trip across the United States, from the Atlantic to the Pacific. Soon afterwards, I became the coffee roaster and delivery driver for Cup-A-Joe. An
opportunity that afforded me exceptional insight into the operations of a small business. During this time, I became inspired by the ideas of Charles Babbage; and so
my interest in microfluidics began. I thank Dr. Soper for accepting me into his
TABLE OF CONTENTS
LIST OF TABLES ... v
LIST OF FIGURES ... vi
INTRODUCTION ... 1
Cancer Disease and Circulating Tumor Cells ... 1
Epithelial Cell Adhesion Molecule ... 2
Microfluidic devices for isolating Circulating Tumor Cells ... 4
Antibodies as an affinity agent ... 11
Other affinity agents ... 13
Synthetic peptides as an affinity agent ... 15
The anti-EpCAM peptide. ... 16
Research objectives and goals ... 17
EXPERIMENTAL ... 18
Evaluation of Peptide-Modified Microfluidic Devices for the Affinity Isolation of EpCAM+ Circulating Tumor Cells (CTCs) ... 18
Materials ... 20
Cell Culture ... 20
Production of the microfluidic devices in Cyclic Olefin Copolymer (COC). ... 21
Attachment of the peptide for affinity selection. ... 23
Blood samples ... 25
Protocol for CTC isolation assay ... 25
Fluorescence microscopy ... 26
High Performance Reverse Phase Liquid Chromatography with Mass Spectroscopy (HPLC/MS) ... 27
Polymeric microfluidic device for CTC isolation ... 28
Modification of the microfluidic device with the anti-EpCAM peptide. ... 31
Isolation of model cell lines ... 34
Isolation of CTCs from clinical samples ... 41
Stability of the peptide and relation to non-specific binding ... 44
Conclusion ... 47
LIST OF TABLES
Table 1. Recovery of SKBR3 cells in PBS as a function of peptide concentration used during preparation of the microfluidic device. ... 38 Table 2. Summary of clinical results for healthy donors and patients diagnosed with EOC
LIST OF FIGURES
Figure 1. The process of intravasation, circulation, and extravasation. Image taken from Meadows, G.; Zhang, H. Alcohol Research Current Reviews 2015, 37(2). ... 1 Figure 2. Space-fill model of the extracellular domain of cis-dimeric EpCAM labeled with
antibodies known to target specific epitopes. Antibodies for which the epitope is unknown are listed below. Image taken from Pavšič, M.; et al. Nature
Communications 2014, 5(4764). ... 3 Figure 3. Confocal laser scanning microscopic image of immunofluorescent labeled
esophageal carcinoma cells. EpCAM is shown in green, the nucleus in blue. Image taken from Tsaktanis, T.; et al. J. Biol. Chem. 2015, 290, 24574-24591. ... 4 Figure 4. Spiral microfluidic devices isolate CTCs by centrifugal forces Beginning at
cross-sectional view X, CTCs are located with WBCs and RBCs. Over the course of 10 cm, the CTCs are shuttled away from the WBCs and RBCs. Image taken from Hou, H. et al.; Sci Rep 2013, 3, 1259. ... 6 Figure 5. Device utilizing 8 µm pores to filter (isolate) CTCs from blood. The device cassette
(A) and filter (B) are shown. Isolated CTCs can be seen in the pores (D). Schematic of the filter (E). Image taken from Yusa, A. et al.;PLoS One 2014, 9(2), e88821. ... 7 Figure 6. Example of a microfluidic device that incorporates an array of microposts to
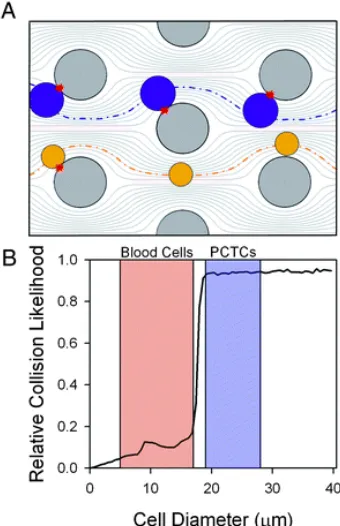
improve the isolation yield of CTCs from blood. Supporting instrumentation (A) creates a pressure difference that pumps blood through the device (B). CTCs are captured by surface immobilized anti-EpCAM antibody (C). Image from Nagrath, S. et al. Nature 2007, 450(7173), 1235-1239. ... 8 Figure 7. The GEDI deisgn. (A) Due to their diameter, CTCs interact with the surfaces of the
pillars, but blood cells remain in laminar and have lower probability of interactions. (B) Graph displaying probabilities of cell-pillar collisions as a
function of cell diameter. Figure from Gleghorn, J. et al. Lab Chip 2010, 10, 27-29. . 9 Figure 8. Example of the Herringbone design. The relief of chevrons produces dual vortices
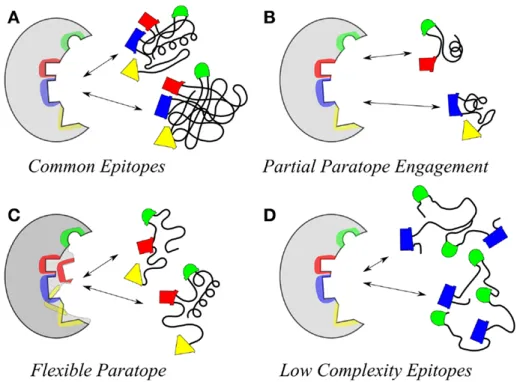
as illustrated by the arrows for forward and reverse flows. Image from Foley, J.; Mashadi-Hossein, A.; Fu, E.; Finlayson, B.; Yager, P.; Lab Chip 2008, 8, 557-564. ... 10 Figure 9. Illustration of conditions that give rise to different specificities between paratope
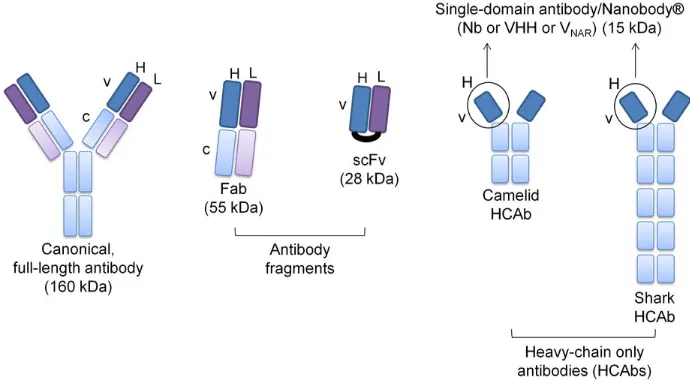
and epitope (left and right in each image, respectively). An exact match between paratope and epitope is possible (A), as well as several truncated epitopes showing cross-reactivity (B). A flexible paratope can fit less-than-perfect epitope orientations (C), or the fit can be a result of flexible antigens presenting the same epitope orientations. Image taken from Kieber-Emmons, T.; et al. Frontiers in Immunology 2014, 5(308). ... 12 Figure 10: Schematic showing distinguishing features of mAb, Fab, scFV and HCAbs. Heavy
chains are shown in blue, light chains in violet. Nanobody fragment of HCAb is circled. Image from Doshi, R.; et al.; Scientific Reports 2014, 4, 6760. ... 14 Figure 11. Primary structure of the anti-EpCAM peptide. From left to right the 23 amino acid
residues are VRRDAPRFSSMQGLDACGGNNCNN. Image created from sequence at <http://www.tulane.edu/~biochem/WW/PepDraw/>. ... 16
Figure 12. The brass mold produces four curvilinear device substrates per cycle. ... 22
Figure 13. Jenoptik Hex 03 hot embossing machine and supporting equipment (A). The brass mold is secured to the top of the chamber (B), a plate of COC is placed on the bottom. A computer script, closes the chamber, begins a vacuum, and
independently heats the mold and COC plate. ... 22 Figure 14. A Hewlett Packard 5890A Gas Chromatograph (A) is used to rapidly raise the
curvilinear devices can be seen in glass sandwich with binding clips. The result is an assembled curvilinear microfluidic device (B). ... 23 Figure 15. Schematic of EDC/NHS coupling. Group 1 represents the COC substrate. Group 2
represents NeutrAvidin. Image taken from "Carbodiimide Crosslinker Chemistry" at www.ThermoFisher.com ... 24 Figure 16. A tumor cell is classified as DAPI and Cytokeratin 19 positive, but CD45 negative
(top row). A white blood cell will stain DAPI and CD45 positive, but Cytokeratin 19 negative (bottom row). ... 27 Figure 17. SEM images of the device hot-embossed into COC. Overview of the curvilinear
channel geometry (image a), view of the channel (image b), the entrance to the curvilinear channels (image c), cut away to reveal the cross-section of the channel (image d). Image taken from Hupert et al. 2014. ... 29 Figure 18. (A) CAD drawing of the module where the large arrow indicates sample flow
direction through the selection channels and (B) SEM of the selection bed showing high aspect ratio sinusoidal microchannels and (C) closeup of one channel. ... 30 Figure 19. (A) Schematic showing attachment strategy for a biotinylated peptide to the
NeutrAvidin-modified COC device. (B) The chemical structure of the EpCAM specific peptide used in the study. ... 32 Figure 20. Verifying NeutrAvidin immobilization experimentally. The COC walls were
activated by UV, then modified by EDC/NHS, either without (A) or with (B) the addition of NeutrAvidin afterwards. A biotin-dye was used to measure
NeutrAvidin binding. Data was analyzed by using the line plot function in ImageJ (C and D). ... 33 Figure 21. One dimensional flow cytometry histogram comparing EpCAM expression in
Hs578T cells (left column) and SKBR3 cells (right column). SKBR3 cells express EpCAM, whereas Hs578T cells do not. ... 35 Figure 22. Fluorescent microscopy of SKBR3 (top row) and HS578T (bottom row) cell lines.
Calcein dye (green) shows good viability following release by trypsin. ... 36 Figure 23. Histogram of data correlating linear flow velocity to CTC recovery efficiency for
both PBS buffer and blood. ... 39 Figure 24. Count of WBCs non-specifically co-isolated on a microfluidic device when either
mAb or peptide was immobilized to the surface. ... 41 Figure 25. (A) Fluorescent microscope images of a CTC isolated with peptide-functionalized
device from a patient with endometrial carcinoma. (B) Images of CTCs isolated with antibody-functionalized device from a patient with epithelial ovarian cancer.
... 43
Figure 26. Degradation of natural aspartic acid (L-Asp) to a succinimide intermediate via nucleophilic substitution. Image from Takahashi, O.; Kirikoshi, R.; Manabe, N. Int J Mol Sci 2015, 16(1), 1613-1626. ... 44 Figure 27. Reverse Phase Liquid Chromatography data at days 1, 9, and 23 for peptide stored
at 22˚C. The anti-EpCAM peptide is initially the dominate biological constituent in solution, but by day 9, degradation leads to competing constituents, and finally by day 23, the peptide is a minor constituent. Mass Spectroscopy data (bottom right image) shows the peptide and 3 major degradation products that form the major biological constituents in solution. ... 45 Figure 28. Histogram of HPLC data showing purity of anti-EpCAM peptide in water at various
INTRODUCTION
Cancer Disease and Circulating Tumor Cells
Tumor metastasis is the predominate cause of mortality in cancer patients (>90% of all deaths).1 The first step in metastasis is invasion of neighboring tissue from cancer
cells emanating from the primary tumor.1 At this point, the tumor is at most 50 to 100
µm from the vasculature, whereupon tumor cells can enter the circulation.2 Such cancer
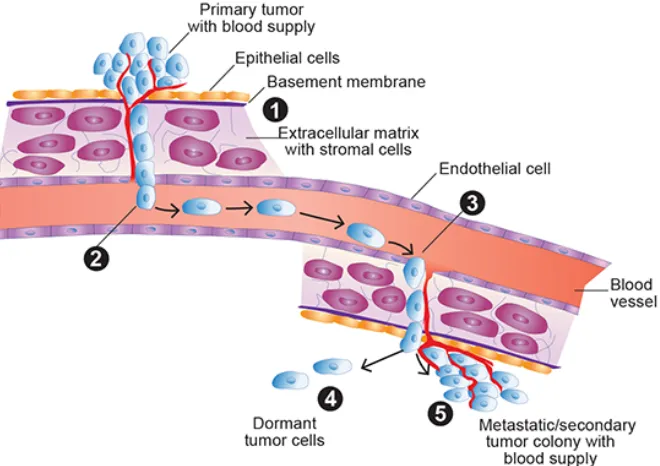
cells are termed circulating tumor cells (CTCs), see Figure 1.
Figure 1. The process of intravasation, circulation, and extravasation. Image taken from Meadows, G.; Zhang, H. Alcohol Research Current Reviews 2015, 37(2).
independent predictor of overall survival rates for cancer patients.4,5 More specifically, a
positive correlation between the number of CTCs, the occurrence of metastasis, and the time until relapse has been observed.6 CTCs have shown prognostic relevance in
metastatic breast, prostate, and colon cancer. Furthermore, xenograft models have demonstrated causation of secondary tumor sites (i.e., metastasis) by CTCs. Isolated CTCs from human breast cancer patients have been shown to be capable of initiating metastasis in mouse models.7
The use of CTCs for the detection of cancer presents many benefits. Because blood collection is a simple and minimally invasive process, isolation of CTCs can be performed frequently, enabling more frequent patient monitoring for disease relapse or effectiveness of chemotherapeutic treatment. In the future, CTCs and other liquid biopsy
markers (i.e., circulating cell-free DNA and exosomes) from whole blood could complement a routine physical exam as a non-invasive way to screen for the potential presence of cancer disease at a very early stage of tumorigenesis.
Epithelial Cell Adhesion Molecule
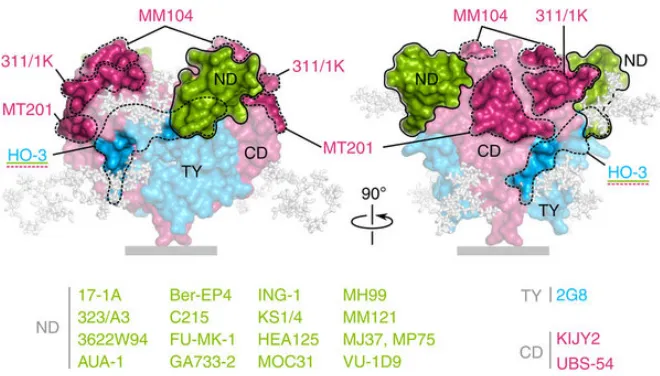
Epithelial Cell Adhesion Molecule (EpCAM) is a 40 kDa transmembrane glycoprotein composed of 314 amino acids expressed by epithelial cells and present in many human carcinomas.8 The extracellular domain of EpCAM (see Figure 2) contains
repeats that are reminiscent of Epidermal Growth Factor, and thereby contribute to reciprocal homophilic cell adhesion.9 EpCAM in normal cells is predominately located in
speculated that EpCAM on normal epithelia is sequestered and less accessible to antibodies than EpCAM in cancer tissue, where it is homogeneously distributed on the cancer cell surface (see Figure 3).10 EpCAM is expressed on essentially all human
adenocarcinomas, certain squamous cell carcinomas, retinoblastoma, and hepatocellular carcinoma.
Figure 2. Space-fill model of the extracellular domain of cis-dimeric EpCAM labeled with antibodies known to target specific epitopes. Antibodies for which the epitope is unknown are listed below. Image taken from Pavšič, M.; et al. Nature Communications 2014, 5(4764).
EpCAM has been shown to be a predictor of poor survival in patients inflicted with bladder, breast, colon, gastric, lung carcinomas, melanoma, and prostate
cancers.11,12,13,14,15,16,17,18,19 Inhibition of EpCAM transcription in breast cancer cells by
siRNA treatment results in a 96% reduction in cellular invasion.20
Figure 3. Confocal laser scanning microscopic image of immunofluorescent labeled esophageal carcinoma cells. EpCAM is shown in green, the nucleus in blue. Image taken from Tsaktanis, T.; et al. J. Biol. Chem. 2015, 290, 24574-24591.
Microfluidic devices for isolating Circulating Tumor Cells
Detection of cancer has traditionally been accomplished by X-ray computed
tomography, ultrasound, and magnetic resonance imaging.21 In order to understand the
genetic composition of the tumor and provide proper treatment, an invasive solid tissue biopsy is typically performed.21 The isolation of CTCs from whole blood allows for a much
less invasive method of collecting tumor cells. Such a test has been termed the “liquid biopsy”.22
CTC detection is a very promising field because many technologies have been developed over the past years to efficiently isolate CTCs directly from whole blood. Detection of CTCs is technically very challenging because these cells occur at very low frequency; 1-1,000 CTCs in a background of millions of white blood cells and billions of red blood cells per milliliter of blood.23 Microfluidic devices have already shown
promising results for the capture of CTCs from whole blood. Such devices are typically judged by three criteria:21 (i) Throughput; the rate at which the sample is processed. (ii)
mode by which microfluidic devices capture CTCs can be divided into two categories: physical and biological selection.24
The physical selection method uses device dimensions to isolate cells by size and
shape. In laminar flow, the path of a cell is determined by its center of mass.25 This
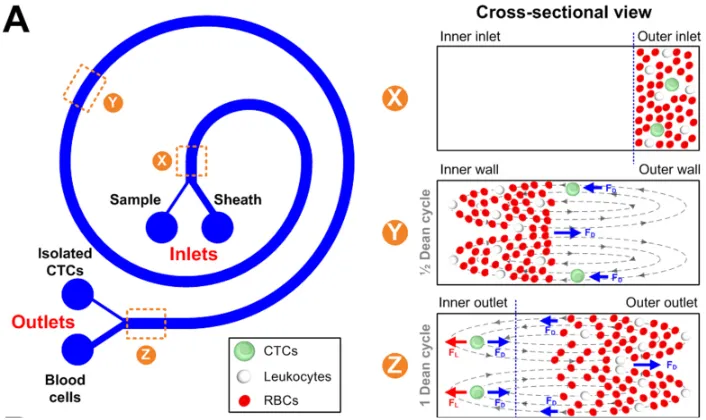
phenomenon allows for size selection based on “pinching” of the microfluidic channel walls. A spiral design (as shown in Figure 4) operating in the inertial regime can take
advantage of counter-rotating vortices, caused by Dean flow, that occur perpendicular to
the direction of bulk flow, to move cells across streamlines based on their size.26 Larger
cells are directed to the interior of the channel wall while smaller cells are directed to the exterior of the channel wall. The spiral device designed by Hou et al. allowed for cell recovery in excess of 85%, but at a purity of only 10% (CTCs/(leukocytes+CTC)). Optimal
Figure 4. Spiral microfluidic devices isolate CTCs by centrifugal forces Beginning at cross-sectional view X, CTCs are located with WBCs and RBCs. Over the course of 10 cm, the CTCs are shuttled away from the WBCs and RBCs. Image taken from Hou, H. et al.; Sci Rep 2013, 3, 1259.
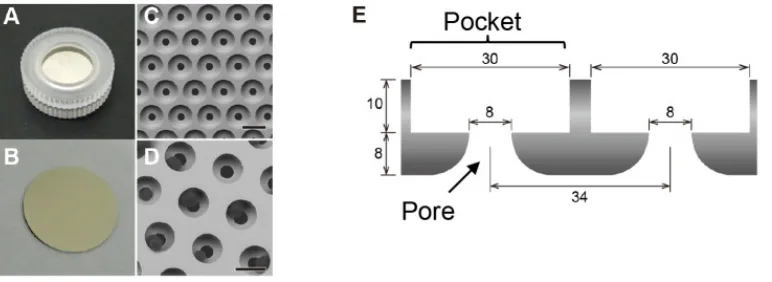
Another form of physical selection called microfiltration utilizes an array of pores to exclude CTCs (15 – 25 µm in diameter) from the sample flow, while allowing the smaller
leukocytes (10 -12 µm in diameter) and erythrocytes (6.2 to 8.2 µm in diameter) to pass.27,28 Such a device (shown in Figure 5) containing 8 µm pores was used by Yusa et al.
to recover CTCs at an efficiency of 90% with a flow throughput of 2 mL/min. The disadvantage of selecting for CTCs by physical parameters is that non-tumorous cells such as mesenchymal stromal cells and large WBC can also be filtered, reducing
Figure 5. Device utilizing 8 µm pores to filter (isolate) CTCs from blood. The device cassette (A) and filter (B) are shown. Isolated CTCs can be seen in the pores (D). Schematic of the filter (E). Image taken from Yusa, A. et al.;PLoS One 2014, 9(2), e88821.
Affinity-based microfluidic devices function by targeting specific antigens on the surface of CTCs.29 Such schemes can broadly be defined as positive selection methods. On
the contrary, negative selection schemes use an affinity agent to target and deplete non-CTC cells from a sample.30 An advantage of negative selection is the potential to enrich
CTCs with uncharacterized immunophenotypes for which the correct positive affinity antibodies are unknown.31 Positive selection assays use affinity agents for the direct
targeting of CTCs.
Separation by affinity most commonly employs antibodies conjugated to magnetic beads or the surface of a microfluidic device.29 CellSearch®, the only US FDA approved
system for CTC selection in metastatic breast, colorectal, and prostate cancer patients, utilizes ferrofluids modified with anti-EpCAM antibodies to isolate CTCs.29 Identification
nanoparticles is that they can become engulfed by phagocytes and other white blood cells, and upon magnetic retrieval, contribute to poor CTC purity.32,33
Figure 6. Example of a microfluidic device that incorporates an array of microposts to improve the isolation yield of CTCs from blood. Supporting instrumentation (A) creates a pressure difference that pumps blood through the device (B). CTCs are captured by surface immobilized anti-EpCAM antibody (C). Image from Nagrath, S. et al. Nature 2007, 450(7173), 1235-1239.
Microfluidic devices, which incorporate an antibody for positive selection of CTCs from a continuous flow of liquid, offer advantages in terms of automation. One such design is the micropost array device as seen in Figure 6. Micropost arrays vastly increase
the surface area available for immobilized affinity agents and also reduce diffusional distances, thereby increasing the probability of interaction between CTC and surface-bound antibodies.33 The cells capture efficiency of 65% was reported, with a purity of
Further work was undertaken to improve the purity of the micropillar array. One such advancement was termed geometrically enhanced differential immunocapture (GEDI). Careful placement and shift of micropillars (see Figure 7) benefitted the capture
of CTCs, while allowing smaller blood cells to remain in laminar streamlines and pass through the device without interacting with surface immobilized antibodies.35 For larger
CTCs, the recovery was improved while significant increase in purity of isolated fractions was observed (Figure 7B).
Figure 7. The GEDI deisgn. (A) Due to their diameter, CTCs interact with the surfaces of the pillars, but blood cells remain in laminar and have lower probability of interactions. (B) Graph displaying probabilities of cell-pillar collisions as a function of cell diameter. Figure from Gleghorn, J. et al. Lab Chip 2010, 10, 27-29.
employs a relief of chevrons to produce chaotic mixing, thereby shifting CTCs out of laminar streamlines, and increasing the probability of interaction between cell and surface immobilized antibody. The Herringbone is easier to manufacture than an array
of microposts and reports improved cell capture efficiency (79%) and purity (14%) when compared to the micropost array design.36
Figure 8. Example of the Herringbone design. The relief of chevrons produces dual vortices as illustrated by the arrows for forward and reverse flows. Image from Foley, J.; Mashadi-Hossein, A.; Fu, E.; Finlayson, B.; Yager, P.; Lab Chip 2008, 8, 557-564.
Positive selection microfluidic devices as mentioned above use different
is fluid shear stress generated on all surfaces that can disrupt non-specific interactions throughout the entire device, thereby increasing the purity of the isolate.28
Sinusoidal microfluidic devices with anti-EpCAM mAb covalently attached to
channel surfaces generate high shear forces that can be used for isolation of CTCs. Kamande and coworkers utilized sinusoidal channels with a recovery of 83% and the purity (calculated using the ratio of the number of CTCs selected to the total number of cells selected) of 86%.37
Antibodies as an affinity agent
Antibodies form strong non-covalent bonds to their respective antigen.38 The
binding site of the antibody is called the paratope, while the corresponding site on the antigen is called the epitope. 39 The paratope consists of ten to twenty amino acids,
resulting in a contact area of 0.4 to 8 nm2.39,40 In general, the paratope and the epitope
associate through ionic and/or hydrophobic forces, which begin to act at a distance of a few nanometers. After water molecules are expelled from the binding pocket, the distance between paratope and epitope atoms is further reduced to the extent that van der Waals forces become significant.41 The sum of van der Waals forces determines the
Figure 9. Illustration of conditions that give rise to different specificities between paratope and epitope (left and right in each image, respectively). An exact match between paratope and epitope is possible (A), as well as several truncated epitopes showing cross-reactivity (B). A flexible paratope can fit less-than-perfect epitope orientations (C), or the fit can be a result of flexible antigens presenting the same epitope orientations. Image taken from Kieber-Emmons, T.; et al. Frontiers in Immunology 2014, 5(308).
When selecting an antibody for affinity selection, it is important to choose an antibody with a favorable equilibrium dissociation constant (KD). This constant is equal
to the rate of dissociation divided by the rate of association.41
𝐊𝐃=
𝐤𝐝
𝐤𝐚
KD forcommercially available monoclonal antibodies is generally in the order of 10-9.43 Monoclonal antibodies produced by the same B-lymphocyte clone will recognize
the same epitope of the monoclonal antibody.44 This occurs because B-lymphocytes from
an inoculated animal are removed from the spleen, immortalized by fusion with a myeloma cell, and then isolated as a single clone and cultured.44 This method of
production enables a consistent supply of antibody reagent, as opposed to polyclonal
the serum of an inoculated animal.40 This difference is important because antigens can
contain multiple epitopes.40
Other affinity agents
Antibodies for toxic and animal-homologous immunogens are not possible to produce.45 For this reason among others, alternative affinity agents are used to bind to
specific antigens, such as aptamers. Aptamers are composed of nucleic acids rather than
amino acids.46 They are created in vitro by a process called systematic evolution of
ligands by exponential enrichment (SELEX).47 The process involves incubation of a
library of random oligonucleotide sequences with an antigen, elution of the bound sequences, amplification of those sequences, and then several more cycles of incubation,
elution and amplificiation.48 The typical size of an aptamer is 5 to 15 kDa, which is about
a tenth of the size of an antibody.48 The smaller size of these molecules in comparison to
antibodies allows them to bind with high resolution to antigens that are expressed at very high density. A disadvantage of aptamers is their susceptibility to nuclease mediated degradation in the absence of stabilizers (i.e., fluorination).48
There also exists alternative affinity agents that are smaller than an immunoglobin G antibody while retaining an amino acid composition. Notable examples include the antigen-binding fragment (Fab) at approximately 50 kDa in size, the single-chain variable fragment (scFv; 30 kDa), and the variable fragment (Fv; 15 kDa).49 As the
Unlike other mammals, camelids and sharks produce a type of immunoglobin G that lacks a light chain; these are called heavy-chain antibodies (HCAbs; 95 kDa).49,51 The
variable domain of the heavy chain on HCAbs (VHH; 15kDa) is often discussed in the
context of Nanobodies®, its trademarked name.52
Figure 10: Schematic showing distinguishing features of mAb, Fab, scFV and HCAbs. Heavy chains are shown in blue, light chains in violet. Nanobody fragment of HCAb is circled. Image from Doshi, R.; et al.; Scientific Reports 2014, 4, 6760.
Nanobodies (VHHs) make use of a single binding domain (linear chain of amino
acids) in order to bind to an antigen, whereas single-chain variable fragments (scFvs) require the fusion of the light and heavy chain variable fragments (Fvs) by a peptide linker to assist the non-covalent bonds in providing the correct spatial conformation of the paratope.52 Thus, with respect to single-chain variable fragments (scFvs), nanobodies
offer the advantage of reduced size for higher resolution antigen binding, and easier isolation from immune libraries by avoiding the scrambling of the two separate binding
Synthetic peptides as an affinity agent
Synthetic peptides can be used as an affinity reagent to bind specifically to an
antigen. The peptide can be identified by rational design or random selection.54 Rational
design requires prior knowledge regarding key amino acids. The random approach uses
in vitro screening methods such as phage display, thereby avoiding the host-related limitations that apply to traditional antibody generation.53 Synthetic peptides are
between 6 and 45 amino acids in length due to the practical limit of bacteriophage surface display. 54 The cost of producing synthetic peptides tends to be lower than the cost of
antibodies.55 Production can be accomplished by in vitro chemical methods such as solid
phase synthesis, allowing for the facile addition of non-natural amino acids and small
molecules.53,56
Arginine-glycine-aspartate (RGD) is a well-known peptide motif with an affinity for the integrin family of cell-surface proteins.57 Beer et al. demonstrated the ability of
immobilized peptides containing the RGD motif to capture human platelets.58 Though
this experiment demonstrated the ability of immobilized peptides containing astute
motifs to capture cells, the integrin receptor was found on a wide variety of cells thereby leading to the broad capture of cell types. Plouffe et al. demonstrated the applicability of short peptides (4 residues) to selectively capture endothelial and smooth muscle cells. 59
The anti-EpCAM peptide.
Bai et al. have reported a peptide of 23 amino acids, which binds to EpCAM with an affinity of KD = 1.98 x 10-9 mol L-1 as compared to the anti-EpCAM antibody, which is
KD = 2.69 x 10-10 mol L-1.60 Surface plasma resonance (SPR) was used to obtain the binding
affinity data, while the sequence (VRRDAPRFSSMQGLDACGGNNCNN) was identified by screening a library of peptides canndidates.60
Figure 11. Primary structure of the anti-EpCAM peptide. From left to right the 23 amino acid residues are VRRDAPRFSSMQGLDACGGNNCNN. Image created from sequence at <http://www.tulane.edu/~biochem/WW/PepDraw/>.
The peptide molecules were conjugated via biotin-streptavidin to 200 nm magnetic nanoparticles. Performance of this peptide-based affinity assay was evaluated with epithelial breast cancer cell lines MCF-7 and SKBR3, prostate cancer line PC3, and liver cancer line Hep G2. Cell lines (i.e., models of CTCs) were pre-stained with Hoechst
Research objectives and goals
The research goal of this thesis was to use a microfluidic device, activated with surface-immobilized peptides, to isolate circulating tumor cells (CTCs) from whole blood.
The peptide of choice was evaluated for specificity and selectivity for receptors present on the surface of the CTCs. The objective of this work was to design an assay enabling isolation of CTCs with high efficiency and purity.
The technology proposed herein would monitor the effectiveness of cancer treatment
EXPERIMENTAL
Evaluation of Peptide-Modified Microfluidic Devices for the Affinity Isolation of
EpCAM+ Circulating Tumor Cells (CTCs)
Clinical tests are evaluated by the sensitivity, specificity, and precision as described in Chapter 1.2. When the clinical test constitutes a screening diagnostic, additional considerations such as reagent stability and cost are important. In this regard,
the typical recognition elements such as monoclonal antibodies (mAbs), oligonucleotides (i.e., RNA aptamers), or molecular imprints each have strengths, but also limitations.61
Monoclonal antibodies are the most commonly used recognition element in immunodiagnostics as their main attributes are high specificity and affinity. However, their lack of long-term stability may affect overall performance of the test. Aptamers are
superior compared to mAbs in terms of stability, yielding a much higher shelf life, and excellent specificity; however, their cost can prohibit their widespread use.62,63
The alternative to the aforementioned recognition elements can be peptides.
Numerous functional groups can be incorporated into peptides at specific locations as a result of chemical synthesis. These functional groups allow for the immobilization of
peptides onto surfaces of many different functionalized solid supports, where peptides can show higher stabilities compared to Abs.64,65,66,67 Because peptides are small
Peptides are known to promote cell adhesion since the discovery of the Arg-Gly-Asp (RGD) domain as the principal integrin-binding domain.66 This discovery initiated
work on designing biomaterials utilizing peptides as adhesive moieties.70 Studies have
demonstrated enhanced adhesion of a variety of cells (e.g., platelets, endothelial and epithelial cells) on surfaces immobilized with different RGD-based peptides.68,71,60
However, the drawback of the RGD-based peptides is the lack of selectivity. RGD has affinity for common cells receptors.68,70 This is the main reason why the majority of
research so far has focused on evaluating biocompatibility of peptide-modified scaffolds, while the selective affinity isolation of specific cells has not been extensively investigated. It is possible, however, to select a peptide harboring a single recognition motif that has a unique affinity for a target protein, allowing for a selective complex formation with a
specific cell receptor.60 Proof of concept research on selective isolation of endothelial and
smooth muscle cells using microfluidic device decorated with selective non-RGD–based peptides was demonstrated.72,73,74 High purities (86%) of cell fractions isolated from
heterogeneous population were reported, suggesting very high specificity of the assay. Based on these studies it is believed that if designed properly, peptides can provide
similar affinities and specificities to mAb, superior long-term stability, and a reduction in cost allowing for their widespread use.72,74
This study is the first to show enrichment of CTCs by a microfluidic device modified
Materials
Peptide were purchased from Bio-Synthesis Inc (Texas, USA). Formaldehyde
(≥38% in H2O), Triton X-100, Bovine Serum Albumin (7.5% in DPBS), and Fetal Bovine
Serum were bought from SIGMA. PBS (no CaCl2, no MgCl2), McCoy’s 5A 1X (with
L-glutamine), MEM Alpha 1X (with L-L-glutamine), Trypsin (0.05% with EDTA), and NeutrAvidinTM were purchased from Thermo Fisher Scientific. Anti-human CD45 (clone
HI30) conjugated to FITC, and anti-human Cytokeratin 19(clone BA17) conjugated to eFlour® 615 were purchased from eBioscience Inc (San Diego, USA).
Cell Culture
SKBR3 and Hs578T cells were used for modeling positive and EpCAM-negative circulating tumor cells, respectively. Hs578T is a mammary gland breast carcinosarcoma cell with mesenchymal and luminal type morphology, characterized by a fibroblast origin. It is typically used as a model for triple negative breast cancer, specifically Claudin-low molecular subtype. SKBR3 is an epithelial breast
adenocarcinoma cell type that is derived from a metastatic site and represents the HER-2 enriched molecular subtype.
Both cell lines are adherent, and were cultured in 25 cm2 flasks with 8 mL of
culture media. The flasks were then kept inside an incubator, maintained at a temperature of 37ºC and an atmosphere comprising 5% CO2. Both cell lines were split
old media, wash in 5 mL of PBS, aspirate PBS, add 500 µL of 0.05% trypsin, incubate for 5 minutes at room temperature, deactivate trypsin with media containing 10% fetal bovine serum. SKBR3 cells were cultured in McCoy’s 5A media with 10% FBS, similarly,
Hs578T cells were cultured in MEM Alpha media with 10% FBS. To estimate the number of cells in a solution, the Nexcelom Cellometer® Auto T4 was used. This process required
20 µL of cell-containing solution to either chamber of the proprietary slide.
Production of the microfluidic devices in Cyclic Olefin Copolymer (COC).
Cyclic Olefin Copolymer (COC) was purchased as a plate (15 cm x 15 cm x 3.175 mm), cleaned with micro90 detergent, rinsed with isopropanol, rinsed in water, and finally dried with compressed air. The microfluidic devices were hot embossed into the
substrate (polymer plate) at the CHANL facility at the University of North Carolina at Chapel Hill (Figure 13). In the vacuum chamber, the brass mold (Figure 12) and polymer
substrate were subjected to a constant force of 300 N until a temperature of 160°C is reached, at which point the brass mold is pressed into the polymer substrate at 12.5 µm/s until a final pressure of 30 kN is achieved. This pressure and temperature was held for
200 s, after which time, both the brass mold and polymer substrate were cooled to 60°C. At 130°C, the pressure was released by having the pistons separate the brass mold from the polymer substrate by 2 mm at a rate of 10 µm/s. Upon separation of the mold from the substrate, the vacuum chamber (Figure 13) opened for removal. The hot embossing
Figure 12. The brass mold produces four curvilinear device substrates per cycle.
Figure 13. Jenoptik Hex 03 hot embossing machine and supporting equipment (A). The brass mold is secured to the top of the chamber (B), a plate of COC is placed on the bottom. A computer script, closes the chamber, begins a vacuum, and independently heats the mold and COC plate.
Each embossing cycle produced four devices, which were diced using a band-saw. General purpose scotch tape was used to cover the embossed features and prevent debris contamination. The diced devices were then cleaned using Micro-90 detergent, isopropyl
alcohol, water and compressed air. Cover plates were harvested from a roll of thin (250 µm) COC film, and cleaned using the same procedure as stated above for the COC plates. Flexible fused silica capillaries with an external diameter of 360 µm and an internal
diameter of 150 µm were inserted into the inlet and outlet features of the embossed polymer substrate. The COC cover slip was placed over the embossed-side of the COC substrate, and the entire device placed between two glass plates, creating a sandwich.
Pressure was applied to the sandwich using binding clips. Bonding of the cover plate to the embossed substrate was accomplished by exposure to 130°C in a convection oven (Figure 14-A) for a duration of 60 min. The final step required for completion of the
microfluidic device (Figure 14-B) was epoxying the capillary-COC junction to prevent leaks
during operation.
Figure 14. A Hewlett Packard 5890A Gas Chromatograph (A) is used to rapidly raise the temperature to 130˚C then maintain that temperature for 60 minutes. Inside, four curvilinear devices can be seen in glass sandwich with binding clips. The result is an assembled curvilinear microfluidic device (B).
Attachment of the peptide for affinity selection.
Sealed devices were exposed to broadband UV light at ~16 mW/cm2 (254 nm) for
15 min using a low pressure Hg lamp (GLF-42, Jelight Company Inc., Irvine, CA). Exposure of the polymer surface to UV radiation produced carboxylic acid groups, which were subsequently used as anchors for the covalent attachment of biologicals.
A solution of 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, 40 mg/mL) and N-Hydroxysulfosuccinimide (NHS, 4 mg/mL) in 1 mL of 2-(N-morpholino)ethanesulfonic acid (MES buffer, 0.1 M, pH 4.8) was infused into the device.
The EDC activates the carboxyl moieties, but is susceptible to hydrolysis. This issue was overcome by the NHS in solution. The NHS stabilizes the ester intermediate needed for a linkage to a primary amine, as shown in Figure 15.
Figure 15. Schematic of EDC/NHS coupling. Group 1 represents the COC substrate. Group 2 represents NeutrAvidin. Image taken from "Carbodiimide Crosslinker Chemistry" at www.ThermoFisher.com
After 25 min of incubation, the EDC/NHS solution was removed from the device by pushing air through a syringe. After removal of the EDC/NHS solution, NeutrAvidin in PBS pH7.4 was immediately infused into the device at a concentration of 2 mg/mL and
allowed to incubate for 0.5 - 3 h. A second infusion of biotinylated peptide at the same concentration was then introduced without introducing air following the first infusion.
Blood samples
BD Vacutainer® tubes containing EDTA were used. Blood samples were kept on a rocker and used within 3 h post-draw. Healthy donor’s blood samples were obtained from the UNC Cancer Hospital Blood Bank. Blood from patients diagnosed with cancer
were collected at the UNC Oncology Clinic or the operating room according to an approved UNC Institutional Review Board procedure.
Protocol for CTC isolation assay
All infusions into the microfluidic device are accomplished by syringes affixed to
a multi-channel syringe pump manufactured by New Era Pump Systems Inc. During the post-modification process, the chip was washed with PBS/0.5% bovine serum albumin (BSA) to remove unbound peptide remaining in the device from the preparation protocol and passivate and protect the microfluidic walls from non-specific adsorption. Next, a syringe containing a pre-mix of cells and buffer (PBS or blood) was infused into the device
at 25 µL min-1 (2 mm/s). Following the infusion of cells in either PBS or blood medium, a
syringe containing 2 mL of 0.5% BSA in PBS is infused at 50 µL min-1 (4 mm/s), to aid in
the removal of non-specifically bound cells. All subsequent infusions from this point in the process were accomplished at 25 µL min-1. The captured cells were stained with
Incubation with anti-CD45 occurred for 30 min at room temperature. The device was then briefly washed with PBS to remove unbound anti-CD45 antibody. In order to stain the interior of the captured cells, a fixing agent and detergent must be used. The fixing
agent, 2% formaldehyde in PBS, was infused and allowed to incubate for 30 min. Next, the non-ionic detergent, 0.1% Triton X-100 was infused and allowed to incubate for 15 minutes. At this point, the cellular membrane of captured cells was crosslinked and permeated. The final step is to add DAPI for nuclear staining, and cytokeratin 19 or
pan-CK for epithelial cytoplasmic staining, in water. This mixture was allowed to incubate for 1 hour on ice or overnight at 4˚C, after which point in time, the device was washed with PBS. The capillary ends were taped to prevent evaporation.
Fluorescence microscopy
Once the devices with captured cells had been stained, CTCs or model CTCs were enumerated to determine cell capture efficiency and purity by fluorescent microscopy. A Zeiss Axiovert 200M microscope with a Photometrics Cascade® 1K camera was used to
count CTCs and white blood cells (WBCs). Images were captured with the DAPI, FITC, and
Cy3 filters (see Figure 16 for example). CTCs were classified as being events positive for
exposure for FITC, 2000 ms exposure for Cy3. Examples of SKBR3 cancer cells and T-cells are shown in Figure 16-A and Figure 16-B, respectively.
Figure 16. A tumor cell is classified as DAPI and Cytokeratin 19 positive, but CD45 negative (top row). A white blood cell will stain DAPI and CD45 positive, but Cytokeratin 19 negative (bottom row).
High Performance Reverse Phase Liquid Chromatography with Mass Spectroscopy
(HPLC/MS)
Peptides were evaluated for purity and stability in the High-Throughput Peptide Synthesis and Array Facility at the University of North Carolina at Chapel Hill. HPLC/MS
Peptides submitted to the Chapel Hill core facility were 10 mg/mL aliquots in water. Storage of the peptide at room temperature occurred in the dark. At regular intervals, peptide aliquots of 30 µL were moved to storage at -20˚C. Upon freezing of the
final sample, all were taken to the core facility for analysis.
Polymeric microfluidic device for CTC isolation
The microfluidic device has been designed to be single use in order to prevent
cross-contamination. Real world considerations for a clinically used disposable device thus requires that production occur at high volume and low cost per unit, which certainly can be realized using the fabrication modality we adopt herein.75 For example, injection
molding allows for fabrication of microfluidic devices in polymer substrates with a cycle
time of 1 to 3 min.76 In a research setting, hot embossing is very effective in terms of
replication fidelity, but has a production cycle time of about 20 min, and thus, this method is of lower production compared to injection molding. Hot embossing was used for the fabrication of the devices used for all experiments recounted herein. The master mold used for hot embossing was made in brass via high precision micro-milling, which is
suitable for machining structures as small as 10 µm and aspect ratios less than 20:1 (depth / width).77,78 The brass mold was previously developed in the Soper lab, and
Figure 17. SEM images of the device hot-embossed into COC. Overview of the curvilinear channel geometry (image a), view of the channel (image b), the entrance to the curvilinear channels (image c), cut away to reveal the cross-section of the channel (image d). Image taken from Hupert et al. 2014.
As presented in Chapter 1, positive selection microfluidic devices utilize various microchannel geometries to optimize CTC recovery. By increasing the likelihood of CTCs interacting with affinity reagent-modified surfaces, the probability that the interaction leads to successful CTC-affinity reagent binding increases. The design of microfluidic
geometry is critical to generate efficient recovery of rare cells while simultaneously disrupting nonspecific interactions to assist in producing a CTC isolate of high purity.
shown to be affinity captured using mAb covalently attached to the COC surfaces and demonstrated high recoveries and exquisite purity.65,66 Detailed evaluation of the design
and uniform filling of all sinusoidal channels for this device has been reported.70
In sinusoidal-based devices (Figure 18), CTCs traverse about the channel’s
curvature (radius of curvature, rc = 125 μm) and experience a centrifugal force (Fc) that propels them towards the channel walls where they can associate with surface-tethered affinity selection agents (i.e., antibody, peptide, aptamer).
Figure 18. (A) CAD drawing of the module where the large arrow indicates sample flow direction through the selection channels and (B) SEM of the selection bed showing high aspect ratio sinusoidal microchannels and (C) closeup of one channel.
mm long channel, a centrifugal velocity on the order of 1 µm/s is significant enough to initiate CTC-wall interactions. Smaller cells, such as 8 μm leukocytes, experience a centrifugal velocity four times smaller than 16 μm CTCs limiting their interactions with
channel walls and while Vc can be enhanced by increasing the cell’s forward velocity, the probability of a successful binding event generally decreases as centrifugal velocity increases.79
The Chang-Hammer modelcan beused to describe the CTC-affinity reagent binding,
and is applicable for antigen-peptide binding.60 For affinity reagent-target binding,
kinetics are balanced by the transient motion of the cell’s surface target as it passes over the surface-confined affinity reagent, thereby limiting the associated residence time for a binding event to occur, which becomes less probable at higher linear velocities and low
cell target expression. Due to the small width of the sinusoidal channels, the average fluid shear stress observed in these devices is 13.3 dynes/cm2 for blood, which is
approximately an order of magnitude larger than micropillar-based CTC devices. This results in high purities >80% for the sinusoidal architecture and mAb modified COC surface.72
Modification of the microfluidic device with the anti-EpCAM peptide.
from the antigen’s epitope. Chemical synthesis of peptides allows for the precise placement of chemical groups that can be used for specific orientations of the affinity reagent to the surface. We modified the 23-amino acid anti-EpCAM peptide discovered
by Bai et al. 2014, by adding a lysine residue to the N-terminus, upon the side chain of which a biotin molecule was added (Figure 19-B). The lysine chain allowed for a flexible
linkage between the peptide and device surface, and reduced the cost of synthesis in comparison to poly(ethylene glycol) linkers. With biotin/lysine linker, the molecular
weight of the peptide was 2.8 kDa.
The density of biotinylated peptide immobilized to the COC channels is limited by NeutrAvidin (NA), as show in Figure 19-A. Each NA molecule can bind a maximum of four
biotin molecules with a dissociation equilibrium coefficient on the order of 10-15.80 This
ensures a stable bond between the biotinylated peptide and the COC device. Unlike
Streptavidin, NA lacks the Arg-Tyr-Asp (RYD) sequence that mimics RGD and would cause non-specific cell adhesion.81
The conjugation of NA to the COC surfaces was thus a critical step that must be verified to understand the efficiency of surface loading. Fluorescently-labeled biotin molecules were used to determine the extent of NA immobilized as a result of NA
concentration and correct UV-activation and EDC/NHS chemistry. The results were implemented into the protocol for device assembly and preparation. Figure 20-B
demonstrates the immobilization of NA to the channel walls, versus the control image (Figure 20-A). The intensity plot for successful NA attachment (Figure 20-D) shows much
higher intensity than the control (Figure 20-C).
The theoretical load of peptides in the selection bed can be determined as follows. A -COOH surface concentration of 9.5 ± 2.3 nmol/cm2 on COC, and a total device surface
area of 6.32 cm2 allows for ~6.32 × 1014 NA molecules (area of 100 Å2 each) for a full
monolayer coverage. This equates to 1.05 nmol of NA molecules. Because each NA molecule can bind with up to four biotin molecules, it is expected that between 1.05 nmol and 3.15 nmol of biotinylated-peptide can be attached to the COC surface.
Isolation of model cell lines
Two breast cancer cell lines were used to optimize the assay for the isolation of CTCs. SKBR3 serves as the positive control; it highly expresses Epithelial Cell Adhesion Molecule (EpCAM) and originates from the pleural effusion of a patient with malignant
adenocarcinoma of the breast.82 Hs578T serves as a negative control; it is a mammary
gland breast carcinosarcoma cell with mesenchymal and luminal type morphology, characterized by a fibroblast origin. Hs578T expresses no EpCAM.82
For both cell types, EpCAM expression was characterized by flow cytometry (Figure 21). Staining of EpCAM on Hs578T cells gave comparable signal to staining with
the immunoglobulin control (IgG), demonstrating the use of Hs578T as a negative control for the CTC capture assay. The right column of Figure 21 shows high expression of EpCAM
Figure 21. One dimensional flow cytometry histogram comparing EpCAM expression in Hs578T cells (left column) and SKBR3 cells (right column). SKBR3 cells express EpCAM, whereas Hs578T cells do not.
In order to test the application of trypsin for release of the cells from culture and
before cell spiking experiments, our model cell lines were tested for their viability. Both cell lines were incubated in calcein acetoxymethyl, which permeates through the membrane. If the cell is viable, intracellular esterases will cleave the acetoxymethyl group of calcein-AM, enabling the calcein dye to fluoresce green. As seen in Figure 22,
Figure 22. Fluorescent microscopy of SKBR3 (top row) and HS578T (bottom row) cell lines. Calcein dye (green) shows good viability following release by trypsin.
Experiments required that EpCAM+ (SKBR3) cells be spiked into either PBS buffer
or healthy blood, then run through a series of three connected devices. The
self-referencing method was used to calculate CTC recovery. This value was obtained by dividing the number of CTCs captured on the first device by the total number of CTCs captured on all devices in the series. The equation for recovery is listed below:
𝐂𝐓𝐂𝐝𝐞𝐯𝐢𝐜𝐞 𝟏 𝐂𝐓𝐂𝐚𝐥𝐥 𝐝𝐞𝐯𝐢𝐜𝐞𝐬=
𝐂𝐓𝐂𝐝𝐞𝐯𝐢𝐜𝐞 𝟏
𝐂𝐓𝐂𝐝𝐞𝐯𝐢𝐜𝐞 𝟏+ 𝐂𝐓𝐂𝐝𝐞𝐯𝐢𝐜𝐞 𝟐… + 𝐂𝐓𝐂𝐝𝐞𝐯𝐢𝐜𝐞 𝐍
experiment was produced by serial dilution of the cultured cells after estimation by an automated cell counter (Cellometer®). The self-referencing method ensures that cell
recovery is determined solely by the peptide and the channel design; free from
confounding factors such as cell loss to the syringe chamber and dead volumes. This method enables devices with high recovery (70-100%) to be operated in a series of two while maintaining an error less than 7%.83 If the recovery efficiency of the device is below
70%, a third device is needed to reduce error. The advantage of this approach is that
knowledge of the exact number of input cells is irrelevant, as the recovery was calculated from the numbers of cells that entered the device and isolated on chips connected in series. The self-referencing technique can be applied to evaluate cell recovery from clinical samples as well.
The recovery of SKBR3 cells from PBS as a function of peptide concentration used for device preparation was evaluated. As shown in Table 1, three different concentrations of peptide were used to prepare the devices. Infusion of peptide into the device at 4 mg/mL was not sufficient for saturation of the COC surfaces. With infusion of 11 mg/mL, cell recovery improved significantly. A second application of peptide at 11 mg/mL (22
Table 1. Recovery of SKBR3 cells in PBS as a function of peptide concentration used during preparation of the microfluidic device.
Peptide Concentration (mg/mL) Recovery (%) of SKBR3 cells from PBS
No peptide (NA only) 2%
4 mg/mL 23%
11 mg/mL 69%
22 mg/mL 92%
The ultimate aim of this research was to allow for processing whole blood directly
without requiring either blood dilution and/or removal of the red blood cells prior to processing using the microfluidics. But, we first optimized various experimental parameters using PBS for better control over certain flow parameters. For example, the effect of flow rate for PBS and whole blood mediums on cell recovery was investigated.
The former is a Newtonian fluid while the latter is a non-Newtonian fluid. The product of fluid velocity and fluid viscosity is shear stress. Shear stress modulates antibody/antigen dissociation rates.84 As blood is subjected to higher velocities, higher stresses will reduce
its viscosity. For this reason, it was predicted that the flow rate for optimal cell recovery in whole blood would be different than the flow rate for optimal recovery in PBS. This
was confirmed by experimentation, whereby it was found that cell recovery in PBS was most efficient at 2 mm s-1, whereas whole blood performed 9% more efficient at 1 mm s
-1 than at 2 mm s-1. Devices immobilized with antibody also showed maximum cell
recovery at flow rates of 2 mm/s since centrifugal forces generated by the sinusoidal geometry is responsible for movement of CTCs to the COC surfaces.85 Control
non-specific interactions between cells and the peptide modified surface existed. In comparison, devices immobilized with anti-EpCAM antibody showed less non-specific interactions as indicated by a Hs578T cell recovery of 4.2 ±2.4%.
Figure 23. Histogram of data correlating linear flow velocity to CTC recovery efficiency for both PBS buffer and blood.
Low velocities reduce the encounter rate between reactive species, whereas high
velocities reduce the encounter duration.86 These competing effects give rise to an
target EpCAM to react with the surface-tethered peptide. Interestingly, the optimal flow rate for the case of blood for the EpCAM/peptide complex was 1 mm/s.
As can be seen from the data shown in Figure 23, the recovery at the optimal flow
velocity for PBS was seen to be 92% while for whole blood, the recovery at the optimal flow velocity was only 61%. In the presence of interfering cells, such as red blood cells and/or white blood cells as well as the other constituents found in blood, such as protein albumins, these materials may compete for binding sites on the limited number of surface
tethered peptides, thus dropping the available sites to bind the limited number of tumor cells seeded into the blood sample.
For the sinusoidal design used herein, centrifugal forces are responsible for pitching cells beyond their original laminar flow line and against the outer wall to allow
facile interactions with the tethered surface affinity reagent. At the necessary timescale, diffusion alone would not be sufficient to interrupt such a process. CTC recovery also depends on antigen expression. Reduced antigen leads to reduced isolation efficiency. The SKBR3 cells used herein have high EpCAM antigen expression, which contributed to high cell recovery from PBS.
Healthy donor blood was infused into a peptide-modified and mAb-modified microfluidic chips to determine the selectivity of the assay involving this particular peptide as an affinity reagent for the targeting of EpCAM. Contaminating CD45+ cells were
isolated at an average of 192 per mL of blood as shown in Figure 24. The range for isolation
of CD45+ cells was 0 to 900 WBC/mL. For comparison, the non-specific isolation of WBCs
the mAbs were better equipped to inhibit non-specific interactions of WBCs with the affinity surface compared to the peptide coated surface.
Figure 24. Count of WBCs non-specifically co-isolated on a microfluidic device when either mAb or peptide was immobilized to the surface.
Isolation of CTCs from clinical samples
Whole blood samples from patients with endometrial (EndC) and epithelial ovarian carcinoma (EOC) were processed using the sinusoidal device with immobilized
anti-EpCAM peptide and anti-EpCAM antibody. The results are summarized in Table 2,
Table 2. Summary of clinical results for healthy donors and patients diagnosed with EOC and EndC.
Pt ID Anti-EpCAM CTC Affinity Selection CTCCK+/mL CTCVIM+/mL
EOC#1 peptide (4 mg/mL) mAb (0.5 mg/mL) 55 12 - -
EOC#2 peptide (11 mg/mL) mAb (0.5 mg/mL) 147 95 30 21
EOC#3 mAb (0.5 mg/mL) 631 -
peptide (22 mg/mL) 761 -
EndC#4 peptide (22 mg/mL) mAb (0.5 mg/mL) 4 1 200 40
EndC#5 peptide (22 mg/mL)mAb (0.5 mg/mL) 2 0 692 45
EndC#6 peptide (22 mg/mL)mAb (0.5 mg/mL) 0 - 1036 -
Isolated CTCs from the clinical samples were stained for cytokeratins (CK), vimentin (VIM), CD45, and nuclear DNA (DAPI), enumerated with a fluorescent microscope and normalized per 1 mL of blood (see Table 2). Purity was not determined. Cancer cells of epithelial origin harbor intermediate filament proteins of the cytokeratin
Figure 25. (A) Fluorescent microscope images of a CTC isolated with peptide-functionalized device from a patient with endometrial carcinoma. (B) Images of CTCs isolated with antibody-functionalized device from a patient with epithelial ovarian cancer.
CTC clinical yields from EOC patient samples using the peptide-modified device at high (22 mg/mL) concentration was comparable to the CTC recovery achieved by the antibody-modified device (Table 2. Summary of clinical results for healthy donors and patients
diagnosed with EOC and EndC.
). Presence of CTCs was detected in 1 of 3 EndC patient samples. CTCs were more likely to stain positive for Vimentin as shown by Table 2. Summary of clinical results for healthy donors and patients diagnosed with EOC and EndC.
Stability of the peptide and relation to non-specific binding
Several non-enzymatic degradation pathways are known for peptides, the implications for which is most likely a loss of specific binding as an affinity agent. The
degradation of aspartic acid (Figure 26) is well established.87
Figure 26. Degradation of natural aspartic acid (L-Asp) to a succinimide intermediate via nucleophilic substitution. Image from Takahashi, O.; Kirikoshi, R.; Manabe, N. Int J Mol Sci 2015, 16(1), 1613-1626.
Naturally occurring L-aspartic acid can undergo nucleophilic substitution whereby the nitrogen molecule of the C-terminal-adjacent amide bond attacks the carboxyl moiety of the L-Asp side chain, resulting in a cyclic succinimide. Rearrangement of the bonds and subsequent hydrolysis can lead to any of several aspartic acid analogues and loss of function. Asparagine is also known to undergo degradation via a cyclic
succinimide intermediary step.88
sulfoxide by either oxygen transfer from hydrogen peroxide or a singlet oxygen. Cysteine can undergo spontaneous oxidation, after which the sulfur molecules of two oxidized cysteine residues can engage in the formation of covalent disulfide bonds, resulting in an
irreversibly altered peptide structure.
To assess the degradation of the EpCAM peptide empirically, aliquots of anti-EpCAM peptide (10 mg mL-1 in water) were stored in the dark at room temperature.
Samples were moved from 22˚C to -20˚C at regular intervals over the course of 23 days,
then analyzed by HPLC using an EMD Chromolith Performance RP-18e 100-2 mm column at 1 ml/min flow of 5-25% (20 min gradient) acetonitrile in water; 0.1% TFA in both
solvents. Examples of the MS data can be seen in Figure 27.
Peak 1 (m/z=2847) corresponded to the anti-EpCAM peptide. Peaks 2, 3, and 4 corresponded to K(Biot)VRRDAPRFSMQGLD (m/z=2002), APRFSMQGLDACGGNNCNN
(m/z=1996), and PRFSMQGLDACGGNNCNN (m/z=1896), respectively. These degradation products are likely the result of degradation at the aspartic acid residue by way of a succinimide intermediate.
A summary of peptide degradation is presented in Figure 28. The HPLC data
obtained revealed that the anti-EpCAM peptide saw a 20% reduction in purity when stored at -20˚C in water (pH=7.4). When stored at room temperature (22˚C) in water, the peptide experienced an increased rate of degradation; a 15% loss of purity within the first two days, culminating in an ultimate 7% purity by day 23. The stability at room
temperature is relevant because the addition of peptides to the microfluidic device during preparation occurred over the course of a day and all at room temperature, and depending on confluency of cell lines or availability of patient samples, devices have been kept for a day or two beyond final preparation prior to sample being run. The effect of peptide degradation is likely a loss of specificity for extracellular EpCAM protein, and
Figure 28. Histogram of HPLC data showing purity of anti-EpCAM peptide in water at various storage temperatures and durations of time.
Conclusion
The ability of an anti-EpCAM peptide to isolate CTCs in a continuous flow sinusoidal microfluidic device was evaluated. CTC recovery from blood was lower in devices employing peptides than in devices employing mAb, but similar in PBS buffer. The reduced recovery was attributed to the peptide’s lower KD in comparison to the
antibody’s KD as well as the possibility of different interfering species binding to the
limited number of sites provided by the surface tethered peptides. The number of WBCs co-isolated on peptide modified devices is higher than the number of WBCs co-isolated on mAb modified devices as well. Finally, devices modified with peptides detected CTCs in 3 of 3 EOC patients, and 1 of 3 EndC patients.
Storage at -20˚C
Future work
Poor peptide stability lead to degradation products that are unlikely to bind
EpCAM positive cells. Truncated peptides may likely bind to non-EpCAM antigens, resulting in reduced cell purity. For this reason, investigating alternative peptide storage methods would be a priority for future experiments. It should be investigated if the peptide can be reconstituted in an aqueous buffer exceeding 20 mg/mL by using dimethyl
sulfoxide. This would minimize degradation of the peptide by shortening the duration of peptide incubation at room temperature. Peptide stability once immobilized in the microdevice should also be investigated. Dry peptides are more stable than peptides stored in buffer. If however the aqueous storage method is to be retained, use of organic
solvents in the aqueous buffer are known to reduce isomerization and deamination by reducing the dielectric strength of the solution.90
It would be possible to improve the capture efficiency of EpCAM expressing CTCs by increasing the surface density of bound affinity agents. Implementing the Huisgen cycloaddition reaction involving azide and alkyne functional groups instead of
NeutrAvidin / biotin could reduce the molecular weight of the linker strategy by approximately 59 kDA.