0022-538X/81/120936-06$02.00/0
Acyclovir-Resistant
Mutants of Herpes Simplex Virus Type 1
Express
Altered DNA Polymerase or Reduced Acyclovir
Phosphorylating Activities
PHILLIP A. FURMAN,'* DONALD M. COEN,2 MARTY H. ST. CLAIR,' AND PRISCILLA A. SCHAFFER2
Wellcome Research Laboratories, ResearchTrianglePark, NorthCarolina27709,' and The Sidney Farber CancerInstitute, HarvardMedical School, Boston, Massachusetts 021152
Received 4 June1981/Accepted 8 August 1981
The biochemical properties offouracyclovir-resistant mutants are described. Twoofthese mutants, PAAr5 and
BWr,
specified nucleotidyl transferase (DNApolymerase) activities which were less sensitive to inhibition by acyclovir
tri-phosphatethan their wild-typecounterparts. Another mutant, IUdRr, exhibited
reduced ability to phosphorylate acyclovir. The fourth mutant, ACGr4, both
induced an altered DNA polymerase and failed to phosphorylate appreciable amounts of acyclovir. BWr,a new acyclovir-resistant mutant derived from the
Patton strain of herpessimplex virus type 1, induced a DNA polymerase resistant toinhibition by acyclovirtriphosphate, but, unlike the polymerases induced by PAAr5andACGr4, stillsensitive to phosphonoacetic acid. Resistance of BWr to
acyclovirmapped close tothe PAArlocus and was separable from mutations in
the herpes simplex virus thymidine kinasegene by recombination analysis.
The nucleoside analog 9-(2-hydroxyethoxy-methyl)guanine (acyclovir, acycloguanosine) is aspecific and effective inhibitor ofherpes
sim-plex virus (HSV) replication (8,25)and
demon-strateslittlecytotoxicitytouninfected
cells
(25).There has accumulated aconsiderable amount
ofevidence indicating that acyclovir exerts its
antiviral effect after conversiontoacyclovir
tri-phosphate (acyclo-GTP), which inhibits the
viral nucleotidyl transferase (DNA polymerase)
more efficiently than does the host cellaDNA
polymerase
(8,12).
Biochemical evidenceindi-catesthatHSVthymidine kinase (HSV-TK) is
the enzyme
responsible
for phosphorylationofacyclovirtoitsmonophosphate (8, 13). Host-cell
enzymes are
apparently
responsible for thephos-phorylation
ofacyclovir
monophosphate(acy-clo-GMP) (11, 21).
Parallel with biochemical studies are the
re-sults of genetic experiments which have impli-cated theHSV-TKand DNApolymerasegenes aslociwhich,whenmutated,canconfer resist-ance toacyclovirinthecell culture (4, 5, 7, 27). Withregardtothe TK gene, severalHSV mu-tants lackingTK
activity
exhibit resistance to acyclovir (4,5,8, 9,27),andthedegreeof resist-ance generally corresponds to the level of TKactivity(4,5).
With
regard
to the DNA polymerase gene,severalmutantswhich areresistant to phospho-noacetic acid (PAA), a recognized marker for
the HSV
DNA
polymerase
gene(2, 3, 16, 17,
22-24), are also resistant to
acyclovir,
yet exhibit wild-typelevels of TK activity (5, 27). Recom-bination andcomplementation
analyses
ofone of thesemutants,PAAN5,
showed that itdefmes
acodominant locus(termed
ACGr-PAA)
distinct from the recessiveacgr-tk
locusandmuchmoreclosely
linkedtothe PAA' locus than it istotheacgr-tk
locus.Complementation
analysis
indi-cated that another mutant,
ACGr4,
was apre-sumptive double mutant
containing
mutationsatboth loci that leadto a
highly
resistant phe-notype(5).Subsequent
intertypic
andintratypic
markerrescue
experiments
with otherPAA
mu-tantshavealsodemonstrated
linkage
ofacyclo-vir resistance with the
PAAr
locus and withtemperature-sensitive
mutations within theHSV DNApolymerasegene
(7;
D.M.Coen and P. A.Schaffer,
unpublished
data;
D.Knipe,
per-sonal
communication).
Toconfirmthe
implications
that theHSV-TKandDNA
polymerase
genesarelociwhich,
whenmutated,
can confer resistance toacyclovir
incell culture, we examinedfour
acyclovir-resist-antmutantsderivedfrom the KOS and Patton strains of HSV type 1
(HSV-1). First,
thesensi-tivity of thesemutants toinhibition
by
acyclovir
and PAAwas examined
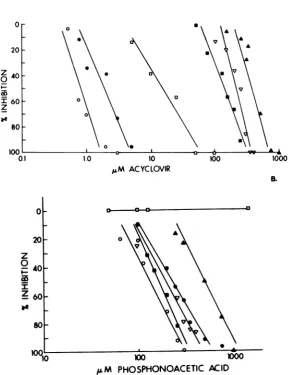
(Fig.
1A andB).
Both the PAA-resistant mutant PAAr5and thepre-sumptive double mutant ACGr4
(5)
were lesssensitivetoinhibition
by
acyclovir
thanwasthe936
on November 10, 2019 by guest
http://jvi.asm.org/
937
wild-type virus KOS
(Fig.
1A). Fifty percenteffective
dose(ED50)
values for PAAr5 andACGr4 were 20- and 490-fold greater,
respec-tively, than that for KOS. PAAr5 and ACGr4
also showed much less susceptibility to
inhibi-tion by PAA, with ED50 values more than
10-and 5-fold greater, respectively, than that ob-tained forKOS
(Fig.
1B). Mutants of the Patton strain ofHSV-1, IUdRr (amutantcharacterized by Smithetal.[28]asbeing resistanttoacyclo-vir and iododeoxyuridine) and
BWT
were alsofoundto be much less susceptibletoinhibition
byacyclovir thanwasthe wild-typevirus. The
ED50 values for IJdRr and BW'were
approxi-mately
100and 200timesgreater, respectively,than that for their wild-typecounterparts. How-ever, sensitivity of these viruses to PAA was
considerably different from thatobservedforthe mutants derived from KOS. Both
Patton-de-rived mutants gave dose-response curves with
PAA comparable tothat.of the Patton strains. In fact, wild-type Patton consistently gave
higherED50 valueswith PAA than did the two mutants.
Cells infected with mutantviruseswere then
tested for their abilitytophosphorylate acyclo-vir.Acyclo-GTP levelsincells infectedwith the mutantsACGr4 and
IUdRF
were 0.3and4.0%of thelevels found in cells infected with theirwild-type counterparts (Table 1). The levels of
acy-A.
0
20
z 40
0
0-1: 60
z
801
0.1
a
1.0 10 100
AM
ACYCLOVIR1000
p O
0
S%I
100 100
pM
PHOSPHONOACETICACID
FIG. 1. Plaque inhibition dose-responsecurvesfor acyclovir (A) and PAA (B) in Vero cells, determined by
using wild-typeKOS(0)andPatton(0) andacyclovir-resistant PAAr5 ([1), IUdRrr(), BW' (A), and ACGr4 (A) viruses. Plaquereduction assaysto determineED50 values foracyclovir and PAA wereperformedas
describedbyCollinsand Bauer(6). Virusstockswerepreparedaspreviously described (8).
0
20
40
z
0
-z
601-80
I 0% .1
2^t%i-1
40,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.500.101.392.242.618.2]clo-GTP in cells infected with the resistant mu-tants PAAr5 andBWrwere comparable to the leveLs in cells infected with wild-type viruses. Moreover, the total amountofphosphorylated acyclovir (all forms: mono-, di-, and triphos-phates)wasconsiderably higher in cells infected
with wild-type viruses, BWr, or PAAr5than in
cells infected with ACGr4 andIUdRr (datanot
shown). Similarly, extractsprepared from cells infected with IUdRr and ACGr4 contained much less acyclovir-phosphorylating activity and HSV-TK activity than did extracts from cells infected with wild-type virus, whereas BW0 and PAAr5 induced acyclovir-phosphorylating and HSV-TK activities comparabletothose of their wild-typecounterparts(P. Keller, personal
com-munication). Thus, the acyclovir resistance of BWr and PAAr5 cannotbeattributedtofailure of these mutants to phosphorylate acyclovir. The lack of acyclovir phosphorylation (TK expression) by IUdRr probably explains the cross-resistancetoacyclovir and IUdR observed for this virus(28).
The inhibitory effect of acyclo-GTP on the
DNA polymerase of mutant and wild-type
vi-ruses was examined by using [3H]dTTP
incor-poration as a measure ofenzyme activity.
En-zyme inhibition curves (Fig. 2) demonstrated
thatDNA polymerases ofthe viruses couldbe separated intotwoclasses, asensitive class and aresistantclass,onthebasis oftheirsensitivities toinhibition byacyclo-GTP. The sensitive class, having I50 (50% inhibition) values of about0.2
,uM acyclo-GTP (Table 1), was comprised of
bothwild-type strainsand theTK-deficient mu-tant derived from Patton (IUdRr). The DNA
polymerases induced by the KOS-derived mu-tants,PAAr5andACGr4,and thePatton-derived
mutant,BWr,wereless sensitivetoinhibition by
acyclo-GTP than their respective wild types. h5o valuesfor PAAr5, ACGr4, and BWrwere
approx-imately 5-, 9-, and 25-fold higher, respectively,
thanthe
Lo
values obtained for their wild-typecounterparts (Table 1). These data confirm the
suggestionofCoenandSchaffer (5) that PAAr5 andACGr4 contain mutationsatthe DNA
po-lymerase locus conferring acyclovir resistance. The sensitivities of the DNA polymerases of PAAr5,ACGr4, andBWrtoPAA inhibitionwere
alsodetermined (Table 1). The DNA
polymer-aseofPAAr5 and ACGr4 werefoundto be
ap-8100
60
20
0.01 0.1 1.0 10
rIMACYCLO-GTP
FIG. 2. Inhibition of wild-type and mutant virus
DNA polymerases by acyclo-GTP. Virus-induced
DNApolymerasewasisolatedandidentifiedas
de-scribedpreviously (11, 29). DNA polymerase assays werecarriedoutasdescribedby Elionetal. (8) and
Furmanetal. (12). ThesubstratesdATP,dCTP, and
dTTPwerepresentataconcentrationof100pM,and
dGTPwaspresent ataconcentration of5p.M.
Sym-bolsfor polymerases: KOS (0), Patton (O), PAAr5
(0),ACGr4(A),B W (U), and IUdRr(A).
TABLE 1. Summary of the biochemical properties of acyclovir-resistant mutants and their corresponding wildtypes'
Virus Acyclo-GTP Polymerase sensitivity (range)(I50
(,4M])^
Viral
sensitivityVrs levels(pmol/ __
ED_____o______M
____106 cells) Acyclo-GTP PAA Acyclovir PAA
KOS 90.7 0.23 (0.11-0.42) 0.50 (0.21-0.86) 0.7 138 PAA`5 55.4 1.17(0.82-1.62) 3.57(2.36-4.71) 14.3 >1,400d
ACG`4 0.3 2.14(1.47-3.33) 3.78(2.43-5.07) 346 >750 Patton 52.8 0.15(0.09-0.24) 1.93(0.43-4.43) 1.4 260 BWr 121.8 3.71 (3.36-4.08) 0.97(0.55-1.50) 224 195 IUdRr 2.3 0.23 (0.04-0.89) 0.50(0.01-1.93) 136 166
Experimentaldetails may be found in thelegendstoFig. 1and2.
bConcentrations of substrates for theacyclo-GTPinhibitionassayaredescribed in thelegendtoFig.2.
Ih0
values were calculatedbyusingthe Probit computer program, whichplacesmoreweightonthosepointsneartheI50 point (10).For the PAA inhibition assay, theconcentration of all fourdeoxynucleoside triphosphateswas
100/AM.
TheED0ovaluesweredeterminedby Probitanalysis(10).
dNoinhibitionatthese concentrations.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.500.67.459.487.590.2]NOTES
proximatelyseventimesmoreresistantto inhi-bition by PAA than was wild-type KOS DNA
polymerase. Incontrast,BWrDNApolymerase
wasfoundtobeno moreresistanttoinhibition
by PAA than was the DNA polymerase of its
parental virus, strain Patton. The DNA
polym-erase induced by IUdR' showed notonly wild-type sensitivity to acyclo-GTP but also
wild-typesensitivitytoPAA (Table1).Theapparent
Kmvalues for thefour natural deoxynucleoside
triphosphates ranged from 1 to 4 ,uM (unpub-lished data). All viral DNA polymerase
prepa-rations exhibitedafourfold stimulation of
activ-ity in thepresenceof 50mMammonium sulfate,
whereas cellularaDNA polymerase activitywas
reduced by 50%,indicating that the polymerase preparationswere virus specific (20,29). In
ad-dition, the DNApolymerases induced by KOS, Patton, IUdRr, and BWrwere50-foldmore
sen-sitive than the a cellular DNA polymerase of
HeLa S-3 cellsto inhibition by PAA ata
con-centration of 5,uM, thus confirming their viral
origin.
Genetic experiments previously identified PAAr5as amutantwhose resistancetoacyclovir
was separable by recombination from the
acy-clovir resistance mutations in acgr-tk mutants
and closely linked tothe PAA resistance locus (5). To determine whether themutantBWr be-haved similarlyinrecombination tests,we
per-formed crosses between BW' and the acg'-tk mutant, ACGr35, which is partially acyclovir
resistant owingtoamutation whichreduces TK
activitytoabout 15% ofwild-type levels (5). The abilitytomeasurerecombination between these
twovirusesdependeduponthe factthatneither plated efficiently in400 1LMacyclovir (Table 2). However, when BW' and ACGr35werecrossed,
3.3% of the resulting progeny wereresistantto
400,uM acyclovir (Table 2). These dataimplya
recombination frequency of 6.6%, whichismuch
greaterthananyfound betweenmutantswithin thesamecomplementationgroupwhichmapin
theuniquesequencesofthe HSV-1genome(R. A. F. Dixon and P. A. Schaffer, unpublished data). Similar results (not shown)wereobtained
when BWT was crossed with the conditionally
resistant acgr_tkmutant,KG-ill,whichexhibits thermolabileTKactivity (4).
To determine whethertheacyclovirresistance
ofBWrwaslinkedtothe PAAresistance locus, we crossed BWr with PAAr5, and the progeny
were examined for their plating efficiency in
both PAA at1.4 mM and acyclovirat 100 tiM.
Each parent used in the crosses wasrelatively
resistanttooneof thesedrugsbutquitesensitive
tothe otherortothe combination of bothdrugs
at these concentrations (Table 2). A
recombi-nant of thesetwoviruses would be expectedto
plate efficiently in both drugs. However, only 0.08% of the progeny were resistant to both
drugs, implying a recombination frequency of
only 0.16% (Table 2). In contrast, in a parallel experiment, when the acgr_tk mutant, ACGr35,
TABLE 2. RecombinationofBW,ACGr35, and PAAr5a
PFU/ml EoPb
NorusdrIn
acyclovir In acyclovir RF- RF-P+Nou
drug In acyclovir (100,uM)and Inacyclovir (100uM)and Ad(%) (400ItM)
PAA (1400 (400jiM) PAA(1,400JIM) AM)
ACGr35 1.1X107 <5.0X 102 <5.0X 102 <4.6X 10-5 <4.6X 10-5
BWr
2.6X107
1.6x105
1.0X104
6.2x10-3
3.8x10-4
PAAr5 2.4X107 1.0X103 5.0x 102 4.0X 10-5 2.0x 10-5
BWTxACGr35 3.3X 107 1.1x 106 3.3x 10-2 6.6
PAAr5x
BWT
3.2x107 2.5X 104 8.0x 10-4 0.16PAAr5xACGr35 7.2x106 7.5x 104 1.0x 10-2 2.0
aRecombinationanalysiswasperformed
essentially
asdescribedby Schafferetal.(26),exceptthat Verocells
wereused instead ofHELcells, and recombinationwasperformedat37°C. Duplicatetubecultures of Vero cells containingapproximately2x105cellsperculturewereinfected either withpairsof mutants, eachat acalculatedmultiplicityof2.5plaque-formingunits(PFU)per cell inatotal volume of0.2ml,orwithsingle parentalvirus controls at a multiplicity of5 PFU per cell in 0.2 ml. Simultaneous assays of inoculum suspensions were
performedtoconfirmcalculatedinput multiplicities; if the actualmultiplicity variedmorethantwofold from the calculatedmultiplicity,results oftestswiththesemutantswereexcluded.
bEOP, Efficiency ofplating.EOP= (PFUpermilliliter in presence ofdrug)/(PFUpermilliliter in absence ofdrug).
'RF - A, Recombination frequency. RF- A = [(PFU per milliliter in presence ofacyclovir)/(PFU per
milliliterin absenceofacyclovir)]x 2x 100%.
dRF - P + A, Recombination frequency. RF -P + A = [(PFU per milliliter in presence of PAA and
acyclovir)/(PFUpermilliliter in absence of PAA andacyclovir)] x2 x100%c.
VOL. 40,1981
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.500.49.445.423.537.2]andPAA'5were crossed andthe progenywere analyzed under identicalconditions,the
recom-binationfrequencywasmorethan 12-foldhigher
(Table 2).
Thus,bothgenetic and biochemicalevidence supportthenotion thatPAAF5, ACGr4,andBWT contained mutations in their DNApolymerase genes which conferred resistance to acyclovir. The resultsofthisstudyindicate thatmutations
can occurintheDNApolymerasegenethatwill confer resistance to bothacyclovirand PAA or
to acyclovir but not PAA. The latter result would beexpected ifthe HSV DNApolymerase conforms to the model proposed by Kornberg
(18) for other DNApolymerases;i.e.,DNA po-lymerase hasanactive center that iscomposed
ofmultiple sites, eachwithadifferentfunction.
Therefore, amutation which affects theprotein
atasiteother than thepyrophosphateexchange
site (the presumptive site of PAA inhibition
[19]) will not necessarily affect the pyrophos-phate exchange site (resistance to PAA). Nev-ertheless, thesimplest explanationforthedata
obtained for the mutantsPAAr5 andACGT4is
thatasinglemutationcanaffectmorethanone site.
Thenewmutantdescribedhere, BWr,which was acyclovir resistant but PAA sensitive,
de-fines yet another phenotype within the DNA
polymerase locus and separatesthe domain of
the DNA polymerasemolecule whichspecifies acyclovir sensitivity from the domain which
specifies PAA sensitivity. Thus, mutants asso-ciatedwith the HSV DNApolymeraselocuscan be temperatureresistant, drugresistant,orboth
(1, 14, 15, 17, 23), the degree of resistance to
both PAA and acyclovir varying. A detailed understanding of the molecular basis for the
widerangeofphenotypeswithin the HSVDNA
polymerase locus awaits further fine-structure
mapping and additionalbiochemical studies of
itsgeneproduct(s).
(This work waspresented in partat the 5th
Cold Spring Harbor Workshop on Herpes
Vi-ruses, ColdSpring Harbor, N.Y., on 31 August
1980.)
Wethank C.Lubbers,P. A.Temple,L. B.Sandner,and P. T. Gelep for excellent technical assistance, J. A. Fyfe for valuable discussion, G.B. Elionfor criticalreading of the manuscript and for continuous support andinterestduring thiswork,andK.0.Smithforsograciously providinguswith hismutants.
ThisstudywassupportedinpartbyPublic Health Service researchgrantCA20260 andprogramprojectgrantCA21082 fromtheNationalCancer Institute. D.M.C.wastherecipient ofpostdoctoralfellowship AI05817from the National
Insti-tutesof Health.
LITERATURE CITED
1. Aron, G. M.,D. J. M.Purifoy, and P. A. Schaffer. 1975.DNAsynthesis andDNApolymeraseactivity of
J. VIROL.
herpes simplexvirus type 1temperature-sensitive mu-tants.J.Virol. 16:498-507.
2. Chartrand, P.,C. S.Crumpacker,P. A. Schaffer, and N. M.Wilkie. 1980. Physical and genetic analysis of
herpessimplex virus DNA polymerase locus. Virology 103:311-326.
3. Chartrand, P.,N. D.Stow,M.C.Timbury, and N. M. Wilkie. 1979. Physicalmapping of paa' mutations of
herpessimplex virus type 1 and type 2 by intertypic markerrescue.J.Virol. 31:265-276.
4.Coen,D.M., R. A. F. Dixon, S. W. Ruby, and P. A. Schaffer. 1980.Genetics of acycloguanosine resistance andthethymidinekinase gene in HSV-1, p. 581-590. In B.Fields,R.Jaenisch, and C. F. Fox (ed.), Animal virus
genetics, ICN-UCLA Symposium on Molecular and CellularBiology, vol. 18. Academic Press, Inc., New York.
5. Coen, D. M., and P. A.Schaffer. 1980. Two distinct loci conferresistance toacycloguanosine in herpes simplex virus type 1. Proc. Natl. Acad. Sci. U.S.A. 77:2265-2269.
6. Collins, P., and D. J. Bauer. 1977. Relative potencies of anti-herpes compounds. Ann. N.Y. Acad. Sci. 2:49-59. 7. Crumpacker, C. S., P. Chartrand, J. H.
Subak-Sharpe, and N. M. Wilkie. 1980. Resistance of herpes
simplexvirus to acycloguanosine-genetic and physical analysis. Virology 105:171-184.
8. Elion,G.B.,P. A.Furman, J. A. Fyfe, P. de Miranda, L.Beauchamp, and H. J. Schaeffer. 1977. Selectivity of action ofanantiherpetic agent 9-(2-hydroxyethoxy-methyl)guanine. Proc. Natl. Acad. Sci. U.S.A. 74:5716-5720.
9. Field, H. J., G. Darby, and P. Wildy. 1980. Isolation andcharacterization of acyclovir-resistant mutants of herpes simplex virus. J. Gen. Virol. 49:115-124. 10. Finney, D. J. 1971. Probit analysis, 3rd ed. Cambridge
University Press, Cambridge.
11. Furman,P.A.,P. V.McGuirt, P. M. Keller, J. A. Fyfe, andG. B.Elion. 1980.Inhibition by acyclovir of cell growth and DNA synthesis of cellsbiochemically trans-formed with herpes virus genetic information. Virology 102:420-430.
12.Furman, P. A., M. H. St. Clair, J. A. Fyfe, J. L.
Rideout,P. M. Keller, and G. B.Elion. 1979. Inhi-bition ofherpessimplex virus-induced DNA polymerase activity and viral DNA replication by
9-(2-hydroxy-ethoxymethyl)guanine and itstriphosphate. J. Virol. 32:72-77.
13. Fyfe, J. A., P. M.Keller, P. A. Furman, R.L.Miller,
and G. B.Elion. 1978.Thymidine kinase from herpes simplex virus phosphorylates the new antiviral com-pound, 9-(2-hydroxyethoxymethyl)guanine. J. Biol. Chem.523:8721-8727.
14. Hay, J., H. Moss, A. T. Jamieson, and M. C. Timbury. 1976. Herpes virus proteins: DNApolymerase and py-rimidinedeoxynucleoside kinase activities in tempera-ture-sensitive mutants ofherpes simplex virus type 2. J. Gen.Virol. 31:65-73.
15. Hay, J., and J. H.Subak-Sharpe. 1976. Mutations of herpes simplex virus types 1 and 2 that are resistant to phosphonoacetic acid induce altered DNA polymerase activities in infected cells. J. Gen. Virol.31:145-148. 16.Honess, R.W., andD.H.Watson.1977.Herpes simplex
virus resistance andsensitivity tophosphonoacetic acid. J.Virol. 21:584-600.
17.Jofre, J. T., P. A.Schaffer,and D. S. Parris. 1977. Genetics of resistancetophosphonoacetic acidinstrain KOS ofherpes simplex virus type 1.J.Virol. 23:833-836.
18. Kornberg,A. 1969.Active center of DNApolymerase. Science 163:1410-1418.
19. Mao, J. C. H., E. E.Robishaw,andL.R.Overby.1975. Inhibition of DNA polymerase activity from herpes
on November 10, 2019 by guest
http://jvi.asm.org/
VOL. 40, 1981
simplexvirus-infectedWI-38cells byphosphonoacetic
acid. J. Virol. 15:1281-1283.
20. Miller, R. L., and F. Rapp. 1976. Distinguishing cyto-megalovirus, mycoplasma, and cellular DNA polymer-ase. J.Virol. 20:564-569.
21. Miller, W. H., and R. L. Miller. 1980. Phosphorylation ofacyclovir(acygloguanosine) monophosphate by GMP kinase. J.Biol. Chem.255:7204-7207.
22. Parris,D.S., R.A. F. Dixon, andP.A.Schaffer. 1980. Physical mapping of herpes simplex virus type 1 ts mutants bymarkerrescue:correlation ofphysical and genetic maps. Virology100:275-287.
23. Purifoy,D. J.M., R.B.Lewis, and K. L. Powell. 1977. Identification of the herpes simplex virus DNA polym-erasegene. Nature(London) 269:621-623.
24. Purifoy, D. J. M., and K. L. Powell. 1977. Herpes simplex virus DNApolymeraseasthe site of phosphon-oacetatesensitivity: temperature-sensitive mutants.J. Virol. 24:470-477.
25. Schaeffer,H.J., L.Beauchamp,P.deMiranda, G. B.
Elion, D. J. Bauer, and P. Collins. 1978.
9-(2-hydrox-yethoxymethyl)guanineactivity against viruses of the herpes group. Nature(London) 272:583-585. 26. Schaffer, P. A., M. J. Tevethia, and M.
Benyesh-Melnick. 1974. Recombination between temperature-sensitive mutants of herpessimplex virus type 1. Virol-ogy58:219-228.
27. Schnipper,L.E., and C. S.Crumpacker.1980. Resist-anceof herpessimplex virustoacycloguanosine: role of viralthymidine kinase and DNA polymerase loci. Proc. Natl. Acad. Sci. U.S.A.77:2270-2273.
28. Smith, K. O., W. L. Kennell, R. H. Poirier, and F. T. Lynd. 1980.In vitro and in vivo resistanceofherpes simplex virus to9-(2-hydroxyethyoxy-methyl)guanine
(acycloguanosine). Antimicrob. Agents Chemother. 17: 144-150.
29. Weissbach, A., S. Hong, J. Aucker, and R. Muller.
1973.Characterizationofherpessimplexvirus-induced deoxyribonucleic acid polymerase. J. Biol. Chem. 218:
6270-6277.