[beta]2-adrenergic receptor modulation of macrophage inflammatory mediator production
Full text
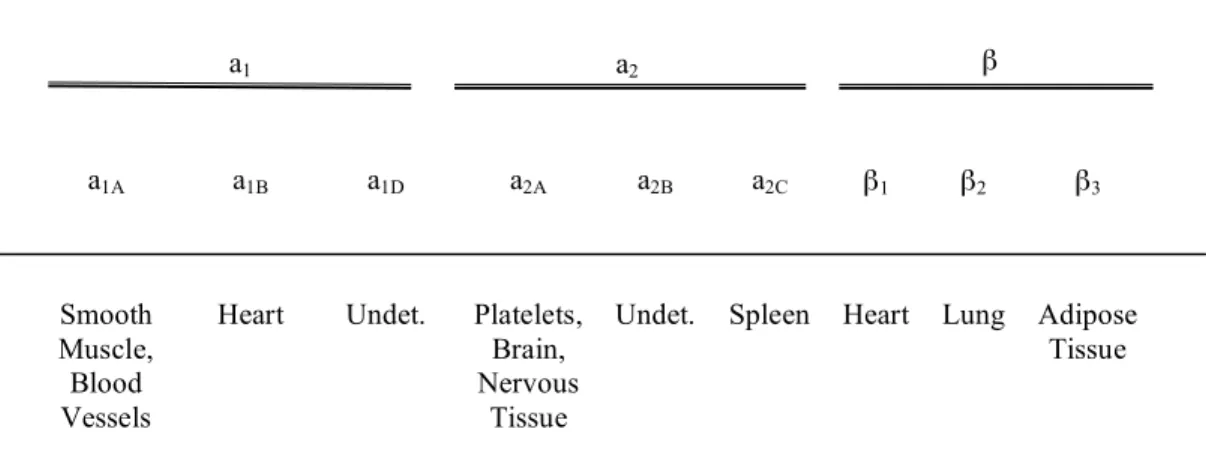
Figure

Related documents
Bologna: Pàtron editore.. Bologna: Pàtron
The flowchart of the proposed algorithm is shown as Figure 3:Face detection modules again separated in to five modules 1(a)Skin Detection: Using color
This affects the performance of final denoised image hence to remove noise affecting approximate coefficients non-linear diffusion filtering is applied on these
A consolidated solution integrating message validation and dissemination using PBFT based consensus, blockchain based incentives and reputation management is presented in [41] and
From there, a combination of metrics are calculated, including the gross economic benefit shareholders are deriving from the reputation asset, the location of value across
Financial barriers to small firms have almost twice the impact on annual growth have is these financial barriers to large businesses.This difference is even more powerful in the
7 However, some children with persistent asthma who developed one severe exacerbation in the prior year have a two folds increased risk of subsequent
Rapid identification of synthetic cannabinoids in herbal samples via direct analysis in real time mass spectrometry. Rapid Commun
