0022-538X/92/116338-15$02.00/0
Copyright © 1992, AmericanSociety for Microbiology
Characterization of Nuclear Proteins That Bind the
EFII
Enhancer
Sequence
in
the
Rous
Sarcoma
Virus
Long
Terminal
Repeat
ROSALIE C. SEARS1ANDLINDA
SEALY',2*
Departmentof Cell
Biology'
andDepartmentof MolecularPhysiology
andBiophysics,2Vanderbilt
University
Schoolof
Medicine, Nashville,
Tennessee 37232Received22April 1992/Accepted 23 July1992
The EFII cis element is a 38-bp sequence at the 5' end of the Rous sarcoma virus long terminal repeat, extending from nucleotides -229to-192(with respecttothe viraltranscriptionstartsite),which isrecognized
bysequence-specific DNA-binding proteinsinavian fibroblast nuclearextracts(L.
Sealy
and R.Chalkley,Mol.Cell. Biol. 7:787-798, 1987). Wedemonstrate thatmultiple copies of the EFII cis element stronglyactivate transcription ofa reporter gene invivo. We correlate the region of the EFII cis element which activates transcriptioninvivo with thein vitro binding siteforthree nuclearfactors, EFIIa, EFIIb, and EFIIc.The
sequencemotifrecognized byEFlIa,-b, and-cis also found inconsensusbindingsites for members ofa
rapidly
growing
family
of transcription factors related to theCCAAT/enhancer-binding protein (C/EBP).EFTIa,
-b,and -care present in fibroblast andepithelial cell lines from various species but aremuch less abundant in differentiated rat liver and kidney cells. The EFlIa bindingactivityis
particularly
abundantin anavianB-cell lymphoma line. Asjudged from molecular weight analysis, cell type distribution,and sequence recognition properties, the EFII factors under study appear todiffer frommostof thepreviously
describedC/EBP-related factors and thus may expand thediversity
oftheC/EBPfamily.
The information accumulated about the regulation of gene expression atthe level oftranscription initiation in eukary-otic cells has revealedanimpressive degreeofcomplexity.
Among the cis-acting DNA sequences that regulate
tran-scription, enhancersare nowknown tobe mosaics of
mul-tiplesequencemotifs. Each of these sequence motifs
repre-sents a discrete binding site for one or more trans-acting
factors. All of these cis-acting elements and regulatory
proteinfactorsworkinginconcertspecify appropriatelevels
oftranscription (31, 44, 49, 57). Manyviruses have evolved
to efficiently utilizethese hostcell mechanisms topromote
high-level expression of theirown genomes. For example,
the avian retrovirus Rous sarcoma virus (RSV) contains strong cis-acting enhancer sequences in the long terminal repeat(LTR)regions flanking either end of theproviralDNA
(13, 14, 23, 25,38,42, 52, 69,71).The potenttranscriptional
activity of the RSV LTR enhancer makes it an attractive
model system forstudyingthe moleculareventsinvolved in
theregulationoftranscriptioninitiation.Thus,wehave been
identifyingandcharacterizingnuclear factorswhich bind in
a sequence-specific manner to the RSV LTR enhancer.
Althoughthetypicaltargetcells for transformationbyRSV
and its relative, Rous-associated
virus-i,
are avianfibro-blasts and lymphoid cells, respectively, the RSV LTR
en-hancer is activeinawidevariety of vertebrate cell types (23, 38,71).This suggests that many of thetrans-actingproteins which mediate the LTR enhanceractivity are common host celltranscription factors.
The major transcriptional control regions for RSV have been localized by deletion mutagenesis (13, 25, 38, 42, 52) and enhancer trap experiments (71) to the U3 region of the RSVLTR, extending from the 5' end of the LTR at position
-229(relativetothetranscription start site) to position -54.
* Correspondingauthor.
We have so far described three nuclear factors, enhancer factors I, II, and III (EFI, EFII, and EFIII, respectively),
which recognize specific nucleotide sequences within the RSV LTR enhancer diagrammed in Fig. 1 (4, 5, 17, 64).
Furtheranalysis of the EFIproteinfactor hasdemonstrated that it specifically recognizestwo inverted CCAAT motifs
(17). The EFIII factor recognizes two sites (4, 5) which
contain a commonsequencemotif knownastheCArGbox
(48, 66). By its
sequence-specific
DNA-binding propertiesand antibody recognition, EFIII has been shownto
repre-sentthe avianhomologtotheserumresponsefactor(4, 5).
Wenow reportthe further characterization ofanumber of
protein factors present in chickenembryofibroblast (CEF)
nuclei which bind to the EFII region of the RSV LTR enhancer.
Other investigators have also observed nuclear proteins
which bindspecificallyinvitrotothe EFIIregionof the RSV
LTR(22, 61)andtorelated butnotidentical sequences in the
Rous-associated virus-2 LTR(59, 60). However, when this
workwasbegun, the transcriptional relevance of the EFII
cis element (RSVLTRsequences from -229to -192) had
not been directly addressed. Mutagenesis experiments had
demonstratedthat removal of the EFII sequencesaspartof
larger deletions crippledenhancerfunction(13, 42, 52),and smaller deletions demonstrated thatatleasteightnucleotides
(-201to -208)within theEFII ciselementarerequiredfor
maximal enhancer activity (25). We present evidence here that an isolated EFII cis element, when multimerized,
strongly enhances transcription invivo. This finding is in
agreement with recent experiments performed in GC rat
pituitarytumorcells, which show that the EFIIciselement
(RSV LTR sequences -231 to -193), linked to aminimal
heterologous promoter, is transcriptionally active, and its
activityis increasedbyexpressionofaconstitutive,calcium/
calmodulin-independentmutantof type II
calcium/calmodu-lin-dependent protein kinase (32). We further demonstrate
6338
on November 9, 2019 by guest
http://jvi.asm.org/
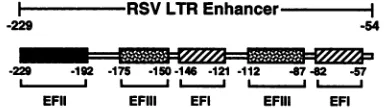
I RSVLTR
-229 -54
-229 -192 -175 -150 -146 -121 -112 -6782 -57
_JL ''_JL1__ L_ _J
EFII EFIII EFI EFIII EFI
FIG. 1. Schematic representation of the RSV LTR enhancer. Bindingsites forthree nuclearfactors, designatedEFI, EFII, and EFIII, are shown. The EFI factor has been foundto specifically recognizeinverted CCAAT motifspresentwithin eachof itsbinding
sites (17, 64).The EFIIbindingsite has beenmapped byDNase I footprintingat the 5' end of the RSV LTR(64).TheEFIIIfactoris suggestedto be the avianhomologto theserumresponsefactor,and
it has been foundtospecifically recognizetheCArG motifpresentin
both itsbindingsites(4, 5).
that theupstreammost oftwo near-directrepeatsequences
presentin the EFII cis element isresponsibleformediating strong transcriptional activityinvivo. At least three heat-stable protein complexes,referred to asEFIIa, EFIIb, and
EFIIc, are identified that specifically bind the EFII DNA
sequencein vitro. Thesethreecomplexesbindselectivelyto the upstream repeatwith high affinity. Thus,the
transcrip-tionally active sequences in the EFII cis element in vivo correspond to the
EFIla,
EFIIb, and EFIIc bindingsite in vitro.All three EFII-binding factors appear to recognize the same nucleotides within the upstream repeat of the EFII DNA sequence. It is now well documented that many
transcription factors occur in families which share DNA sequence recognition properties (30). The nucleotide se-quence recognized by
EFIla,
-b, and -c is also found inconsensusbinding sites for members of arapidly growing family of transcription factors related to the previously
describedCCAAT/enhancer-binding protein (C/EBPa) (1, 7, 10, 15, 27, 29, 34, 39, 56, 58, 61). C/EBP-related family
members belong to the basic region-leucine zipper (bZIP)
class oftranscriptionfactors. bZIPtranscriptionfactorsare
characterized by a conserved DNA-binding domain,
con-taining clusters of basic amino acids, immediately adjacent
to a conserved dimerization domain containing a leucine residue at nearly every seventh position, known as the leucine zipper (36, 40, 41, 62). Three reported members of theC/EBPfamilyoftranscription factors, C/EBPa, IBF,and
Ig/EBP-1(C/EBP-y),have been shownto beable tobind the
EFII region of the RSV LTR in vitro (33, 58, 61). We
demonstrate, based on different sequence-specific DNA recognition properties and apparent-molecular-weight anal-yses, that EFIIa, -b, and -c are probably distinct from
C/EBPa and IBF. Moreover, expression of most of the
identifiedC/EBP-relatednuclearfactors isparticularly abun-dant in differentiatedliver, lung, andadipose cells (1, 3, 7, 10, 11, 15, 18, 56, 73),whileEFIIa, -b,and-caremuch less abundant indifferentiated rat liver andkidneycells than in
numerous fibroblast and epithelial cell lines from various species. However, Ig/EBP-1, EFIIa, andEFIIb havevery similarsequencerecognition characteristics,asjudgedfrom theirabilityto bindboth the EFIIsequenceandthe E site in themurine immunoglobulin heavy-chainenhancer withhigh affinity (58). We also notethat thereisalarge excessofthe EFIIa factor in an avian B-cell lymphoma line. Thus, it is possiblethat the EFIIaprotein playsarole inB-cell-specific gene expression, and further characterization of the three EFII-bindingfactorsmayexpandthediversityofthe C/EBP-relatedfamilyoftranscriptionfactors.
MATERIALS AND METHODS
Cell culture. CEFwere
prepared
from10-day-old
embryos(SPAFAS Inc., Preston, Conn.)
and maintained in medium199
supplemented
with 10% tryptose phosphate broth, 5%fetal calfserum,and 1% chickenserum.Thefollowingwere
also addedtothe medium: 25 U of
penicillin
G sodiumperml,
25 ,ugofstreptomycin
sulfateperml, and 0.14%sodiumbicarbonate. Dulbecco's modified
Eagle's
medium (DMEM)(high glucose) supplemented
with 10% calfserumwas usedfor all othercell
lines, including
A431epidermal
carcinomacells, BALB/c-3T3 fibroblasts,
NIH 3T3fibroblasts,
babyhamster
kidney (BHK) cells,
Rat-1fibroblasts,
avianB-celllymphoma
lineBk3A,
and Coscells,
with the followingexceptions.
BHK cellsweremaintained ina 1:1 mixture ofDMEMand Ham's F12
medium;
DMEMforRat-1cellswassupplemented
with 10% fetal calfserum; DMEM for Bk3Acellswas
supplemented
with10%tryptosephosphate
broth,5% calf serum, and 1% heat-inactivated
(60°C,
30 min)chickenserum. BHK cellswerea
gift
fromRoger
Chalkley(Vanderbilt University), BALB/c-3T3
and NIH 3T3 cellswerea
gift
from JackPledger (Vanderbilt
University), Rat-1cellswere a
gift
fromMike
Bishop (University
ofCaliforniaatSan
Francisco),
Cos and Bk3A cellswere agift
from SteveHann
(Vanderbilt University),
and A431 cells were a giftfrom Graham
Carpenter (Vanderbilt
University).Plasmid construction.
pSRA-CAT
was generated in thislaboratory
as described before(4).
pe- CAT wascon-structedasfollows.Viral sequences in
pRSV-LTR
(64)fromtheEcoRIsiteat-54totheBamHIsiteat+524(numbering
with reference tothe start site of
transcription
in the viralgenome)
werecloned into the PvuII siteofpBR322
tocreatepRSV-LTRe-.
pRSV-LTRe-
wasdigested
withBstEII at+103 in the viralsequence,treatedwithDNApolymeraseI
at
4°C
tocreateblunt endsasdescribed before(64),
andthendigested
withBamHIatposition
+524.pSV2-CAT (24)
wasdigested
withHindIII,
treatedasabovetocreatebluntends,and then
digested
withBamHI. The HindIII-BamHIfrag-ment
containing
thechloramphenicol
acetyltransferase(CAT)
genewastheninsertedintopRSV-LTRe-,
replacingthe
423-bp
BstEII-BamHIfragment
to generate pe- CAT. TheHindIII
sitefrom theCATgene,now atposition
+103 in pe- CAT(with
respect tothe start site for transcription), was notregenerated.
The
p(EFII)nCAT
series of reporterconstructs werecre-atedasfollows. The
following
double-stranded44-bpoligo-nucleotide
containing
the38-bp
EFIIcis element (-229 to-192 in theRSV LTRare shownin
capital
letters):5'cogagAATGTAGTCTTATGCAATACTCTTGTAGTCTTGCAACAO
3'cTTACATCATACGTTATGAGAACATCAGAACGTTTggg9ct5'
withAvaI
ends,
wassynthesized
andpreparedasdescribed below. This44-bp
EFIIoligonucleotide
was treated withpolynucleotide
kinase and 1 mM ATP andligated
with T4 DNAligase (according
tothemanufacturer'sspecifications)
to create head-to-tail multimers. EFII multimers were
treatedasaboveto createbluntends. pe-CATwas
digested
withAcclata
unique
site 235nucleotides upstream from the start oftranscription
and treated as above tocreate blunt ends. EFIImultimersweretheninsertedatthe AccIsiteto create thep(EFII)nCAT
constructs.p(EFII3'/5')2+CAT
was created as follows. The double-stranded 50-bp EFII3'/5'
mutantoligonucleotide (see
Fig.
3A for sequence),synthesized
andprepared
as describedbelow,
was treated withpolynucleotide
kinase and 1 mMATP andpurified by
polyacrylamide gel electrophoresis, electroelution,
andon November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.89.284.82.136.2]gen chromatography (Qiagen Inc., Chatsworth, Calif.) prior to incubation with T4 DNA ligase at 16°C. The ligated EFII
3'/5' mutant oligonucleotide was then inserted into the
vector pe- CAT, which had been previously digested with AccI and treated as above to generate blunt ends.
The reporter plasmids p(EFII5')4-CAT4 and p(EFII3') 2-CAT4 were created as follows. The following
double-stranded 20-bp and 18-bp EFII oligonucleotides, containing
the 5' EFII sequences from -226 to -213 (20 bp) or the 3' sequences from -205 to -194 (18 bp) in the RSV LTR
(capitalletters):
20bp:5'ccgagGTAGTCTTATGCAAc3'
3'cCATCAGAATACGTTgggct5' 18 bp: 5'cogagGTAGTCTTGCAAc3'
3'cCATCAGAACGTTgggct5'
withAvaIends, weresynthesized and prepared as described
below. The 20-bp and 18-bp oligonucleotides were then
treated withpolynucleotide kinase and 1 mM ATP prior to incubation with T4 DNA ligase (according to the manufac-turer'sspecifications).The ligated 20-bp oligonucleotide was then inserted into the BamHI site in the polylinker of a modified Bluescript M13+ (Stratagene, La Jolla, Calif.) plasmid created in this laboratory which lacks both XhoI and
SmaI sites, after treatment of both the ligated
oligonucleo-tide and themodified Bluescript vector as described above to create blunt ends. A clone containing four copies of the 20-bp oligonucleotide in the modified Bluescript vector,
designated pI120x4, was digested with BamHI and HindIII
to generate afragment of 120 bp containing the four ligated 20-bp oligonucleotides. This fragment was then inserted between the BamHI and HindIII sites of the polylinker at position -105, immediately upstream of the thymidine
ki-nase promoter in the plasmid pBLCAT4 (6), to create
p(EFII5')4-CAT4. The ligated 18-bp oligonucleotide was
treated to create blunt ends and inserted into the plasmid pBLCAT4, which had been previously digested with BamHI
(position -105)andtreated to create blunt ends, to produce
thereporterplasmid p(EFII3')2-CAT4.
TheplasmidpMLS-II, used to make the end-labeled probe
for footprinting, was generated asfollows. The 44-bp EFII
oligonucleotide described above was blunt-ended and
in-serted into the unique SmaI site in the polylinker of a modified Bluescript+ which had been previously digested
withXhoI,blunt-ended asdescribed above, and religated to
destroy the
XhoI
site. The AvaI site on either end of the insert was regenerated.The inserts in all of the above plasmids were sequenced (45)
toconfirm insert number and orientation and the absence of mutations. We noted that the sequence of both copies of the EFII
3'/5'
mutantoligonucleotide in the p(EFII3'/5')2+CAT clone differed from the sequence given in Fig. 3A in that bothare missing the terminal G of the BamHI linker sequence
insertedto disrupt the 3' repeat.
Transfection and enzymatic assays. Transfections were
performed by the calcium phosphate coprecipitation
tech-nique as described by Graham and Van der Eb (26), with 1 ,ugoftest plasmid DNA, 2.5 ,ug ofpSV40-13-gal DNA (43), and 1.5 ,ug of pUC19 DNA per 10 ml of medium in 100-mm tissue culture dishes. Cells were plated the day before transfection and transfected when they were approximately 80 to 90% confluent. One hour prior to transfection, CEF
were refed with fresh medium 199 (supplemented as de-scribedabove) containing 10 mM HEPES
(N-2-hydroxyeth-ylpiperazine-N'-2-ethanesulfonic acid, pH 7.3). Cells were
exposed tothe CaPO4 DNA precipitate for 6 to 8 h, after
which they were washed once with Tris-glucose buffer containing 137 mM NaCl, 5 mM KCl, 5.6 mM dextrose, 25 mM Tris (pH 7.4), 25 U of penicillin G sodium per ml, 25
,ug
of streptomycin sulfate per ml, and 4.5 ,ug of phenol red sodium salt per ml and then refedwith fresh supplemented medium 199. Transfected cells were allowed to grow for an additional 48 h. Cells were then washed twice in cold (4°C)
phosphate-buffered saline (PBS) and incubated for 5min at 37°C in buffer containing 40 mM Tris (pH 7.5), 1 mM EDTA (pH 8), and 150 mM NaCl to loosen cells from the tissue culture dish.
All subsequent steps involving transfected cells were carried out at4°C. Cells were collected by centrifugation (5 min at 3,700 x g) and lysed by sonication, and cell extracts wereprepared as described by Boulden and Sealy (4). CAT assays with these cell extracts were performed by the procedure of Gorman et al. (24). CAT activity was calculated for each test plasmid by cutting out the acetylated and unreacted forms of [14C]chloramphenicol, quantitating the radioactivity in each by liquid scintillation counting, and determining the percent acetylation. Units of 3-galactosi-dase activity in the same cell extracts were assayed and calculated as described by Norton and Coffin (51). CAT activity was normalized by dividing percent acetylation by units of,B-galactosidase activity for each test plasmid.
Nuclear extracts. Nuclear extracts (0.5 M NaCl) were prepared from 14-day-old chicken embryos (CE) (SPAFAS) aspreviously described by Sealy and Chalkley (64), with the modifications described by Boulden and Sealy (4). The following changes were also made. The phosphatase inhibi-tors 3-glycerolphosphate (10 mM), sodium vanadate (100 FM), and sodium molybdate (10 mM) were added to all buffers. The protease inhibitor benzamidine (10 mM) re-placed leupeptin and pepstatin in buffer G. Homogenized embryos were filtered through cheesecloth prior to passage through Miracloth (Calbiochem Corp., La Jolla, Calif.). Nuclei were prepared and sequentially extracted by one washwith buffer A (5 ml perembryo), one extraction with buffer B (1 ml perembryo), and one extraction with buffer B containing 0.63 M NaCl (3 ml perembryo). This yielded an approximately 0.5 M NaCl extract because of the volume of the nuclear pellet. Following thisextraction, the supernatant wascollected byultracentrifugation in an SW27 rotor at 25 krpm for 60 min. The supernatant was dialyzed against 50 volumes ofdialysis buffer containing 10 mM HEPES (pH 8), 1 mM EDTA (pH 8), 50 mM NaCl, 50% glycerol, 7 mM
,B-mercaptoethanol,
1mMphenylmethylsulfonyl fluoride,
10 mMbenzamidine, and the phosphatase inhibitors mentioned above.Nuclear extracts (0.5 MNaCl) from cells grown in mono-layers, including CEF, A431, BALB/c-3T3, NIH 3T3, BHK, Cos, and Rat-1 cells, were prepared as described by Sealy and Chalkley (64), with the following modifications. The phosphatase inhibitors mentioned above were added to all buffers. Nuclei were prepared byonly one wash in buffer A. Thecrude nuclei were then resuspended directly in buffer B containing 0.53 M NaCl (yielding an approximately 0.5 M NaCl extract because of the volume of the nuclear pellet). Thesupernatant was collected at 16,000 x g and dialyzed as described above. Nuclear extracts (0.5 MNaCl) from Bk3A cells were prepared as described for the above monolayer cellsexcept that these suspension cells were pelleted at 750 x g for 10 min, washed twice with cold (4°C) PBS, and resuspended in buffer A to prepare nuclei. The 0.5 M NaCl extraction was performed with 1 ml per 109 cells. Heated nuclearextracts were prepared as described for the
on November 9, 2019 by guest
http://jvi.asm.org/
PROTEINS THAT BIND THE EFII ENHANCER IN THE RSV LTR 6341
layer cells with thefollowingmodifications. After the30-min
0.5 MNaClextraction,the mixturewasheated for5 min at 100°C, placed onice for 10 min, and subjected to
ultracen-trifugation in an SW56 rotor at 30 krpm for 30 min. The
supernatant was dialyzed as described above. Nuclear ex-tracts (0.5 MNaCl) prepared from thefollowing cells were gifts from the indicated individuals and were prepared as describedabove:H4,Tony Ip(VanderbiltUniversity); adult ratliver andkidney, Michelle Cissell (Vanderbilt
Universi-ty). Protein concentrations in extractsweredeterminedby
the method of Schaffner and Weissman (63), with bovine
serumalbumin asthe standard.
Radiolabeled andcompetitor DNAs. Oligonucleotideswere
synthesized(DiabetesResearchTraining CenterDNA core,
VanderbiltUniversity)andpurifiedasdescribed byBoulden
andSealy(4).Equal amountsof eacholigonucleotide,
mea-sured by the
A260,
and its complement were annealed in buffer containing 10 mM Tris (pH 8), 1 mMEDTA(pH8), and from 200 to 400 mM NaCl by placingthe DNAs in a 2-literboiling-waterbath,whichwasallowed to cool slowlyto room temperature overnight. Coding-strand nucleotide
sequences ofthe double-stranded oligonucleotides used for
theDNA-binding studiesaredisplayedinFig.3AandTable
1, except for the oligonucleotides labeled Core, DEI, IBF DNA, and IgH-E, which areshown below:
Core:5'agcttGGGCTGTGGAAAGGAGGGc3' 3'aCCCGACACCTTTCCTCCCgagct5'
DEI:5'togacTATGATTTGTAATGOGGGotga3' 3'agctgATACTAAAACATTACCCCgaget5' IBFDNA:5'CTGTTGGCTGCAATTGCGCCACCGCCACAG3'
3'GACAACCGACGTTAACGCGGTGGCGGTGTC5'
IgH-E:5' TA TGTTGAGTGAGTCAA3'
3'AATTCAAATTTTATAAAAATTTACTTAACTCGTTACAACTCAACTCAGTT5'
TheCoreoligonucleotide representssequencesfrom -48 to -65 from the collagen III promoter, which contains the simian virus 40 (SV40) core enhancer motif (50); linker sequences are indicated by lowercase letters. The DEI oligonucleotide represents sequences from -90 to -107 from the rat albumin promoter (9). IBF DNA represents sequences from the gag gene internal enhancer of RSV, designated OpvuII3' byKarnitz et al. (34). IgH-Erepresents sequences from position301 to position 350 in the murine immunoglobulin heavy-chain enhancer as defined by Ephrussi et al. (16). Since the 44-bp EFII oligonucleotide described above was unable to bind EFII DNA-binding proteins when radiolabeled and used in an electrophoretic mobility shift assay (EMSA) (unpublished results), a 50-bp EFII oligonucleotide containingRSV LTRsequencesfrom -229 to -192 withXhoI andBglIIlinkersonthe5'endand a SmaI linker on the 3' end was synthesized for use in EMSAs(seeFig. 3A).
Radiolabeled probes for EMSAs were prepared as fol-lows.Double-strandedoligonucleotides(200 ng)were radio-labeled with
[y,-32P]ATP
and T4 polynucleotide kinase ac-cording to the manufacturer's specifications. Full-length radiolabeledoligonucleotideswerethenpurifiedbyelectro-phoresis on native 12% polyacrylamide gels containing lx TBE (89 mM Trisbase,89 mMboric acid, 2.5 mM EDTA
[pH8.5]),followedbyelectroelutionin lx TBEandethanol precipitation. Nonradiolabeled competitor oligonucleotides were also purified by polyacrylamide gel electrophoresis (PAGE)in1x TBE,andthe amount of DNArecoveredwas quantitated by A260 measurements. All oligonucleotides were resuspended in 10 mM Tris-100 mM NaCl-1 mM EDTA(pH 8).
Radiolabeled probes for footprinting were prepared as
follows.Theplasmid pMLS-II (see above)wasdigestedwith
eitherBamHI (tolabel thecoding strand)orHindIII(tolabel the noncoding strand) and end-labeled with
[a-32P]dATP,
[a-32P]dTTP,
(a-32P]dGTP, [a-32P]dCrP, and DNApoly-merase I at4°C according to the manufacturer's
specifica-tions. End-labeled, linearized pMLS-II was then digested
with either HindIII (if end-labeled at a BamHI site) or
BamHI (if end-labeledat aHindIIIsite). The resulting
76-bp
end-labeled restrictionfragmentswerepurifiedbyPAGEas
describedabove.
EMSAs.Samplesof0.5 M NaCl nuclear extract,
contain-ingbetween 1 and 10 ,ugof protein, depending onthe cell
type analyzed, were mixed with 0.5 ng of radiolabeled
double-stranded oligonucleotide and 1.3 ,ug of poly(dI):
poly(dC), prepared as described previously (64), in the
presence or absence of nonradiolabeled, gel-purified
com-petitor DNAs. Binding reaction mixes were incubated at
room temperature for 30 min in a final volume of 20 ,ul,
containing7.5 mM HEPES(pH 8), 2.5 mM Tris(pH8), 0.8
mM EDTA (pH 8), 100 mM NaCl, 3.3 mM MgCl2, 5 mM
,-mercaptoethanol, and 20% glycerol. Sampleswere
ana-lyzed on native 4%polyacrylamidegelscontaining 1 xTGE (25 mM Tris, 190 mM glycine, and 1 mM EDTA [pH 8.5]) in
the absence oftracking dye. Gelsweresubjectedto
electro-phoresis inlx TGE for 50minat 200 V andsubsequently
dried for autoradiography. When appropriate, the
protein-DNA complexes were excised from the dried gel, and the
radioactivity in the complexwas quantitated by liquid
scin-tillation counting. Background counts were subtracted by
countingan equivalent section of the driedgel outside the
sample lanes. The relative binding affinityofprotein-DNA
complexes forspecific nonradiolabeled competitor
oligonu-cleotides was determined
by calculating
the percentage ofradiolabeled DNA bound ina specificcomplex in the
pres-ence of competitor DNA relative to that observed in the
absence ofcompetitorDNA, plottedversuslog (competitor
DNA). TheamountofcompetitorDNArequiredtoreduce
the formation ofa specificcomplex by 50%wascalculated
from this plot and divided by the amount of wild-type
competitor DNA
(representing
thesequence of theradiola-beled DNA used in the
EMSA)
neededtoreducetheamountof the same
complex by
50% to obtain relative bindingaffinities.
OP/Cu footprinting. Samples of 0.5 M NaCl nuclear
ex-tractcontaining either 12 or4 ,ug of protein from CEF or
Bk3Acells,respectively,weremixed with100 to 200kcpm
(approximately
2ng)ofend-labeledprobe(see above)in the presence of 1.3 p.g ofpoly(dI):poIy(dC)
and subjected to EMSA as described above withthefollowingmodification:20-,ulbindingreactionmixeswereanalyzedon6%
polyacryl-amide gels containing lx TG (25 mM Trisbase, 190 mM
glycine [pH 8.5]).Thegelsweresubjectedtoelectrophoresis
in1xTG for 1.5hat200 V.Following
electrophoresis,
gelswerewashedoncewith50 mMTris(pH 8)for15 minatroom
temperature, placedin fresh50 mM
Tris,
and treated in situwith
orthophenanthroline-copper
(OP/Cu) endonuclease as describedbyKuwabara and Sigman (37)for 6minatroom temperature. Free and bound complexes, visualized byautoradiography
of thewetgelat4°C,wereexcised from thegelandelectroeluted in 1xTBE.Cleavedend-labeled DNAs
were then ethanol
precipitated
with 5 ,ug of carrier tRNA.Samples
containing equal
counts(defined by
Cerenkovradi-ation),
and the end-labeledprobes
chemically
cleaved atpurine
residues,
wereseparated
on a 20%polyacrylamide
sequencing gel
asdescribedby
Maxam andGilbert(45).
on November 9, 2019 by guest
http://jvi.asm.org/
Apparent-molecular-mass
analysis.
Samplesof 0.5MNaCl nuclear extractcontaining
100 or 30 ,g ofprotein
from14-day
CE or Bk3Acells, respectively,
were subjected tofractionationon a sodiumdodecyl sulfate (SDS)-12%
poly-acrylamide gel,
transferred to an Immobilon-P membrane(Millipore Corp., Bedford, Mass.),
andeluted,
asdescribedby
Faber andSealy
(17),
except that the Immobilon-Pmembrane
corresponding
totheseparating gel
was cutinto 17slices,
eachapproximately
3-mmwide,
and theproteins
in each slice were eluted into 40 ,l of cold(4°C)
elution solution.Samples (10
RI)
of each SDS-sized fraction wereanalyzed by
EMSA. To determine the range of apparent molecularmassesof theproteins
ineachfraction,
individualprotein
standards wereseparated
on the same SDS-12%polyacrylamide gel,
transferred to an Immobilon-Pmem-brane,
andvisualizedby
amido blackstaining
as describedby
Schaffner and Weissman(63).
RESULTS
Specific
sequences within the EFII cis element activatetranscription invivo. We have
previously
demonstratedby
DNase I
footprinting
thatafactor(s) (termed EFII)
in nuclear extracts from avian fibroblastsspecifically
binds to a38-bpsequenceatthe 5' end of the LTR of the Schmidt
Ruppin-A
strain of
RSV, extending
from -229 to -192 nucleotidesupstreamof thestartsite for
transcription (64).
Toassessthe functionalsignificance
ofthisregion
of the LTRenhancer fortranscriptional activation,
weinsertedone ormorecopies
of theEFIIbinding
site into the reporterplasmid
pe-CAT.As shown inFig. 2A,
pe- CAT is similar to theplasmid
pSRA-CAT,
whichwasdescribedpreviously (4),
exceptthat sequencesin U3responsible
for LTR enhanceractivity
have beendeleted, leaving just
54 nucleotides upstream of the start site fortranscription.
These 54nucleotides,
which include a TATA box atapproximately position -30,
are sufficienttodirectaccuratebut very low levels oftranscrip-tion of the CAT gene in vivo
(20). Up
to sixcopies
of theEFII
oligonucleotide
were inserted into pe- CATat aposi-tion
(-235)
comparable
to that found in thewild-type
RSV LTR. Theresulting
constructs,designated
p(EFII)nCAT,
were then tested for their
ability
to activatetranscription
in transient-transfection assays in CEF. As shown inFig.
2B,multiple copies
of theEFIIciselement,
in eitherthesense or antisenseorientation,
wereabletostrongly
activatetranscrip-tion of the CAT gene
compared
with the enhancer-minusconstruct
(pe- CAT).
Sixcopies
of the EFIIciselement in the sense orientation[p(EFII)6+CAT]
were able toactivatetranscription approximately
40-fold.Inspection
of the nucleotide sequence of the EFII ciselement
(Fig.
3A)
reveals that anearly
direct repeat of15nucleotides is present in the LTR sequence from -227 to -213 and from -206 to
-194,
with the 3' repeatlacking
an AT present in the 5' repeat. To determine whether the presenceoftheserepeated
sequences isimportantfortran-scriptional activation,
wecreateda mutantEFIIciselement in which both repeatsweredisrupted by 10-bpBamHIlinker sequences(EFII 3'/5'
mutant; see Fig. 3A for sequence). Twocopies
of thismutantoligonucleotidewerealso inserted into the pe- CAT vector at -235. When the resulting construct,p(EFII3'/5')2+CAT,
was tested in transient-transfection assays, the mutant EFIIcis element failed toactivate
transcription
above the level observed with pe-CAT(Fig. 2C).
Thus,
it appears that one or both of therepeated
sequences intheEFIIciselementarenecessary fortranscriptionalactivation, presumablyby actingas abinding
site(s)forone or moretrans-actingfactors.
In ordertodirectlytestthetranscriptional activity of each near-direct repeat, we analyzed two abbreviated EFII cis elements in which eitheronly the upstream 5' repeat, from -226 to -213, oronly the downstream 3' repeat, from -205
to -194,wasreiterated inareporter gene construct. Specif-ically, four copies of the 5' repeat or two copies of the 3' repeat (each separated by a 6-bp AvaI linker sequence) wereinserted immediately upstream of the thymidine kinase promoterand CAT gene in the vector pBLCAT4(6),creating p(EFII5')4-CAT4 and p(EFII3')2-CAT4, respectively.
In-terestingly, in transient-transfection assays,
p(EFII5')4-CAT4 activated transcription 36-fold relative to pBLCAT4, whilep(EFII3')2-CAT4wasunable to activate transcription above the basal activity of pBLCAT4 (Fig. 2C). Althoughwe
have not compared equal copy numbers because such con-structs were notavailable, it is unlikely that more copies of the3' repeat would leadtotransactivationequivalent to that with the 5' repeat when two copies are inactive. Ingeneral, transactivationby multimerized cis elements increases
incre-mentallywithincreasing copy number, as seenfor the
com-plete EFII cis element (Fig. 2B). Our data suggest that
transcriptionalactivationby the EFIIciselement is mediated
primarily throughthe 5' near-direct repeat sequence. Thus,
wewished to furthercharacterize theprotein factors which
specifically bind this element in vitro and, in particular,
determine whetherthey recognizethe upstream repeat.
Nuclearproteins whichbind totheEFIIciselementinvitro specifically recognize the upstream repeat. In our previous
study,weidentifiedtwoEFIIDNA-bindingfactors present
in0.3to0.5 M NaClextractsofquailfibroblast nuclei which
specificallybound the EFII ciselement contained ina286-bp
restriction fragment encompassing the entire RSV LTR
enhancer (64). In order to facilitate further analysis of
proteins recognizingthe EFII ciselement,werepeated this
experiment with a double-stranded 50-bp oligonucleotide,
referred to as EFII DNA (see Fig. 3A for coding-strand
sequence information), containing the 38-bp EFII cis
ele-mentwith linker sequencesonboth ends. As shown in Fig. 3D, lane 1, two major protein-DNA complexes, which we
refer to as EFIla and EFIIb, are present in anEMSA of radiolabeled EFII DNA following incubation with heated
(100°Cfor 5min)0.5 MNaCl nuclearextractpreparedfrom
14-day-old CE. A third minor protein-DNA complex of
slowerelectrophoretic mobility, designatedEFIlc,wasalso detected. Thetwofaster-migrating protein-DNA complexes
likelyarise from some abundant, nonspecific DNA-binding
proteins in the high-NaCl extract, since they were not
inhibited by the addition of a variety of nonradiolabeled
competitor DNAs (data not shown). Results identical to
thosepresentedfor the14-day-oldCE nuclearextractwere observed with both heated and unheated 0.5MNaCl nuclear
extractsfrom CEF(datanotshown).
To assess whetherbindingof EFIIa, EFIIb, or EFIIc in vitro would correlate withtranscriptionalactivation invivo,
weinvestigatedtherole of thetworepeated sequences in the
EFIIciselement in theformation of the three complexes. In addition to the EFII 3'/5' mutant described above, we
obtained two EFII mutantoligonucleotides in which either the 5' repeat (EFII 5' mutant) or the 3' repeat (EFII 3'
mutant) was disrupted by a 10-bpBamHI linker sequence
(Fig. 3A). Representative competition experiments with
thesemutantoligonucleotides and thewild-type EFIIDNA are shown in Fig. 3B. The decrease in relative binding
affinities of the EFIla, -b, and -c factors for each of the
on November 9, 2019 by guest
http://jvi.asm.org/
r-LTREnhancer-TrI
pSRA-CAT: pEBR322 RSVSgenonioe
PVLJII l-229) (489)
pe-CAT: pBR322
(EFII r)
Accl (-235)
) K
EcoRI l+X)
(-54) Lv
CAT
gene
R U5
-Hindlil <- 103)
r raP CAT
gene
U3 1IH|d5
EcoR l- l Hindlil
(-54)
L,>-
(+10O3)B.
40-30
0
20-LUJ
< 10
-j
LuJ
0
38+4
16+6
d
~~~~1+
.w
go
e
e*
1
'
FIG. 2. Specificsequenceswithin the EFII cis element activatetranscriptioninvivo.(A) pSRA-CATisachimericconstructcontaining
aCATreportergenelinked tothe330-bpRSV LTR and 260bpof upstreamsequencenormally foundatthe 3' end of the RSV provirus
genome.TheRSVLTR enhancer extends from -229to-54 inpSRA-CAT.pe-CAT isanenhancer-minusconstructwhich lacks the RSV genomesequencesfrom the PvuII siteat-489totheEcoRI siteat-54. Thesequencesfrom -54 to +1 in bothplasmidsrepresentaminimal promoter(MP) containingaTATAboxatapproximately -30. (B) Uptosixcopies of the EFII cis elementwereinserted into the plasmid pe- CATatthe indicated AccIsite, creatingthep(EFII)nCATseries,in which1, 2,or6copiesof theEFIIciselementineither thesense
(+)orantisense(-)orientationwereinserted.Samples (1 ,ug)of eitherpSRA-CAT,pe-CAT,orp(EFII)nCATweretransiently transfected
into CEFalongwith 2.5 ptgofpSV40-3-gal, encoding ,-galactosidase, as aninternalcontrol. A representative CATassayis shown for the indicated reporterplasmids. CATactivitywascalculated and normalized asdescribed in Materialsand Methods. RelativeCAT activity,
indicating the foldinduction of normalizedCATactivityfor each reporterplasmid above that attained with the referenceplasmidpe-CAT, isgraphed. Data represent the averages ofat least three separate experiments + standard deviation (SD). (C) The additional reporter constructs p(EFII3'/5')2+CAT, pBLCAT4, p(EFII5')4-CAT4, and p(EFII3')2-CAT4 were also tested in transient transfections as
described forpanelB.p(EFII3'/5')2+CATcontains twocopiesof the EFII3'/5'mutantoligonucleotide (see Fig. 3A for sequence) in thesense
orientation insertedattheAccIsite ofpe-CAT.p(EFII5')4-CAT4contains fourcopiesof the 5' repeat(EFIIsequencesfrom-226to-213;
seeMaterials andMethods),andp(EFII3')2-CAT4containstwocopiesofthe 3'repeat(EFIIsequences -205to -194;seeMaterials and Methods);both inserts areinthe antisenseorientation,and bothwereplaced immediatelyupstreamofthe thymidine kinasepromoterand CATgenein thereporterplasmid pBLCAT4.Relative CATactivity,calculatedasdescribedforpanel B, withpe- CATas areference for p(EFII3'/5')2+CATandpBLCAT4as areference forp(EFII5')4-CAT4 and p(EFII3')2-CAT4, is graphed. Datarepresenttheaveragesof
atleast three separate experiments SD.
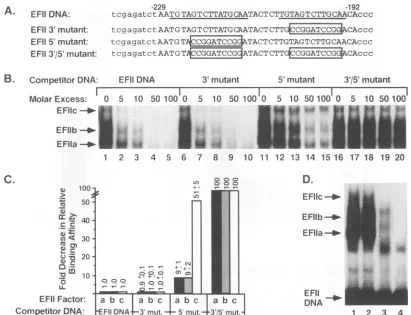
mutantoligonucleotides comparedwith theiraffinities for the
wild-typeEFII DNAsequencearepresentedinFig.3C. We observed that theEFIIa, -b,and-ccomplexescould bindto the EFII 3'mutant,which retains theupstream repeat,with
affinityequaltothatseenwith the EFIIDNA. In contrast, all
three EFIIDNA-binding complexesdemonstratedamarked reduction in relativebinding affinitytothe EFII 5' mutant; EFIIa and EFIIb had approximately 9-fold lower relative
binding affinity, and EFIIc binding affinity was reduced to approximately50-fold below thatseenwiththeEFIIDNA. When the EFII 3'/5' mutant was used as the competitor oligonucleotide, all three EFII factors demonstrated greater than 100-fold-lower relativeDNA-bindingaffinity. Thus, at leastoneintactrepeat mustbe present forEFIIa, -b, and-c complex formation, and all three factors have a higher
affinityfor the5' repeatcontainingtheadditionalAT
nucle-otides.
We alsoanalyzedtheabilityof the threeEFII complexes to directly bind each of the EFII mutant oligonucleotides (Fig. 3D). We observed that all three complexes formed equallywell with the radiolabeledEFIIDNA(Fig. 3D,lane 1) orthe radiolabeled EFII 3' mutant (Fig. 3D, lane 2). In contrast, EMSA with the radiolabeled EFII 5' mutant(Fig. 3D, lane 3) resulted in two less-prominent DNA-binding
activities that displayed proportionally slower migrationin the gel than EFIIa and EFIIb, and no EFIIc-likecomplex waspresent. There is evidence that thesameDNA-binding proteincanhave different mobilities inagelmatrix when it occupiesdifferentpositionsonoligonucleotidesof thesame length (35, 55). Therefore,it ispossible that thetwo DNA-A.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.126.492.76.383.2]A. EFIIDNA:
EFII 3'mutant: EFII5'mutant:
EFII3a/5'mutant:
B. CompetitorDNA:
-229 -192
tcgagatct AAT GTAG T CTTATG CAATACT CTTGTAG T CTTG CAACAccc
tcgagatctAAT GTAG T C TTATG CAATAC TCTTCGTTC CAccc
tcgagatctAAT G
TCCGGT
TACTCTTGTAG TCT TGCAACAc
c ctcgagatctAATGT CGGATCCG ATACTCTCTGTCGCAccc EFIIDNA
Molar Excess: 0 5
EFllc
-40-EFIlb
Ia-EFlla - *12
1 2
10 50 100 0 5 10 50 100 0 5 10 50 100 0
3 4 5 6 7 8 9 10 11 12 13 14 15 16
5'mutant 3'/5' mutant 5 10 50100
M.'
M
tA1
17 18 19 20
3'mutant
C.
D.
100 +*
>
o
Lo
EFIIc-*-
50-FI
~40
b EFDIi
- ACr0*303
EFlIa-mu-'-
.4r
FIG. 3. EFIIa,EFIIb, and EFIIc nuclear factors recognize the upstream repeat in the EFII cis element. (A) Coding-strand nucleotide sequences of double-stranded 50-bp oligonucleotides containing either the wild-type EFII cis element (EFII DNA) or the EFII cis element in which either the 3'repeat (EFII 3' mutant), the 5' repeat (EFII 5' mutant), or both (EFII 3'/5' mutant) have been disrupted by 10-bp BamHI linker sequences, indicated by open boxes. The 38-bp EFII cis element (representing RSV LTR sequences from -229 to -192) is indicated by capital letters. The near-direct repeat sequences present in thewild-typeEFII cis element are underlined. (B) EMSAs were performed as described in Materials and Methods. Briefly, 2,ugof heated 0.5 M NaCl nuclear extract from 14-day-old CE were incubated with 0.5 ng of 32P-labeled EFITDNA in the presence of 1.3 ,ug of poly(dI):poly(dC) and in the absence or presence of a 5- to 100-fold molar excess of nonradiolabeled competitor DNAs. The positions of the retarded EFIIa, EFIIb, and EFIIc protein-DNA complexes are shown. (C) The relative bindingaffinities of EFIIa, -b, and -c for each competitor oligonucleotide were calculated as described in Materials and Methods. The fold decrease in binding affinity relative to wild-type EFII DNA is graphed, and data represent the averages of at least three separate experiments ± SD. (D) EMSAs were performed with 2plgof heated0.5 M NaCl nuclear extract from14-day-oldCE in the presence of 1.3
,ugof poly(dI):poly(dC) and 0.5 ng of the following 32P-labeled EFII wild-type and mutant oligonucleotides: EFII DNA (lane 1), EFII 3' mutant (lane 2), EFII 5' mutant (lane 3), and EFII 3'/5' mutant (lane4). The unbound 32P-labeled EFII DNAs are indicated along with the retarded EFIIa,EFIIb, and EFIIc protein-DNA complexes.
binding activities in Fig. 3D, lane 3, represent EFIIa and
EFIIb, since bothcanbindwith loweraffinitytothe EFII 5'
mutant(Fig. 3B and C). On the other hand, the two
com-plexes inFig. 3D, lane 3, couldrepresentunrelatedproteins,
whicharenormally maskedby the presence of the EFIIa, -b, and -c complexes. EMSA with the radiolabeled EFII 3'/5'
mutant (Fig. 3D, lane4) didnotresult in any specific EFII
DNA-binding activity. The results of both thecompetition
anddirectbinding studies in Fig. 3 are highly consistent with
ouranalysis of thetransactivation properties of the EFIIcis
element invivo. The upstream repeat in the EFII cis element mediates both high-affinity recognition by the EFIIa, -b, and
-cfactors in vitro and strong transcriptional activation in vivo.
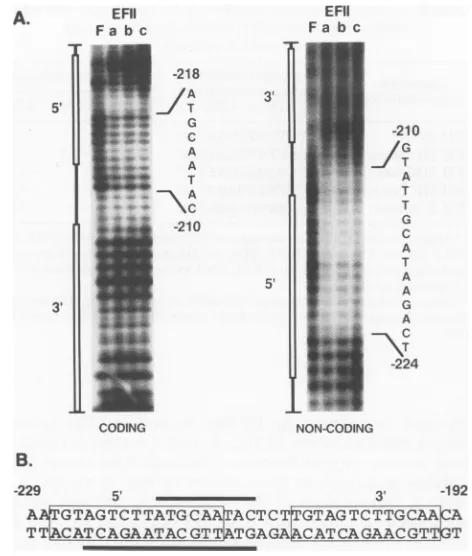
OP/CufootprintingofEFIIa,
EFIIb,
and EFlIc.Inordertofurther define the nucleotides within the EFII cis element which EFlIa, -b, and -c recognize for sequence-specific DNA binding, OP/Cu nuclease footprinting of each DNA-protein complex was performed. The EFIIa, -b, and -c
DNA-protein complexes were resolved by EMSA and
treated in situ with the chemical endonuclease OP/Cu.
Cleaved DNA was then isolated from each complex and
analyzed, along with the cleaved unbound DNA, on a
high-resolution sequencing gel (Fig. 4A). Interestingly, all
three complexes exhibited essentially identical footprints.
Strong protection was observed over 15 nucleotides from
-210to -224onthenoncodingstrand, with weaker
protec-tion of 9 nucleotides from -210 to -218 on the coding strand. The footprints on both strands were observed to
coincide closely but not exactly with the 5' repeat, as
illustrated inFig. 4B.Noprotectionwasobserved with any ofthecomplexesoverthe3' repeatoneitherstrand. These
findingsaresimilartoobservations byGoodwin, who found
by footprintinganalysis that erythroid nuclear proteins
pro-tected sequences from -224 to -215 in the RSV LTR enhancer(22).
Since theEFIIa, -b,and -ccomplexesfootprintthesame
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.104.515.78.393.2]5'
EFII Fa bc
ti
IwF a
-a. m, ..
m;&-
-'W.Z.
81 70 61 52 45 39 34 29 25 22 19 k
EFII Fa bc
-218
/A
G
C
A A T A
-210
CODING
-210 /G
1T T
T
G
C
A T A A G A
C
T\~
-224
EFlic - e ?;
EFIlb
--EFlia
0
jo.MAk
WV
1 2 3 4 5 6 7 8 9 10 11
NON-CODING
B.
-229 5'
3-
3 -192A GTAGTCTTATGCAATACTCT GTAGTCTTGCAA A TT CATCAGAATACGTTATGAGA CATCAGAACGT T
FIG. 4. EFIIa, EFIIb,and EFIIc protect thesamenucleotides within theEFII cis element.(A)EMSAswereperformedwith 2ng
ofa double-stranded end-labeled76-bprestrictionfragment probe, containingthe38-bpEFII ciselement,and 12,ugof CEF 0.5MNaCl nuclearextract.Following EMSA,thegelwastreated withOP/Cu endonuclease. TheEFIIa, -b,and-cprotein-DNA complexes and the freeprobewereexcised from thegel.Thecleaved,end-labeled DNA was recovered by electroelution and analyzed on a
high-resolutionsequencing gel.The nucleasecleavage patternsofthe free probe (lane F)and theprobeboundbyeitherEFIIa(lane a),EFIIb (lane b), or EFIIc (lane c), for both the coding and noncoding strands of theEFIIciselement,areshown. A schematicof the EFII
cis element with the 5' and 3' near-directrepeats indicatedbyopen
bars is shown beside the lanes. Footprinted nucleotides are indi-cated for each strand.(B)Schematicrepresentationof the EFIIcis element, indicatingtheEFIIa, EFIIb, and EFIIcfootprintsonthe coding (overlined) and noncoding (underlined) strands. 5' and 3' near-direct repeatsequencesareboxed.
sequences in the EFII ciselement, it is possible that they
representthesameDNA-binding proteinand that the differ-ences in electrophoretic mobility are due to the fact that
EFIIa and EFIIbare proteolytic products of EFIIc.
How-ever, no increase is observed in the relative amounts of
EFIIa and EFIIb versus EFIIc with increasing age ofthe extract.Alternatively, thesameDNA-binding proteincould
giverisetotheslower-migrating EFIIbandEFlIc complexes
ifitwasdifferentially posttranslationallymodifiedor associ-ated with othertranscriptionfactors. On the otherhand,the
EFIIa, -b, and -c complexes could contain distinct
DNA-binding proteinswhich haveverysimilar,orperhaps identi-cal,sequencerecognitionproperties.This latterpossibilityis consistent with thegrowingnumber oftranscriptionfactors that have been foundtoexist infamilies which demonstrate
nearlyidentical sequence recognition properties (30). EFHa, EFHb, and EFIIc are composed ofmultiple
DNA-binding proteins. To address whetherthe same ordifferent
DNA-binding proteins arepresent in the EFIIa, -b, and -c
* EFII
DNA FIG. 5. Apparent-molecular-mass determination. Fourteen-day-old CE 0.5 M NaCl nuclear extract (120 pg) was subjected to SDS-PAGE and transferredto an Immobilon-Pmembrane, which wascutinto 17 3-mm-wide slices. Theproteins in each slicewere
eluted
separate!y
and tested inEMSAs. EMSAswere performed with0.5ngof31P-labeledEFII DNAand either2,gof14-day-old CE 0.5 M NaCl nuclear extract in the presence of 1.3 ,ug of poly(dI):poly(dC) (lane 1) or 10 p.l of SDS-PAGE-fractionated 14-day-old CEextract, ranging in size from 81 to 19 kDa, inthepresence of 100 ng of poly(dI):poly(dC) (lanes 2 to 12). EFIIa, EFIIb,and EFIIc retardedcomplexesintheunfractionatedextract
(lane 1)areindicated. Sizes areshown above the lanes(in kilodal-tons).
complexes, 14-day-old CE nuclearextractwas subjectedto SDS-PAGE and blottedtoImmobilonpaper.Thepaperwas cut into small slices, and the proteins in each slice were
eluted and assayed for EFII DNA-binding activity. As showninFig. 5, three protein-DNA complexes (lanes 4 and 9) migratedtothepositions correspondingtoEFIIa, -b, and -cobserved inunfractionatednuclearextracts(lane 1). The
dominant shifted band in lane 9 (also found in lane 10) is equivalent in mobility to EFIIa and represents a protein complex composedofone or moresubunits of 22to29 kDa. The two slower-mobility complexes in lane 4 migrate to
positionscorresponding toEFIIb and EFIIcand represent complexesmadeupof subunits of52to61 kDa. These three renatured EFIIa, EFIIb, and EFIIc complexes specifically recognizedthe EFIIcis elementincompetitionassayswith thewild-typeandmutantEFIIDNAs (datanot shown).
In additiontotherenaturedEFIIa, -b, and-ccomplexes, anumber of otherprotein-DNA complexeswereobserved, although most did not correspond in mobility to EFIIa, EFIIb, orEFIIc. The faster-migrating complexes in Fig. 5,
lanes 4 to 6, were found tobe nonspecific in competition
assays (data not shown). Although the complex in lane 3
migrates to a position similar to that ofEFIIa, it did not demonstratehigh affinityfortheEFIIcis element in compe-tition assays(datanotshown). For thisanalysis, aminimal amount of nonspecific competitor DNA was used in the EMSAinordertomaximizerecovery.The addition ofmore nonspecific poly(dI):poly(dC) to the EMSA eliminated the nonspecific complexesmentioned above but hadnoeffecton theEFIIa, -b,and-ccomplexesinFig. 5,lanes 4and 9(data not shown). The complexes in Fig. 5, lanes 2 and 7,
A.
A
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.72.308.80.358.2] [image:8.612.332.566.81.275.2]however, were shown to specifically bind EFII DNA and correspond in apparent molecular mass to one or more subunits of 70to81 kDa and 34to39 kDa, respectively. The significance of the minor complexin lane 2 is not known. However, several of the previously characterized C/EBP family members arein the 35to40 kDa rangeand, through shared sequence recognition properties (see below), would be likelytointeract with theEFII cis element. Interestingly, the complexinFig. 5, lane 7,wasnotobserved when CEF 0.5 M NaCl nuclear extract was analyzed following SDS-PAGE fractionation. Moreover, the complex in lane 7 isnot evident when unfractionated 14-day-old CE extracts are used in EMSAs with 32P-EFII DNA, suggesting that the subunits of 34to39 kDaarenotasabundantasand/or bind theEFIIcis element with lower affinitythantheEFIla, -b, and-cfactors.
Anadditional, slower-migrating complexcanbe detected in Fig. 5, lane 9, which specifically recognizes theEFII cis element by competition analysis (data not shown). This
complex displayed DNA-binding characteristics consistent with the addition ofasecond protein complextoarepeated binding site. For example, we observed that the additional band in lane 9wasonlypresent athigh concentrations of the 25-to29-kDa fraction, and in competition experiments with unlabeled EFII DNA, itwas seento disappear priortoany diminution in the renatured EFIIa band. Moreover, the additional complex in Fig. 5, lane 9,wasabsent when the 25-to29-kDa fractionwas subjectedtoEMSA with the radio-labeledEFII3'mutant,which lacksthelower-affinity down-stream repeat (data not shown). Thus, we interpret the additional shifted complex seen in lane 9 to be a second EFIIa complex, bindingtothe 3'repeatunderconditions of sufficiently highfactor concentration. Thisinterpretation is further substantiated by the footprinting experiments
pre-sented inFig. 7(see below).
Since these molecular mass determinations were
per-formed with the factorspresent ina crudenuclearextract, the potential for proteolysis of the factors is a concern.
However, we performed a similar analysis of the EFII factors afterlysingintact CEF inSDS-containingbuffer and
immediately boiling the total-cell lysate. This approach
should minimizeproteolysisasmuchaspossible. Nonethe-less, results identical to those presented in Fig. 5 were obtained (data not shown). It is entirely possible that
pro-teins in additiontothepolypeptidesdetectedhere afterSDS electrophoretic separation are components of the EFII DNA-binding activitiesin14-day-old CEnuclearextracts.A
polypeptide whose DNA-binding activity cannot be rena-tured after SDS exposure will not be detected, nor will a complex which requires the association of heterologous
subunits for DNA binding, unless all polypeptides in the
native complex have similar molecular masses and are
renatured in thesamefraction. Despitetheselimitations, this analysissuggeststhat, atminimum, twopolypeptides of 22 to29kDa and 52to61 kDa arepresentinthe EFIIa and in
the EFIIb andEFIIccomplexes, respectively.
EFIIa, EFTIb, and EFIHc recognize the same nucleotide sequence. Although EFIIa, -b, and -c protected the same nucleotides withinthe EFII ciselement from OP/Cu cleav-age (Fig. 4), these complexesappear tobecomposed ofat least two polypeptides ofvery different molecular masses which couldberecognizing differentsequencemotifs within the15-nucleotide footprint. Toattempt todiscernany differ-ences inDNA sequence recognition among these polypep-tides,weobtainedaseries ofEFII oligonucleotides harbor-ing small substitution mutations across the 15-nucleotide
TABLE 1. Relative binding affinities of EFIIa, EFIIb, and EFIIc nuclear factors forEFII mutantbinding sites by
competition analysis
Competitor
Footprinted
nucleotide Relative bindingaffinityboligoucletide sequence(codingstrand,
-224 to -210) EFIIa
EFIIb
EFIIc
EFII DNA AGTCTTATGCAATAC 1 1 1
EFIIHL mutant AaaaTTATGCAATAC -1.2 -1.2 -1.3 EFII HM mutant AGTCggggGCAATAC -6 -7 -21 EFII HR mutant AGTCTTATGgggTAC -8 -9 -45 EFII5' mutant AcoggatcoggATAC -9 -9 -51 a Sequence information forcompetitor oligonucleotides: EFII DNA and EFII 5'mutant,Fig. 3A; EFII HL, HM, andHR mutants aredouble-stranded 50-bpoligonucleotides identicaltoEFIIDNA exceptfor the mutations shown
inlowercaseletters.
b RelativebindingaffinitieswerecalculatedasdescribedinMaterialsand Methods.Negative values indicateaffinity lower than that of wild-type EFII
DNA.
sequence footprinted by EFIIa, -b, and -c. The relative
binding affinitiesof theEFIIa, -b,and-cfactorsfor each of
these mutant oligonucleotides (obtained from competition
analyses analogous to those shown in Fig. 3) are given in
Table 1. Substitution of the GTC on the left side of the
footprinted sequencewith AAA(EFIIHLmutant) didnot
decrease the relativebindingaffinities of either EFlIa,-b,or
-c. Thus,we can narrowdown the sequence important for
the DNA-binding activities of EFIIa, -b, and -c to the 11
nucleotides TTATGCAATAC (coding strand). Substitution
ofeitherTTAT withGGGG(EFIIHMmutant)orCAAwith
GGG
(EFII
HRmutant)
reduced thebinding affinitiesofbothEFIIaand EFIIb 6-to9-fold andthebinding affinityof EFIIc
21- and 45-fold, respectively, relative to wild-type EFII DNA. The decrease in the relative binding affinities of all
three complexes for the EFII HM and HR mutantsis very similartothat observed with the EFII 5'mutant(Table1 and
Fig. 3C),inwhichmostof thefootprintedsequence(10of15
nucleotides) has beendisrupted. The only exception is that
EFIIcbinds tothe EFII HM mutantwith a slightly higher
relativeaffinitythantoeither the EFII 5' mutantorthe EFII HR mutant,possiblybecause ofapartial abilitytorecognize
theremainingGCAATACportionof the
li-nucleotide
bind-ing site. The low-affinity binding of EFIla and EFIIb to
oligonucleotides harboring mutations within the
11-nucleo-tidebindingsite islikelyduetotheabilityof these factorsto
recognizesimilar sequences in the 3' repeat.
Atthis level ofresolution, the EFIIa and EFIIb factors appear to have identical sequence-specific DNA-binding
properties, while EFlIc has a more restricted sequence
requirementforthe 5' repeat.Itisinteresting that the EFII
sequences important for high-affinity binding by all three
EFIIDNA-binding factors contain the sequence motif
TT-NNGCAAT. This motif is found inanumberofhigh-affinity
bindingsites for the previously describedtranscription
fac-torC/EBPa(27, 61, 65, 70)aswellasforagrowingnumber
ofC/EBP-related familymembers(1,7,10, 15, 34,56, 58).In
fact,C/EBPahas been showntobind the EFIIciselement in
the RSVLTRinvitro; Ryden and Beemon demonstrated by DNase I footprinting experiments that a heat-stable factor present in chicken liver nuclear extracts and purified
C/EBPotboth bound theEFIIregionof theRSV LTR(61).
Interestingly, unlike the EFII-binding proteins described
here, footprints appeared concomitantlyoverboth repeats in
the EFIIciselement withincreasing concentrationsofeither
on November 9, 2019 by guest
http://jvi.asm.org/
TABLE 2. Relative bindingaffinities of EFIIa,EFIIb, and EFIIc nuclear factors for various binding sites by competition analysis
Competitor Nucleotide Relativebindingaffinityb oligonucleotidea sequence EFIIa EFIIb EFIIc
EFII DNA TTATGCAATAC 1 1 1
IgH-E TTGAGCAATGT -1.6 -1.5 -4
IBF DNA GGCTGCAATTG -3 -3 -4
DE I TTTTGTAATGG -11 -9 -14
Core TGTGGAAAGGA >100 >100 >100
ISequenceinformation forcompetitor oligonucleotides: EFII DNA, Fig.
3A; IgH-E,IBFDNA,DEI,andCore, Materials and Methods. b See Table 1, footnoteb.
the liver nuclear factororpurifiedC/EBPa(61). The murine C/EBP-related factorimmunoglobulin enhancer-binding pro-tein
(Ig/EBP-1)
hasalso been shownby Roman et al. tobindthe EFII cis element inthe RSVLTRinvitroas a
3-galac-tosidase fusion protein encoded in part by the murine Ig/EBP-1cDNA,and the role ofIg/EBP-1 in regulating RSV
transcription inmurine cells wasdiscussed by these
inves-tigators (58). In addition, a putative C/EBP-related factor,
the internal binding factor (IBF), whichwas characterized
andpurified fromBHKcells,wasreported by Karnitzetal.
to bind the EFII cis element in competition analysis (33).
Given that the EFII factors recognize a sequence motif
which fits the consensus binding site for C/EBP-related
transcription factors, we wished to compare the sequence
recognition requirements of EFIIa, -b, and -c in avian
fibroblasts with those of the members of theC/EBP family
which have been reported tobind the EFII cis element in otherspecies and cell types.
Comparison of the DNA-binding properties of EFIIa,
EFIHb,andEFMc with those ofC/EBPa, IBF, andIJEBP-1.
Initially, a series of competition experiments were
per-formed to ascertain whether the EFIIa, -b, and -c factors could also bind to high-affinity recognition sites used to
characterize C/EBPcx, IBF, and Ig/EBP-1. As shown in
Table 2, the EFII factors showednegligible tolow
recogni-tion oftwohigh-affinity C/EBPa binding sites:theSV40core
enhancer sequence found in thecollagenIII promoter(9, 29, 61)and the DE Isite in theratalbumin promotor(18, 50, 73).
In contrast, oligonucleotide DNAscontaining binding sites for either
Ig/EBP-1
or IBF competed relatively well forEFIIa, -b, and-cbindingactivities. EFIIa and EFIIb bound
IgH-E, an oligonucleotide which encompasses the E site
from the murine immunoglobulin heavy-chain enhancer,
with only slightly lower affinity than they did EFII DNA.
The IgH-E nucleotide sequence was used to identify the
Ig/EBP-1 expression clone from murine L-cell and B-cell
cDNAlibraries
(58).
Sequence analysisofIg/EBP-1revealed that it hashighhomology toC/EBPa throughout theDNA-binding domain and leucine zipper region
(58).
EFIIa andEFIIb also bound to IBF DNA, an oligonucleotide which
containssequencesfromaninternalenhancer located within
the gag gene of RSV (2, 33), with threefold lower affinity
thanto EFII DNA. TheIBFnucleotide sequence wasused
to characterize and purify the IBF DNA-binding activity
from BHK cell nuclear extracts (33, 34). IBF has been
reportedtobe relatedtoC/EBPafrom itsoverlappingDNA
sequencerecognition propertiesand heatstability (8, 34, 39).
EFIIcrecognized both IgH-E and IBF DNA with fourfold lower affinitythan the EFII sequence. These differences in the affinities of EFIIa, -b, and -c for the EFII, IBF, and
IgH-E DNAs, although relatively small, were reproducibly
observed.
Comparisonof the celltypedistribution ofEFMIa, -b, and -c
withIBF,
Ig/EBP-1,
andC/EBPa Although the binding sites for IBF and Ig/EBP-1 were recognized by the three EFII factors present inCEnuclearextractswith moderatetohighaffinity, similarDNA-binding specificitycouldbe exhibited
by different proteins that may be selectively present in
different cell types. Thus, we nextcharacterized the EFII
DNA-binding activity in cell types in which IBF and Ig/
EBP-1 expression has been documented. As mentioned
above,
IBFwas purified from BHK cell nuclear extracts,and
Ig/EBP-1
expression, monitored by Northern (RNAblot) analysis, isobserved in allmurine celltypestested but
isparticularlystronginmurineBlymphocytes.We prepared
nuclear extracts from BHKcells and from anavian B-cell
lymphoma line, Bk3A, to testfor EFIIDNA-binding
activ-ity.AsshowninFig. 6A,lane1,andFig. 6C,lane 5, typical
EFIIa, -b,and-ccomplexesareobserved whenradiolabeled
EFIIDNAisincubated withaBHKcell nuclear extract. To
confirm that the BHK extract contained active IBF, we
incubatedtheextractwith radiolabeledIBF DNA. Astriking
difference between the radiolabeled EFII and IBF
DNA-binding
patternswasobserved(Fig.
6A, lanes1and 2).TheIBF DNA-binding pattern more
closely
matched thatre-ported by
Karnitz et al.(34).
We performed competitionanalyses with unlabeled EFII DNA in an EMSA with
radiolabeled IBFDNAandBHKextract.As shown in Fig.
6A,
lanes2to7, byouranalysis,IBFhasnegligible affinityforthe EFII DNA sequence;an80-fold molarexcessof EFII
DNAwasrequired tocompetefor50% of the IBFbinding
activity.
Thus, we conclude that the IBF factorcharacter-ized inBHKcells isunlikelytobeanintegralpartofeither
the EFIIa, -b,or -cDNA-binding complex. Moreover, this
IBFfactor doesnotappear tobe very abundant in nuclear
extracts from 14-day-old CE since we do not observe its
formation in EMSAs with radiolabeled IBF DNA and
14-day-old
CEextract(data
notshown).In contrast, radiolabeled IgH-E and EFII DNAsyielded
nearly identical binding patterns in the Bk3A lymphoma
extractexceptthatnoEFIIccomplexwasobserved to form
with radiolabeledIgH-E
(Fig.
6B). Also, radiolabeledIgH-Ebound EFIIa and EFIIb in EMSAs with 14-day-old CE
extract
(data
notshown).
Thus,it is possiblethat eitherof theavianEFIIaand EFIIb factors ishighlyrelatedtomurineIg/EBP-1.A greatexcessof theEFIIafactor,farexceeding
the level of EFIIb
(or EFIIc),
was detected with eitherradiolabeledEFIIorIgH-EDNAin the Bk3Aextract(Fig.
6B). Although
theproportions
of EFIIa, -b, and -c aredifferent fromthose observed in CEF, 14-day-old CE, and
BHK extracts, allthree complexesin the Bk3Aextract are
heat stable and demonstrated characteristic DNA-binding
specificities
incompetition
analysis (Fig. 6C,lane9,and datanot
shown).
Wealso observed thatanincrease in theamountof Bk3A nuclearextractresultedin thedisappearanceof the EFIIb and EFIIcshifts and theformationofa newcomplex
(Fig. 6C,
lane10),
termedABF(additional
binding factor).The ABF complex appeared onlywith the Bk3A extract;
titration of all otherextractsledto auniformincrease in all three factors
(data
notshown).
As will be discussed later(Fig. 7),
theABFcomplexappearstorepresenttheaddition of a second EFIIa factor to the downstream near-direct repeatin the EFIIciselement,
causedby
thehigh
concen-tration of EFIIa in the B-cellextract.InadditiontoBHKand Bk3A nuclear extracts,wetested anumber offibroblast and
epithelial
cell lines for theEFIIa,
on November 9, 2019 by guest
http://jvi.asm.org/
A. BHK extract B. Bk3A extract
EFllc_H ^ ^ ^ F
~~~Fllc--_
EFllb _-o
WV,
EFllb_-p
EFll_IIFIa
DNA ; X _ IBF EFII _ IgHE
DNA DNA DNA
1 2 3 4 5 67 1 2
molar excess: 0 1 5 10 50100I
competitor: EFII DNA
C. Ligprotein: 3 2 3 3 9 7 4 10 1 2
I- ABF
4- EFIIa
6 7 8 9 10
> a) r X OD 3I
[image:11.612.61.286.79.420.2]wLX
FIG. 6. Comparingthe celltypedistribution ofEFIIa, -b,and-c
with that ofIBF,Ig/EBP-1,andC/EBP.(A)EMSAswereperformed
with 9 ,ug of BHK 0.5 M NaCl nuclear extract and 0.5 ng of 32P-labeled EFII DNA(lane 1) or IBF DNA (lanes 2 to7) in the presenceof 1.3 p,gofpoly(dI):poly(dC)and in the absence(lanes1 and2)orpresence(lanes3 to7)ofa1-to100-fold molarexcessof nonradiolabeled competitor EFII DNA. (B) EMSAs were per-formed with 1,ugofheated Bk3A 0.5 M NaCl nuclear extract and 0.5 ng of32P-labeled EFII DNA(lane 1)orIgH-EDNA(lane 2)in the presenceof 1.3p,gofpoly(dI):poly(dC). (C)EMSAswereperformed
withsamplesof 0.5 MNaCl nuclearextractsfrom the indicatedcell
typesand0.5 ng of32P-labeled EFII DNA in the presence of 1.3,ug
ofpoly(dI):poly(dC). Lanes: 1and2, 14-day-old (14d) CE; 3,A431
epidermal carcinoma cells; 4, BALB/c-3T3 fibroblasts; 5, BHK
cells; 6,rat H4hepatoma cells; 7,adult ratliver;8,adultratkidney;
9 and10, avianB-celllymphomalineBk3A. The amountofprotein
included ineachEMSA is listedabove the correspondinglane.A,
heated extract. EFIIa, EFIIb, EFIIc, and ABF retarded
protein-DNAcomplexesareindicated.
-b, and -c DNA-binding pattern (Fig. 6C). We observed a patternofbinding activitysimilartothat ofEFlIa,-b, and-c
in nuclearextractsfrom human epidermal carcinomaA431 cells (Fig. 6C, lane3) andmurinefibroblasts(BALB/c-3T3; Fig. 6C, lane 4) as well as several other fibroblast lines
(Rat-i, NIH3T3, andCoscells;datanotshown). The EFII
DNA-bindingactivities inthese extracts,asinourstandard CEextract,wereheatstable (Fig. 6C,comparelanes 1 and
2, also lane 3; data not shown). We also obtained nuclear extracts fromadifferentiated ratliverheptomacellline,H4,
and adult rat liver and kidney tissue. Reproducibly less (hepatoma and liver; Fig. 6C, lanes 6 and 7,respectively) or negligible (kidney; Fig. 6C, lane 8) EFII DNA-binding activ-ity was detected in these extracts. Furthermore, retarded complexes corresponding in mobility to EFIIa were not
observed. Although we cannot rule out the possibility that poor extraction or selectiveproteolysis of the EFII factors occurred consistently in the H4, liver, andkidney extracts, all extracts tested were prepared by the same method, and analysis of proteins that bind a hypersensitive site 4,800bp upstream of the phosphoenolpyruvate carboxykinase gene demonstrated that the H4, liver, and kidney extracts we analyzed were not significantly proteolyzed (12). Although the tissues and cell types tested were from variousspecies, itappears fromthis preliminarysurvey thatthe EFIIa, -b, and -c binding activities are not uniformly present in all cell types.
EFIIa binds to the 3' repeat sequenceinthe EFII cis element
athigh concentrations. Wewere intrigued by the unusually large amount of the EFIIa factor present innuclear extracts from Bk3A cells(Fig. 6B and C), as well as the presence of theABF complex when higher concentrations of the extract wereused in theEMSA (Fig. 6C, lane 9). We analyzed Bk3A nuclearextractby SDS gel electrophoresis and Immobilon transferand obtained results identicaltothose shown for the 14-day-old CE extract in Fig. 5 except that in the 25- to
29-kDa fraction, greater amounts of both the EFIIa-like factor and the slower-mobility complex were seen. As shown in Fig. 7A, when we compared the mobility shift
pattern seen in the Bk3A 25-to29-kDa fraction(lane2)with
theEFII DNA-binding pattern of unfractionated Bk3A ex-tract, in which the ABF complex is observed (lane 1), we
found that the ABF factor was identical in mobilityto the slower complexin the 25- to 29-kDa fraction. This slower
complex was previously interpreted to represent an
addi-tional EFIIacomplex binding to the 3' repeat in the EFIIcis
element. We therefore suggest that the ABF complex also represents the binding of a second EFIIa factor to the 3' repeat in the radiolabeled EFII DNA, because of the abun-dance of EFIIa inthe Bk3A cells. In support ofthis,wealso noted that the ABFcomplex did not form inanEMSAwith
the radiolabeled EFII 3' mutant, which contains only one
intact repeat, and that ABF demonstrated thebinding kinet-ics expected for the addition ofasecond factorto arepeated binding site by competition analysis (data not shown) and extracttitration (Fig. 6C, lanes 9 and 10).
In order to directly analyze the possible presence of an
additional proteincomplex bound to the 3' repeat of the EFII cis element in the ABF complex, we performed OP/Cu nuclease footprinting of both the EFIIa protein-DNA com-plex and the ABF protein-DNA comcom-plex present in the Bk3A nuclear extract. Comparison of the EFIIa footprints with the ABFfootprints (Fig. 7B) revealed that proteins in the ABFcomplex protected the same sequences in the 5' repeat onthecoding and noncoding strandsastheproteins in the EFIIa complex, although protection was enhanced in theseregions. However, only proteins in theABFcomplex protected similar nucleotides in the 3' repeat. The additional
ABF footprints in the 3' repeat protected 12 nucleotides from -203 to -192 on the noncoding strand and 10 nucleo-tides from -201to-192 on thecoding strand. A diagram of the ABF footprint along with the EFIIa footprint is pre-sented inFig. 7C.