0022-538X/90/094288-08$02.00/0
Copyright ©1990,American Society forMicrobiology
Quantitative Polymerase Chain Reaction Analysis of Herpes
Simplex Virus DNA in Ganglia of Mice
Infected with
Replication-Incompetent
Mutants
JONATHAN P. KATZ, ETHAN T. BODIN,AND DONALD M. COEN* Department of Biological Chemistry and Molecular Pharmacology,
Harvard MedicalSchool,Boston, Massachusetts 02115 Received 19April 1990/Accepted6June1990
To study the rolesofviralgenesintheestablishmentandmaintenance ofherpessimplexvirus(HSV)latency, we have developed apolymerase chain reaction assaythatisbothquantitativeand sensitive.Using this assay, weanalyzedthelevels ofviral DNA intrigeminalgangliaof mice inoculatedcorneally with HSV mutants that aredefective for virus replicationat one or moresites in mice and for reactivation uponganglionic explant. Ganglia frommiceinfected with thymidine kinase-negative mutants, which replicate at the site of inoculation andestablishlatency butdo notreplicateacutely ingangliaorreactivate uponexplant,contained arangeof levelsofHSV DNA that overlapped with the range found inganglia latently infected withwild-typevirus. On average, these mutant-infected ganglia contained one copy ofHSV DNA per 100 cell equivalents (ca. 10 molecules), which was 50-fold less than theaverage for
wild-type
virus. Ganglia from mice infected witha ribonucleotide reductase deletion mutant, which is defective for acute replication and reactivation upon ganglionic explant, also containedonaverageonecopyof HSV DNA per 100 cellequivalents.We also detected substantial numbers of HSVDNAmolecules (upto ca. 103)ingangliaof mice infectedwithanICP4deletion mutantandotherreplication-negativemutantsthat areseverelyimpairedforviral DNAreplicationandgene expression. These results raise the possibility that such mutants can establish latency, which could have important implications for mechanisms of latencyandforvaccine andantiviraldrugdevelopment.Herpesviruses, like retroviruses, papillomaviruses, and hepadnaviruses, establish latent infections in their hosts, forming lifelong reservoirs of recurrent disease that resist cure (1). Herpes simplex virus (HSV) latency (12, 27) is preceded by productive infection at the periphery of a
mammalian host. The virus enters nerve endings and mi-grates to ganglionic nuclei, where productive infection can again ensue. With time, latency develops, in which viral DNA (9, 23) but no infectious virus is present. However, infectious virus can be reactivated by certain stimuli or by explant of ganglia.
The roles of productive infection processes in the estab-lishment and maintenance of HSV latency have not been determined. Considerable understanding of the productive infection cycle of HSV and the roles of specific genes has been gained through the use of engineered mutants with defects in functions important for virus replication and gene expression (24). When such replication-defective mutants have been tested in a mouse model of latency, they have failedtoreactivate from explanted ganglia (14, 16). It has not been possible to determine whether the failure of these mutants toreactivate is due solely to the replication defect duringreactivation or also to a requirement for specific viral productive infection processes in the establishment and maintenance of latency. Such processes could include DNA replication or steps in the regulatory cascade of virus gene expression.
Certain mutants that are replication competent in cell culture fail to reactivate from explanted ganglia in a mouse model. These include thymidine kinase-negative (tk-) mu-tantsand certain thymidine kinase-deficient mutants. These mutantsdo establish and maintain latency, as evidenced by
* Correspondingauthor.
expression of
latency-associated
transcripts (5, 17,29)
and their ability to rescue replication-negative virus following superinfection of dissociatedganglia
(5)or tobe rescuedby superinfection with wild-type virus (8), demonstrating the presenceofbiologically active virus genomes. However,in at least two cases (8; K. Hicks, D. Yager, and D. Coen, unpublished results), tkmutantgenomes were not detected reliably by blot hybridization methods.To quantify the numberof viral genomesin gangliafrom mice infected with various HSV mutants, we have devel-opeda quantitative polymerase chain reaction (PCR) assay forHSV DNA. Thishas allowedus tobegintoaddress the question ofwhether certain productive infection processes arerequired for establishment and maintenance oflatency by determining howmuch, if any, viralDNAisresident in ganglia of mice infected with replication-incompetent mu-tants.
MATERIALS AND METHODS
Cells andviruses. Theviruses used in this studyarelisted inTable 1. Thewild-type strain was HSV type1KOS,which was propagated and assayed on Vero cells, maintained as described previously (31). Mutants dlsptk and dlsactk (5) engineered to contain360- and 4-base-pair deletions in the HSV tk gene,respectively, werepropagatedand titerswere determined on Vero cells. Mutant ICP6A (11) contains a 2.9-kilobase-pair deletion in the gene encoding ICP6, the large subunit of HSV ribonucleotide reductase. This mutant and the D14 cells(10)onwhich it was propagated and its titer determined were kindly provided by D. Goldstein and S. Weller. High-titer stocks of mutants d120 and n12, which containa4.1-kilobase-pairdeletion and a nonsense mutation in the geneencodingICP4, respectively (6, 7), were gener-ously provided by N. DeLuca and P. Schaffer. A high-titer 4288
on November 10, 2019 by guest
http://jvi.asm.org/
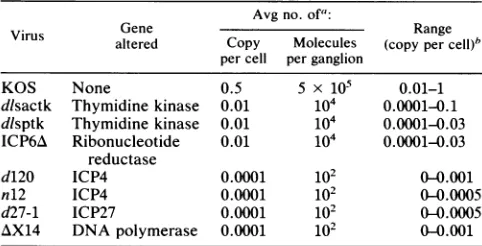
TABLE 1. Amount of HSV DNA in ganglia of mice infectedwith HSV mutants
Avg no. of':
Virus Gene Range
altered Copy Molecules (copy percell)"
percell perganglion
KOS None 0.5 5 x 105 0.01-1
dlsactk Thymidine kinase 0.01 104 0.0001-0.1 dlsptk Thymidine kinase 0.01 104 0.0001-0.03 ICP6A Ribonucleotide 0.01 104 0.0001-0.03
reductase
d120 ICP4 0.0001 102 0 0.001
n12 ICP4 0.0001 102 0(0.0005
d27-1 ICP27 0.0001 102 00.0005
AX14 DNApolymerase 0.0001 102 0-0.001
a Values shown are the averages of measurementsanalyzedas inFig.1 to 4 on atleast sixgangliafrom miceinfected with each virus.Measurements
wereincludedonly when they came from experiments in which all of at least
threenegativecontrols yielded noHSV-specificsignal. The average number
ofmolecules per ganglion value wascalculatedon the basis of about106cells
perganglion.
bValues represent the ranges ofmeasurements obtainedfrom individual
gangliaanalyzed asdescribedinfootnotea.
stock of mutant d27-1 (22), which contains a 1.6-kilobase-pair deletion in the gene encoding ICP27, was graciously provided by S. Rice and D. Knipe. Mutant AX14 (18), which containsa1.2-kilobase-pairdeletionin the gene encoding the HSV DNA polymerase, and the DP6 cells on which it is propagated and its titer determined (18) were kindly pro-vided by A. Marcy.
Infections of mice. Seven-week-old CD-1 mice (Charles River Breeding Laboratories, Kingston, N.Y.) were mock inoculated or inoculated at thecornea with wild-type virus or the mutants as described before (16, 28)at adose of2 x 106 PFU/eye with the exception of mutant d120, which was inoculated at 107 PFU/eye.
Isolationofganglionic DNA.To prepare ganglionic DNA, gangliawere removed from mice >30 days after infection, using instruments that had been treated with DNase and autoclaved to avoid introduction of contaminants. Only ganglia from animals infected with a given mutant were taken on any given day. Methods for DNA extraction, including additional precautions to avoid contamination, have beendescribed in detail(3). Briefly,eachganglionwas
placed in a screw-cap microcentrifuge tube and digested overnight at 50°C with 100
pug
of proteinase K per ml in proteinasedigestion buffer (20mMTrischloride[pH7.4],20 mM EDTA [pH 8], 0.5% sodium dodecyl sulfate). The samplewasgently mixedand phenol-chloroform extracted, the organicphase wasback-extracted oncewithproteinase
digestionbuffer, and the pooledaqueousphases were chlo-roform extracted twice. Ammonium acetate was added to the final aqueous phase to 2.5 M, and the DNA was
precipitated with ethanol, washed once with 70%
ethanol,
andsuspendedin10mMTrischloride-1mMEDTA, pH7.5. The intactness and concentration of the DNA were esti-mated by agarose gel
electrophoresis
alongside
known amountsof standard DNA. Byperforming
extraction proce-dures gently, the vastmajority
of the DNAmigrated
ashigh-molecular-weight species.
PCR.ThestandardPCR,using TaqDNApolymerase
(25),
was modified to permit quantification of HSV DNA. The details of the
procedure,
including
measures topreclude
contamination with exogenous DNA sequences, are pre-sented elsewhere (3). Briefly, 100-ng
samples
ofganglionic
DNAor, as standards, 100-ng samples of mouse tail DNA
spiked with knownamounts of HSV DNA were mixed with 50pmoleach oftwo pairs of primers. One primer-pair, CT TAACAGCGTCAACAGCGT and CAAAGAGGTGCGGG AGT, was specificfor the HSV tk gene (13, 19, 30) andthe
other, AGTGTGCGGGGATGCAGT and ACGCGAGAGC CCCACGTA,wasspecificfor thesingle-copymouseadipsin gene (20). These DNAs were then assembled into 100-,l reactions containing50mM KCl, 10mM Tris chloride (pH 8.4), 4.5 mM
MgCI2
(whichwasfoundtobeoptimalfor this primer-template combination), 100 ,ugofgelatin perml, 200 ,uM concentrations of each deoxynucleoside triphosphage, and 4 U of Taq DNA polymerase. PCR amplification was thenperformed for30cycles,with denaturation for 1minat94°C, annealingfor 2 minat55°C, and extensionfor3 minat
72°C, withafinal additional extension of7 min.
Analysisof PCRproducts. A
10-pl
portionfrom each PCRamplification
was electrophoresed on a12% nondenaturingpolyacrylamide
gel,visualizedbyethidiumbromidestaining, transferredtoanylonfilter(Gene-Screen Plus;NewEngland Nuclear-Dupont), and UV cross-linked as described before (2). The filter was thenprehybridized at 50°Cfor2 hin 6x SSPE (0.54 M NaCl, 60 mM NaH2PO4 [pH 7.4], 60 mM EDTA [pH 7.4])-1% sodium dodecyl sulfate-Sx Denhardt solution-200 ,ugofnative salmon spermDNAperml-200pLg
of denatured salmon sperm DNA per ml-10% dextran sul-fate. The filterwasthenhybridizedin thesamesolutionwith an oligonucleotide probe specificfor the HSV PCRproduct, CAGATCTTGGTGGCGTG, radiolabeled with T4 polynu-cleotide kinaseas
described,
for2 h at 50°C. Thefilterwas washed fourtimes at room temperature (5 min each time) with 6x SSPE-1% sodium dodecyl sulfate, four times at50°C (10mineach
time)
with 6x SSPE-1% sodiumdodecyl
sulfate, and once at roomtemperature with 6x SSPE. The filterwas thenexposedto Kodak XAR-5 film. When inten-sifying screens were used, the film was
preflashed.
For certainexperiments, nylon
filterswerestripped
oftheHSV-specific
probe byboiling
in 0.015MNaCl{-.0015
Msodium citrate-1% sodiumdodecyl
sulfate for 20 min and thenreprobed
asabove with radiolabeledoligonucleotide
specific
forthemouse
adipsin
PCRproduct,
AGTCGAAGGTGTGG TTAC.Autoradiographic signals
wereanalyzed by
densito-metry with anLKB laserscanner.RESULTS
Development ofa quantitative PCR assay. Initial slot blot
hybridization experiments
toquantify
HSV DNAinganglia
from mice infected with certain HSV mutants
proved
to be tooinsensitivetodetermine whether theseganglia
contained HSV DNA(16;
Hicks etal.,
unpublished results).
We thereforeconverted thePCR,
an assaycapable
ofdetecting
single
moleculesofDNA(25),
to anassay thatcanquantify
rareDNAs.Thiswas
accomplished by
varying
severalofthe parametersof the standard PCR assay.Figure
1 shows the resultsobtained ina reconstructionexperiment
designed
to test thequantitative
aspects of the PCR assay. Various amounts of HSV DNA were mixed with 100 ng ofmouseDNA andtwo
pairs
ofprimers.
Oneprimer
pair
wasspecific
for the HSV tk gene
(13, 19,
30)
and the otherwasspecific
forthe
single-copy
mousegeneadipsin
(20).
The mixturesweresubjected
to 30cycles
ofPCR,
and theresulting
products
were
separated
onapolyacrylamide gel
andtransferredtoanylonfilter. When thefilterwas
probed
witha radiolabeledoligonucleotide
specific
for the HSV tk PCRproduct,
theautoradiographic
signal
of thisproduct
decreasedmonoton-ically with
increasing
dilution of the amountofvirus DNAon November 10, 2019 by guest
http://jvi.asm.org/
Copies per Cell HSV DNA
in 100 ng Mouse DNA
H x
Hinfl I 10--1 1 0-2 10-3 10-4
100 ng
Ganglion DNA from
Mock-infected
0
Mice
-HSVtk Product
1_0.1w,401W
.--Mouse
Adipsin
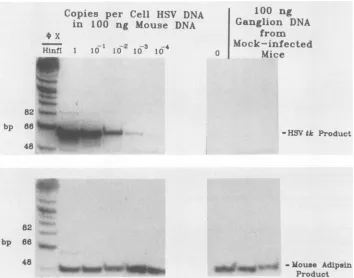
ProductFIG. 1. QuantitativePCRanalysisof HSVDNA.(Top) Quantitativedetection ofHSVDNA.Tenfold dilutions of HSV-1 strain KOSDNA
weremixedwith 100ngofDNAfrom the tails of uninfectedmicesothat the viralDNAwaspresentat 1copy permousecellequivalent(0.33
pgof HSVDNA), 0.1 copy, etc.,asindicatedatthetopof each lane. Asnegativecontrols, 100ngofmousetail DNA alone and 100ng of DNA from each oftwo trigeminal ganglia of mock-infected mice were also included. These samples were amplified by PCR, using oligonucleotide primers specificforaportionof theHSV tkgeneanda second set ofprimersspecificfor themouseadipsingene.ThePCR
productswereelectrophoresedon apolyacrylamide gel alongsideradiolabeledHinfi-digested(X174DNA,which servedassize markers.The DNAwastransferredtoanylonfilterandprobedwitharadiolabeledoligonucleotide specificfor the HSV PCRproduct.(Bottom)Detection of the internal controlmousePCRproduct.The PCRproductsshownwereanalyzed bystrippingthe filter andprobingwithanoligonucleotide
specificfor themouseadipsingenePCRproduct.
(Fig. 1, top). In contrast, the PCR signal corresponding to the mouse adipsinproduct, which could be visualized
rou-tinely by ethidium bromide staining (not shown), remained relatively constant. This is shown as detected by probing with an appropriate radiolabeled primer (Fig. 1, bottom).
Whendensitometric analysisof theHSVsignalfrom similar reconstruction experiments was performed, a nearly linear
log-logrelationship between signal intensityand theamount of viral DNA was obtained (see Fig. 3B for an example).
This empirically derived relationship was convenient in permitting analysis of a wide range of amounts of HSV DNA.
The assay could readily detect amounts of HSV DNA mixed with mouse DNAat 10-4 copies percell equivalent
(mostobvious in Fig. 2B), which correspondsto two mole-cules of HSV DNA (based on Poisson distribution
princi-ples,it isfairertosaythat theamountof HSVDNApresent
in this mixture is one or a few molecules). A zero-copy
reconstruction control and ganglionic DNA prepared from twomock-infected mice yielded similar mouseadipsin
sig-nals, but no detectable HSV-specific signal (Fig. 1). These
threenegative controlsvalidated the rigorousmeasuresused
toprecludecontamination (3) andweresimilarly negativein
allexperiments reported below. The mouseadipsin internal
control both verified true negatives and allowed standardi-zation ofPCRs thatvaried in efficiency oramount ofinput DNA. Indeed, increasing or decreasing the amount of a
mixture of HSV andmouseDNAasmuchas10-fold didnot change the ratio of the HSV and mouse PCR signals (not shown).
Quantification of DNA from wild-type and tk mutant la-tently infected ganglia. We next used the quantitative PCR
assay to measure the amounts ofHSV DNA in trigeminal ganglia from CD-1 mice following corneal inoculation with wild type HSV-1 strain KOS and various mutants ofthis strain. Thirty days ormoreafter inoculation, atwhichtime the wild-type virus has established a reactivatable latent infection (16), gangliawere harvested. DNAwas prepared
and measured for its content of HSV DNA by using the
assay. Ganglia from mice latently infected with wild-type virus contained on average 0.5 copy per cell equivalent of HSV DNA (ca. 5 x 105 molecules per ganglion), although
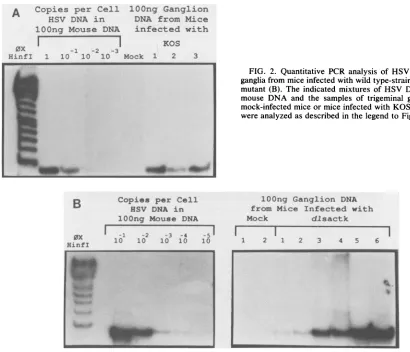
the amount of HSV DNA perganglion ranged from below 0.1copy percelltoabout 1copypercell in individualganglia (Fig. 2A,Table 1). This variationevidentlyisapropertyof the gangliathemselves; itcouldnotbe ascribedtovariability in the PCRassayasrepeatassaysofdifferentsamples from individualganglia yielded similar results (not shown). Both theaverage value and the range were very similartothose obtained forgangliaofCD-1 micefollowing corneal inocu-lation withwild-type strain KOS, using slot-blot hybridiza-tion methods (16).
Figure 2B shows the results obtained with ganglia from mice infected withmutant dlsactk. This mutantand mutant 82
bp 86
48 +u^.
82 bp 668
48
_S_11_
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.129.483.77.355.2]lOOng Ganglion DNA from Mice infected with
KOS
-1 -2 -3
1 10 10 10 Mock 1 2 3
B
Copies
per CellHSV DNA in lOOng Mouse DNA
0X
HinfI
-I -2 -3 -4
10 10 10 10
-51
10
MM W
dlsptk, which contain deletions in the HSV tk gene and do not express thymidine kinase enzyme, grow to wild-type titers in the mouse eye following corneal inoculation, but do notreplicatedetectablyintrigeminal ganglia (5). They do not reactivate upon ganglionic explant, but do establish latent infections,asevidenced by expression of latency-associated transcripts and their ability to rescue replication-negative virus following superinfection of dissociated ganglia (5). As expected from these results, ganglia from mice latently infected with the tk-mutantscontained substantial numbers of HSV DNA molecules, ranging up to 0.1 copy per cell (ca. 105molecules perganglion) (Fig. 2B). Although, as was the case withwild-type virus, therewas substantial variation in the amountsofHSV DNA in individualganglia,the average amount ofHSV DNA present in ganglia of mice infected with either tk- mutant was 0.01 copy per cell (Table 1). Thus, the average amount of DNA in ganglia from tk-mutant-infected micewasless than thatfrom KOS-infected mice, butthe ranges ofvaluesoverlapped.
Quantification of DNA from mice infectedwith a ribonucle-otide reductase mutant. Figure 3 shows the resultsobtained with ganglia from mice infected with mutant ICP6A. This deletion mutant fails to specifyHSV ribonucleotide reduc-taseactivity(11).Althoughitcanreplicatein many cell types in culture (11), it ishighlydefective forreplicationinmouse
cells at 38°C (14). Following corneal inoculation, ICP6A replicatesverypoorlyin the eye,achieving barelydetectable titers.Itfailstoachieve detectable titers in
trigeminal
ganglia
duringthe4daysfollowinginoculationortoreactivatefrom these ganglia upon explant 30 days after inoculation
(14).
FIG. 2. Quantitative PCR analysis of HSV DNA in trigeminal ganglia from mice infectedwith wild type-strain KOS (A) anda tk-mutant(B). The indicated mixtures of HSVDNA and uninfected
mouse DNA and the samples of trigeminal ganglion DNA from
mock-infected miceormiceinfected with KOS andmutantdlsactk
wereanalyzedasdescribed inthe legendtoFig. 1.
lOOng Ganglion DNA from Mice Infected with
Mock dlsactk
2 1 2
..3
I
1 2 1 2 3 4 5 6
.i
-.-Despite the replication defects of ICP6A, levels of HSV DNAaveraging 0.01copy percellequivalent (ca. 104 mole-cules per ganglion) could be detected in ganglia of mice
infected with this mutant (Fig. 3, Table 1). Thus, minimal replication at the site of inoculation suffices for tens of thousands of viral DNA molecules to reach the trigeminal ganglion and bemaintained there stably.
ICP4, ICP27,andpolmutants.Basedontheseresults, we next asked whether any viral replication is necessary for viral DNA to reach the trigeminal ganglion and be main-tained there. We therefore tested several mutants that are
replication negative becauseof defects in viralgene
expres-sion and DNAreplication. These mutants include d120(6), whichcontainalargedeletion in thegeneencodingthemajor regulatory protein, ICP4;n12(7),whichcontainsanonsense
mutation in the ICP4 gene; d27-1 (22), which contains a
deletion in thegeneencodingtheregulatory protein,ICP27; and AX14(18), which containsadeletionin the HSV DNA polymerase gene. TheICP4 mutantsare severelyrestricted in the expression ofearlyand late genes, while the ICP27 andDNApolymerasemutants areseverelyrestricted in the expressionof lategenes.All ofthe mutants fail tosynthesize detectable levels of viral DNA except the ICP27 mutant, which nonetheless is very restricted in its DNA synthesis (22). These replication-incompetent mutants are routinely
propagated on cell lines containing viral genes that can
complementthe mutations; importantly, mutantsd120, and d27-1 carry deletions that cannot be rescued by the genes resident in the cell lines. Asaresult,nodetectablewild-type
A Copies per Cell HSV DNA in lOOng Mouse DNA
0x
HinfI
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.77.487.72.424.2]A Co pies per- Cell HSV DNA in
lOOng
Mouise
DNA¢~~
~~~4-HinfI 10- -2 03
-14
100 ng Ganglion DNA from Mice Infected
with ICP6A
1 2 3 4 - 5
S -4 -3 -2 -1 0
[image:5.612.163.458.74.489.2]HISVDNAmoleculezpercell (log)
FIG. 3. Quantitative PCR analysis of HSVDNA intrigeminal gangliafrom mice infected witharibonucleotide reductasemutant.(A)The indicated mixtures of HSV DNA and uninfectedmouse DNAand the samplesoftrigeminal ganglionDNA frommice infected with HSV
mutantICP6Awereanalyzedasdescribed in thelegendtoFig. 1.(B)DensitometricanalysisofquantitativePCR.Theautoradiographsshown inpanel Awerescannedon anLKB laserscanner,andintegralsof thesignalswerecomputed.Thelogsof thecomputed signalswereplotted
relativetothelogs of theamountsof HSV DNApresentin the mixtureswithmouseDNA,asindicatedbytheopen squares.Thesignalsfrom thegangliaareshownasclosed triangles. Themouseadipsin signalswereindistinguishable in all samples.
virus ispresentinstocks of thesemutants(6, 22;N.DeLuca, S.Rice, and D. Knipe, personal communications).
Despite the severe defects of the mutant viruses, HSV DNAcouldbedetected readily inganglia from mice infected 30daysor more previously with themutants(Fig. 4, Table 1). As stated previously, datawere used only from
experi-ments such as that shown in Fig. 4 in which all negative
controls(atleastthreeperexperiment)gave noHSVsignal,
butgavesimilarmouseadipsin signals(notshown). Different aliquots ofganglionic DNA from mice infected with these mutants could contain no detectable DNA or varying
amountsofDNA.In particular,upto0.001copy percell(ca.
103moleculesperganglion)wasfound in the ganglia of mice
that hadbeeninfected with d120 (Fig. 4).Repeated analyses of DNAfrom specific ganglia revealed that certain samples that scorednegative inonePCRassayoften scoredpositive
inothers, which would beexpectedfromaPoisson distribu-tion of HSV DNA molecules among aliquots ofganglionic DNA. Takingintoaccount alloftheassays, levels of HSV DNA averaging 0.0001 copy percell (ca. 102 molecules per
ganglion) were detected in ganglia of mice that had been
infected with anyof the replication-negative mutants. DISCUSSION
We describe here the developmentofa quantitative PCR
assay for HSV DNA and its use in quantifying DNA in
gangliaof miceinfected with HSVmutants.Assummarized in Table 1, wefound substantialamounts ofHSVDNAnot only inganglia of mice infected with tk- mutants knownto
establish latentinfections(5) but,moresurprisingly, in mice infected with the replication-defective mutant, ICP6A, and
A w...P* w f
B
C'
fr
00-I
I
on November 10, 2019 by guest
http://jvi.asm.org/
Copies per Cell
HS'V DNA in
lOOng Mouse DNA
100 ng Gaiiglion DNA from Mice Infected with:
lock d120(ICPF4
nu12(ICP4a&m)
d27-1(ICP277d)sX II
IEinfI 1 10 10 10-4 10S 0 1 2 1 2 3 4 5 8 1 2 3 1 2 3
FIG. 4. QuantitativePCRanalysisof HSV DNA intrigeminal gangliafrom miceinfected withimmediate-early regulatory genemutants. Theindicatedmixturesof HSVDNA anduninfectedmouse DNA and samples of trigeminal ganglion DNA from mock-infected mice or mice infectedwithHSV mutant d120, n12, ord27-1were analyzed as described in the legend to Fig. 1.
even in mice infected with replication-negative mutants.
Below we discuss aspects of the quantitative PCR assay,
how the amounts of HSV DNA found in ganglia relate to
biologicalproperties of the tk- and ICPA mutants, and the possible implications for mechanisms of latency and for vaccine and antiviral drug development of our finding of
HSV DNA in ganglia of mice infected with replication-negative mutants.
Quantitative PCR assay for HSV DNA. To develop the
quantitative PCRassay, themostimportantmodificationsto
the standardassay(25)were optimizing the MgCl2
concen-tration, increasing the amount of DNA polymerase, and limiting the amount of template DNA. These and other parameters are discussed in more detail elsewhere (3).
Under the conditions described here, the nearly linear log-log relationship between autoradiographic signal and amountof DNA in the rangeofroughly 10-4 and 1copy per
cell sometimes breaks down above 10' copy per cell;
however, decreasing the number of PCR cycles allows linearityinthe range of10-1 through 10 copiespercell (E.
Pelosi andD. Coen,unpublished results).
Forgangliafrom miceinfectedwith wild-type virus(Fig. 2A, Table1)andforICP0deletionmutants(notshown), we
obtained values forHSV DNAcontent with thePCRassay
that were very similar to those obtained with slot blot hybridization (16). These results also help establish the validity of the PCRassay. The PCRassaywas nevertheless
farmoresensitivethan slot blothybridization, withwhich it
was difficult to distinguish signal from background below 0.01to0.1 copy percell(16) and which failedto determine whether ganglia from mice infected with mutant n12
con-tained HSV DNA (16). The PCR assay answered that question (Fig. 4).
The use of PCR to detect viral nucleic acids in clinical specimens is growing rapidly. Two features of the assay
described heremaybeuseful in clinicalsettings.Theinternal control (in clinical specimens this could be a single-copy
humangene)isinvaluableboth ineliminatingfalse-negatives and inaidingquantification.Thequantitativeassayishelpful in assessing the presence ofcontaminating sequences and thelikelihood thatapositiveresult isduetocontamination. Inour hands, when contamination occurred, itwas usually
at the level ofone or a few molecules per sample, which would be easy to distinguish from true positives in many
clinical situations.
After the present study was completed, a PCR assay to
quantifyhumanimmunodeficiencyvirusDNAwasreported
(21). This assay, whichuses end-labeled PCR primers and
thus does not require blotting and hybridization steps, ap-pearedtobe asquantitativeasthe assaydescribed here, but somewhat less sensitive and more likely to detect nonspe-cificproducts.
Levels of HSV DNA in ganglia latently infected with tk-mutants.Our finding that the ranges of the amounts of DNA found ingangliaof mice infected with wild-type strain KOS and the tk- mutants overlapped supports our previous conclusion (5) that these mutants establish latency. Never-theless, there was on average 50-fold less DNA per ganglion fromthe mutant-infected micethanfromwild-type-infected mice. In contrast, in situhybridization experiments detected three- to fivefold fewer
latency-associated
transcript-posi-tive cells in tk-mutantlatently infectedgangliathan in those of wild type (5). The autoradiographic signal per cell wassimilar between KOS and tk- mutant-infected ganglia (5); still, there could be fewer HSV genomes per cell in mutant-infected ganglia. It is alsopossible that more HSV DNA is foundinnon-neuronal cellsinKOS-infected gangliathanin mutant-infected ganglia.These possibilitiesareunder inves-tigation.
Twoother reports have estimatedthe amountofDNA in
ganglia
latently infected with tk mutants. Efstathiou et al. (8), in agreement with our findings, reported that withSouthern
blothybridization
such ganglia contained much less HSV DNA(undetectable)
than didganglia
from wild-type-infected mice. In contrast, Leist etal. (17),using
slot blothybridization,
reported that suchganglia
whenpooled
containedamounts ofHSV DNA similar to thosefound in wild-type-infected ganglia, although a comparison of the
hybridization
signalswith knownamountsofHSV DNAwas notreported.The differences between these results could be due to differences in mouse strains, routes of
inoculation,
and/or thetkmutantsused. Efstathiouetal. (8)and weengineered
deletions in tk
protein-coding
sequences away from the UL24 gene, which can beimportant
for virusgrowth
(15).
ThemutantsdidnotexhibitdetectableTKactivities
(<0.4%
of wild type). The mutant
analyzed by
Leist et al.(17)
deletedthetkpromoter and muchof the UL24 gene without deletingtk
protein-coding
sequences. TheTKactivity
of this mutant could not bedistinguished
from that of mock-in-fectedcells;however, in the assayused,
thiswas14%thatofwild-type-infected
cells.WenotethatlevelsofTK thatare5 to 10% thoseofwild-typevirusaresufficienttoallowacutereplication
inganglia
and otherbiological
activities(4).
HSV DNA in ganglia of ICP6A-infected mice. We were
surprised
tofindlevels of HSV DNAaveraging
0.01 copy peron November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.112.503.75.201.2]cell in mice infected with mutant ICP6A, a mutant whose replicationat thesite of inoculation isbarelydetectable(14).
This average amount of DNA is similar to that of the tk-mutants,whichreplicatetowild-typetitersin themouseeye
andestablish latency. Thiscomparisonraisestwo
possibili-ties: (i) barely detectable levels ofreplicationat the site of
inoculationdeliver HSV DNA togangliaaswellaswild-type
levels ofreplication, and (ii) the ICP6A mutant establishes
latent infections despite its replication and reactivation
de-fects. StudiestoexamineICP6A-infectedgangliafor biolog-ically activegenomesare underway.
HSV DNA is resident in ganglia of replication-negative
mutants: possible implications for viruslatency and for
vac-cine and antiviral drug development. The results of Fig. 4
show that neither productive infection, viral DNA replica-tion,northenormalregulatorycascade ofgeneexpressionis
requiredfor hundredsorthousands ofviral DNAmolecules
to reach the trigeminal ganglion and be maintained there
stably, acritical component ofthe establishment and
main-tenance of latency. Although we cannot comment on the
physical state or biological activity of the HSV DNA in
ganglia from mice infected with replication-negative
mu-tants, ourresults raise the possibility that mutants that are
severelyrestrictedforproductivevirusreplicationandgene expression can establish latency. This is consistent with
recentstudies(M. Kosz-Vnenchak, D.M. Coen,and D. M.
Knipe, submitted for publication) that show that HSV tk
deletion mutantscanestablishlatencyinthe faceofseverely
restricted lytic gene expression. They also are consistent
with results from Steiner et al. (26), who studiedan HSV
mutantdefective inthe viriontransactivatingfactor Vmw65
(ot-transinducing factor, VP16). This mutant replicated poorly in the mouse eye (but evidently not as poorly as
ICP6A[14])and notdetectablyinmouseganglion, yetcould
reactivate fromlatencywith fairefficiency. Keepingin mind
the reservations stated above, these studies taken together
suggest that establishment and maintenance of latency do
notrequire lytic pathwaysofgeneexpression and thatviral replicationitselfmerely permitsanincreaseinthenumber of
virusgenomesthatcangain accessto neuronal nuclei. This
contrasts with models in which latency is established by a
change in the balance ofregulatory activities that operate during productive infection. We think that consideration
should be given to two hypotheses. (i) As yet undescribed
viral regulatory genes promote the latency pathway. (ii)
Establishment of HSV latency is a passive process that is governed by neuronal factors rather than by any de novo
synthesizedviralgene product.
There isabundant evidencethat preventionofHSV
rep-lication by vaccines or antiviral drugs can greatly reduce
reactivatablelatent infections(12). Based onthediscussion
above, we suggestthat this is due solely to decreasingthe
number of virus genomes that are capable of establishing
latent infections. Our results therefore lead to the
specula-tion that chemotherapeutic or immunoprophylactic strate-gies targeted against lytic functions of the virus may be
unable to prevent latency completely.
ACKNOWLEDGMENTS
We thank N. DeLuca, D. Goldstein, D. Knipe, D. Leib, A. Marcy, S. Rice, P. Schaffer,and S. Weller forsupplying high-titer
stocksofmutantsand forhelpful discussions;C.Bogard, D. Frazier (and his finger), D. Leib, and J. Jacobson for help in inoculating
mice and harvesting ganglia; H.-Y. Minand B. Spiegelman for
mouseadipsinprimers; and K. Hicksforinitialwork in settingup
thePCRassay.
This work wassupportedbyPublic Health Servicegrants BRSG S07RR05381 (D.M.C.) andP01 A124010 from the National
Insti-tutesof Health(P. Schaffer,D. Knipe,andD.M.C.).
LITERATURE CITED
1. Ahmed, R., and J. G. Stevens. 1990. Viral persistence, p. 241-265. In B. N. Fields,D. M. Knipe,R. M. Chanock, M. S. Hirsch, J. L. Melnick, T. P. Monath, and B. Roizman (ed.), Virology, 2nd ed. Raven Press, NewYork.
2. Coen,D. M. 1990. Transfer of DNA frompolyacrylamidegelsto nylon filters by electroblotting, p. 2.9.14-2.9.17. In F. M. Ausubel, R. Brent, R. E. Kingston, D. D.Moore,J. A. Smith, J. G. Seidman, and K. Struhl (ed.), Current protocols in molecularbiology.JohnWiley&Sons,Inc., New York. 3. Coen, D. M. 1990. Quantitationof rare DNAsbythepolymerase
chain reaction, p. 15.3.1-15.3.8. In F. M. Ausubel, R. Brent, R. E.Kingston, D. D. Moore, J. A. Smith, J. G.Seidman,and K. Struhl (ed.), Current protocols in molecularbiology. John Wiley&Sons, Inc.,New York.
4. Coen, D. M., A. F. Irmiere, J. G. Jacobson, andK. M. Kerns. 1989. Low levelsofherpes simplexvirusthymidine-thymidylate kinaseare notlimitingforsensitivityto certain antiviraldrugsor
forlatency ina mousemodel. Virology168:221-231.
5. Coen, D. M., M. Kosz-Vnenchak, J. G. Jacobson, D. A. Leib, C. L. Bogard, P. A. Schaffer, K. L. Tyler, and D. M. Knipe. 1989.Thymidine kinase-negative herpes simplexvirusmutants
establishlatency inmousetrigeminal gangliabut do not
reacti-vate.Proc. Natl.Acad. Sci. USA86:4736-4740.
6. DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants ofherpes simplex virus type 1 in the gene encoding immediate-early regulatory proteinICP4. J. Virol. 56:558-570.
7. DeLuca, N. A., and P. A. Schaffer. Physical and functional domains of the herpes simplex virus transcriptionalregulatory proteinICP4. J. Virol. 62:732-743.
8. Efstathiou, S., S. Kemp, G. Darby, and A.C.Minson.1989. The role ofherpes simplex virustype 1thymidine kinase in patho-genesis. J. Gen. Virol.70:869-879.
9. Efstathiou, S., A.C.Minson,H.J. Field, J. R. Anderson, and P. Wildy. 1986. Detection of herpes simplex virus-specific DNA sequences in latently infected mice and in humans. J. Virol. 57:446-455.
10. Goldstein,D.J., and S. K.Weller. 1988. Herpessimplexvirus type1-inducedribonucleotidereductaseactivity isdispensable for virusgrowth and DNA synthesis: isolation and characteri-zation ofanICP6lacZinsertionmutant. J.Virol. 62:196-205. 11. Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in
herpes simplex virustype 1-infected cells can compensate for the lossof the large subunit of theviral ribonucleotide
reduc-tase: characterization of an ICP6 deletion mutant. Virology
166:41-51.
12. Hill,T.J.1982.Herpessimplex virus latency,p. 175-240. In B. Roizman (ed.), The herpesviruses, vol. 3. Plenum Publishing Corp., New York.
13. Irmiere,A.F.,M.M. Manos, J. G. Jacobson, J. S. Gibbs,and D. M. Coen. 1989. Effect ofanamber mutationin the herpes simplexvirus thymidine kinasegene on polypeptide synthesis andstability.Virology168:210-220.
14. Jacobson, J. G.,D. A.Leib, D. J. Goldstein, C. L. Bogard, P.A. Schaffer,S. K.Weller,and D.M.Coen.1989. A herpessimplex virusribonucleotide reductasedeletion mutant is defective for productiveacuteandreactivatablelatent infections of miceand forreplication in mouse cells. Virology173:276-283.
15. Jacobson, J. G., S. L. Martin, and D. M. Coen. 1989. A conserved openreading framethat overlaps the herpes simplex virusthymidine kinasegene is important for viral growth in cell culture. J.Virol.63:1839-1843.
16. Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager,D.M.Knipe, and P. A. Schaffer. 1989. Immediate-early
gene mutants define different stages in the establishment and
reactivation ofherpes simplex virus latency. J. Virol. 63:759-768.
17. Leist, T. P., R. M. Sandri-Goldin, and J. G. Stevens. 1989.
on November 10, 2019 by guest
http://jvi.asm.org/
Latentinfections in spinal ganglia with thymidine kinase-defi-cientherpessimplex virus. J. Virol. 63:4976-4978.
18. Marcy, A. I., D. R.Yager, and D. M. Coen. 1990. Isolation and characterization of herpes simplex virus mutants containing engineered mutations at the DNA polymerase locus. J. Virol. 64:2208-2216.
19. McKnight, S. L. 1980.The nucleotide sequence and transcript of theherpessimplex virus thymidine kinase gene. Nucleic Acids Res.8:5949-5964.
20. Min, H.-Y., and B. M. Spiegelman. Adipsin, the adipocyte serine protease: gene structure and control of expression by tumornecrosisfactor. Nucleic Acids Res. 14:8879-8892. 21. Pang, S., Y. Koyanagi, S. Miles, C. Wiley, H. V. Vinters, and
I. S. Y. Chen. 1990. High levels ofunintegrated HIV-1 DNA in brain tissue of AIDSdementia patients. Nature (London) 343: 85-89.
22. Rice, S. A., and D. M. Knipe. 1990. Genetic evidence for two distinct transactivation functions oftheherpessimplex virus a protein ICP27.J. Virol. 64:1704-1715.
23. Rock, D. L., and N. W. Fraser. 1983. Detection of HSV-1 genomein thecentral nervoussystemoflatentlyinfected mice. Nature(London) 302:523-525.
24. Roizman, B., and A. E. Sears. 1990. Herpessimplex viruses and theirreplication, p. 1795-1841. In B. N. Fields, D. M. Knipe, R. M.Chanock,M.S.Hirsch,J. L.Melnick,T. P.Monath,and B. Roizman (ed.),Virology, 2nd ed. Raven Press, NewYork. 25. Saiki, R. K., D. H.Gelfand,S. Stoffel,S.J.Scharf,R.Higuchi,
G. T. Horn, K. B. Mullis, and H. A. Erlich. 1988. Primer-directed enzymatic amplification of DNA with athermostable DNApolymerase. Science 239:487-488.
26. Steiner, I., J. G. Spivack, S. L. Deshmane, C. I. Ace, C. M. Preston, and N. W. Fraser. 1990. Aherpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein estab-lishes latentinfection in vivo in the absence of viralreplication andreactivatesefficientlyfromexplanted trigeminal ganglia.J. Virol.64:1630-1638.
27. Stevens, J. G. 1989. Human herpesviruses: aconsideration of the latent state.Microbiol. Rev. 53:318-332.
28. Tenser, R. B., and M. E. Dunstan. 1979. Herpes simplex virus thymidinekinase expression in infectionof thetrigeminal gan-glion.Virology 99:417-422.
29. Tenser, R. B., K. A. Hay, and W. A. Edris. 1989. Latency-associated transcript but not reactivatable virus is present in sensoryganglionneuronsafter inoculationofthymidine kinase-negative mutants of herpes simplex virus type 1. J. Virol. 63:2861-2865.
30. Wagner, M. J., J. A. Sharp, andW. C. Summers. Nucleotide sequenceofthethymidine kinasegene of herpessimplex virus type-1. Proc.Natl. Acad. Sci. USA 76:3683-3687.
31. Weller, S. K., D. P. Aschman, W. R. Sacks, D. M. Coen,and P. A.Schaffer. 1983. Geneticanalysis oftemperature sensitive mutants of HSV-1: the combineduse ofcomplementation and physical mapping for cistronassignment. Virology 130:290-305.