MOLECULAR PROFILING OF CLINICAL DRUG RESISTANCE
Roshawn Watson
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the School of Pharmacy.
Chapel Hill 2010
Approved by:
ABSTRACT ROSHAWN WATSON:
Molecular Profiling of Clinical Drug Resistance (Under the direction of Howard McLeod, Pharm.D.)
One of the greatest challenges in oncology is drug resistance. 5-Fluorouracil (5-FU) is the third most commonly used anti-neoplastic, so lack of initial or continued response to 5-FU represents a big clinical problem. Despite its prominence in cancer treatment, the mechanisms for its resistance remain largely undefined. The third leading cause of cancer-related deaths is colorectal cancer for which 5-FU is an essential part its therapeutic backbone. Resistance to 5-FU is a primary cause of treatment failure. Developing models explaining 5-FU resistance is imperative to advancing care.
iv
colorectal cancer cell lines. DUT and UCK2 knockdown decreased IC50 by >2-fold in two or more cell lines while DPD knockdown yielded decreased IC50 by >2-fold in one cell line and nearly 2-fold in the others. This mechanistic validation supports overexpression of these targets as a mechanism for 5-FU resistance. Additionally, copy number gains in TYMS occurred 5 times more frequently in exposed compared to unexposed patients, suggesting that TYMS gains is also a mechanism for 5-FU resistance.
To my wife, family, and friends who provided the support system necessary
vi
ACKNOWLEDGEMENTS
It is my pleasure to honor the many people who helped bring this endeavor to fruition.
I would like to thank my advisor, the incomparable Dr. Howard Mcleod, who provided the framework, expertise, and insight that helped me to excel. His unique style of advising allowed me much freedom to develop an agenda and identify my own research interests while working on a phenomenal project. I deeply valued his remarkable leadership skills because he always had timely advice, constructive criticism, and deep insights in both professional and personal matters. It is an honor to work with such an expert and someone who embodies excellence. I typically felt motivated every time I left his office, which was invaluable.
I must also acknowledge the guidance and support from the past and present faculty and staff and the entire Eshelman School of Pharmacy. I would like to particularly thank Amber Allen, Angela Lyght, Kathy Maboll, Pam Griffin, Becky Eatmon, Peggy Quinn, Tania Cherry, Tejinder Rakhra-Burris, and Sherrie Settle for their help with administrative concerns throughout the years. Additionally, I need to thank Allison Deal because without her support and commitment, this project would not be possible.
I have received an exceptional amount of guidance and support from past and present members of the McLeod lab. In particular, I am thankful to Janelle Hoskins, Tammy Havener, Cindy Lodestro, Filipe Muhale, Michael Wagner, and Todd Auman. Thanks to Pei-shi Ong, Kristi Richards, and Janna Hutz for insightful discussions about science, professionalism, and other matters.
I would like to thank the many people with whom I’ve shared meaningful life experiences who set me on this path. From Dr. Buck Hales, who guided my first research efforts at the University of Illinois and helped me strongly consider attending graduate school, to those whose gift of companionship made my days at UNC more enjoyable. Brandon Swift, Sarai Smallwood, Dave Harbourt, Courtney Boyd, Mary Peace Mcrae, Diedre Washington, Julie and Mike Dumond, Amanda Corbert, Angela Kashuba, Dhiren Thakker, and Amica Simmons Yon deserve thanks for so much needed laughter and friendship. I also thank the Royster’s Society of Fellows and the GI Spore both for funding and much professional and social enrichment.
viii
strive to excel at all I do, and without her pulling for me, I would not persevered. I also thank my brother who always believed me. To my phenomenal wife, Venita, your support and wisdom has made the difference. During difficult times, you were always there to commiserate, motivate, challenge, and console. I am truly blessed, and I love you.
TABLE OF CONTENTS
LIST OF FIGURES………... ... x LIST OF TABLES... xii LIST OF ABBREVIATIONS AND SYMBOLS………... xiii
CHAPTER
I. INTRODUCTION: ...1 II. CHARACTERIZATION OF THE RETROSPECTIVE CLINICAL COHORT
AND PHENOTYPING STRATIFICATION ...25 III. QUANTITATIVE DETERMINATION OF EXPRESSION OF 5-FU PATHWAY
PROTEINS AND FUNCTIONAL VALIDATION ...46 IV. COPY NUMBER DETERMINATION OF 5-FLUOROURACIL PATHWAY
TARGETS FOR MECHANISTIC ELUCIDATION OF METASTATIC
COLORECTAL CANCER RESISTANCE ...80 V. CONCLUSIONS AND FUTURE STUDIES: MOLECULAR
HETEROGENEITY AND 5-FLUOROURACIL RESISTANCE IN
METASTATIC COLORECTAL CANCER ...104 APPENDICIES
x LIST OF FIGURES
Figure 1-1 5-Fluorouracil Structure...15 Figure 1-2 5-fluorodeoxyuridine Monophosphate (FdUMP) – the active metabolite of
5-fluorouracil ...16 Figure 1-3 5- Fluorouracil Pharmacokinentic and Pharmacodynamic Pathway ...17 Figure 2-1 Model for an ‘enriched’ approach for genomic discovery...39 Figure 2-2 Kaplan-Meier curve depicting the overall survival of the entire clinical
cohort from surgical resection of liver metastases...40 Figure 2-3 Kaplan-Meier curve depicting the disease-free survival of the entire
clinical cohort from diagnosis of liver metastases ...41 Figure 2-4 Kaplan-Meier curve depicting the disease-free survival of the entire
clinical cohort from surgical resection of liver metastases. ...42 Figure 3-1 Distribution of DPD AQUA scores and western blot densitometry values
in cell lines controls...68 Figure 3-2 Representative AQUA of 5-FU pathway target (TS) in colorectal liver
metastases ...69 Figure 3-3 Profiles of cell sensitivity of genes encoding proteins whose expression is
significantly linked to 5-FU exposure...70 Figure 4-1 Histogram of TYMS copy number in tumors from 5-FU exposed and
unexposed patients ...96 Figure 4-2 Histogram of TYMP copy number in tumors from 5-FU exposed and
unexposed patients. ...97 Figure 4-3 Kaplan-Meier analysis of overall survival in patients with metastatic
colorectal cancer receiving neoadjuvant chemotherapy prior to surgical
resection of their metastases ...98 Figure 4-4 Kaplan-Meier analysis of overall survival in patients with metastatic
colorectal cancer receiving adjuvant chemotherapy following surgical
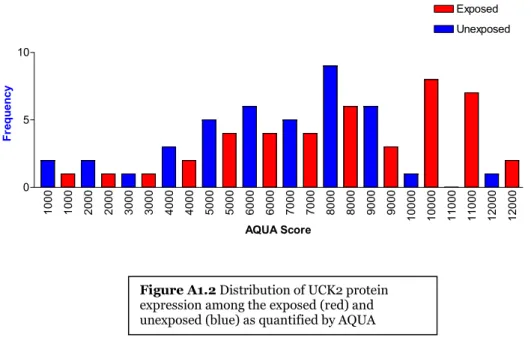
Figure A1-2 Distribution of UCK2 protein expression among the exposed (red) and
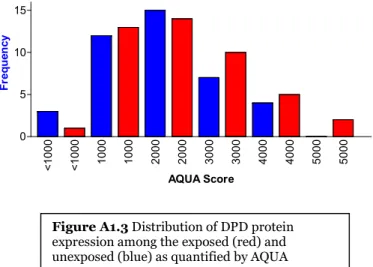
unexposed (blue) as quantified by AQUA...131 Figure A1-3 Distribution of DPD protein expression among the exposed (red) and
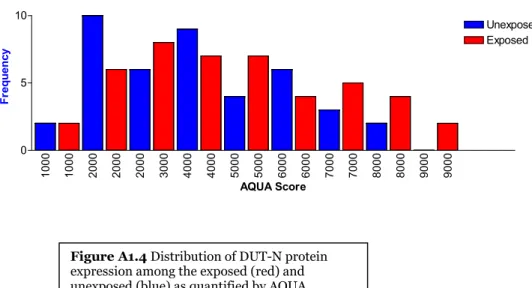
unexposed (blue) as quantified by AQUA...132 Figure A1-4 Distribution of DUT-N protein expression among the exposed (red) and
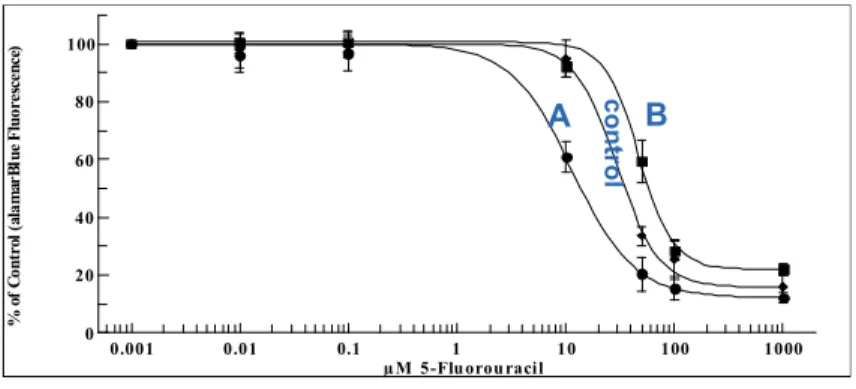
unexposed (blue) as quantified by AQUA...133 Figure A1-5 Example of dose-response curves used to quantify altered 5-FU phenotype 134 Figure A1-6 DPYD mRNA expression as determined by microarray in exposed (red)
and unexposed (blue) samples ...135 Figure A1-7 DUT mRNA expression as determined by microarray in exposed (red) and
unexposed (blue) samples ...136 Figure A1-8 UCK2 mRNA expression as determined by microarray in exposed (red)
xii
LIST OF TABLES
Table 2-1 Clinical cohort demographics and baseline disease characteristics ...43 Table 3-1 Clinical cohort demographics and pathological parameters ...71 Table 3-2 Logistic regression multivariate analysis of clinical features and DPD
expression as determined by AQUA...72 Table 3-3 Logistic regression multivariate analysis of clinical features and DUT
expression as determined by AQUA...73 Table 3-4 Logistic regression multivariate analysis of clinical features and UCK2
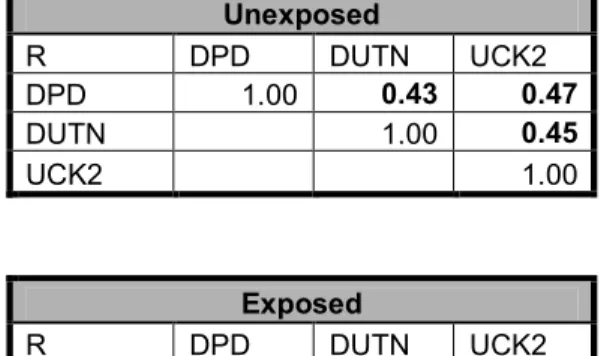
expression as determined by AQUA...74 Table 3-5 Correlation matrix of DPD, DUT, and UCK2 for unexposed and the
LIST OF ABBREVIATIONS AND SYMBOLS
5-FU 5-Fluorouracil
AJCC American Joint Committee on Cancer AQUA automated quantitative analysis
BrdU bromodeoxyuridine
CEA carcinoembryonic antigen
CEPH Centre d’Etude du Polymorphisme Humain
CRC colorectal cancer
Ct threshold cycle
DAPI 4',6-diamidino-2-phenylindole
DHFU dihydrofluorouracil
DNA deoxyribnucleic acid
DPD dihydropyrimidine dehydrogenase (protein) dTMP deoxythymidine monophosphate
dTMP 2V-deoxythymidine-5V-monophosphate
dUDP deoxyuridine diphosphate
dUMP 2V-deoxyuridine-5V-monophosphate
DUT deoxyuridine triphosphatase
ELISA enzyme-linked immunosorbent assay
ERBB2 HER-2/neu
FBS fetal bovine serum
FdUMP fluorodeoxyuridine monophosphate
xiv
HER2 human epidermal growth factor receptor 2
HIPAA Health Insurance Portability and Accountability Act
HR Hazard Ratio
IC50 concentration which inhibits biological process by 50%
IHC immunohistochemical
IRB Institution Review Board
LOH loss of heterozygosity
mCRC metastatic colorectal cancer mRNA messenger ribonucleic acid
N/C nuclear to cytoplasmic protein expression ratios
NCI National Cancer Institute
NME non-metastatic cells
PTEN Phosphatase and Tensin Homolog
qRT-PCR real time quantitative PCR
RECIST Response Evaluation Criteria In Solid Tumors
RNAi RNA interference
RRM1 regulatory unit of ribonucleotide reductase RRM2 catalytic unit of ribonucleotide reductase
shRNA short hairpin RNA
SSDI Social Security Death Index
TMAs tissue microarrays
TSER thymidylate synthase tandem repeat TYMP thymidine phosphorylase (gene) TYMS thymidylate synthase (gene)
Chapter 1
General Introduction
Variability in drug response is one of the most pervasive problems with patient
care, especially variability to chemotherapy and immunologic therapy.1 For instance,
despite the addition of novel agents and better optimization of regimens, over 50% of
patients undergoing chemotherapy for advanced and metastatic colorectal disease will not
receive significant therapeutic benefit.2-4 Many factors have been shown to influence
response to drug therapy including: age, gender, tumor subtype, disease stage, comorbid
diseases, overall performance status, pharmacokinetic and pharmacodynamics factors.5-7
Unfortunately, our knowledge of these factors typically does not translate into
development of predictive models for chemotherapy response. Our inability to
discriminate between patients who will derive benefit from chemotherapy and those who
will not causes non-responders to be exposed to unnecessary toxicity and expenses.
Identifying genetic contributors to variability in chemotherapy response allows for the
appropriate agents to be selected at the initiation of chemotherapy and when the regimen
must be modified. Presently, there are several examples where interindividual differences
in patient response have been linked to sequence variation and differences in expression
of drug-metabolizing enzymes, drug transporters, and drug targets.5,6,8 Genetics is
believed to account for between 20 to 95% of the variability in drug disposition and
effects.9 Such variability forms the basis for tumor heterogeneity and normal germline
differences between cancer patients. It is often a lack of an understanding of patient
genomic diversity that makes it particularly difficult to predict chemotherapy response
This challenge has given rise to pharmacogenomics, a branch of personalized
medicine that has made progress in dissecting the diverse responses to chemotherapy.
Pharmacogenomics is the study of genetic differences underlying interindividual
variability in drug responses.5,10 It includes the studies of variation in RNA expression,
somatic mutations, and germline DNA.11 It aims to identify genetic changes in
drug-metabolizing enzymes and other molecules influencing drug activity.11
Pharmacogenomics is particularly important to oncology because systemic toxicity and
unpredictable efficacy often characterize chemotherapy. Additionally, the high costs of
chemotherapy make selection of the appropriate agent financially prudent. Incorporating
pharmacogenomics into treatment decisions has been shown to improve patient
outcomes. This is particularly true for colorectal cancer (CRC). In this chapter, we will
discuss CRC, resistance to its treatment, targets associated with its resistance, and the
methodologies used to identify the determinants to CRC chemotherapy resistance.
Colorectal Cancer and Resistance
Colorectal cancer is currently the third leading cause of cancer-related deaths in
the US.12Despite some recent advances, such as the development of new treatments and
regimens, nearly all patients with metastatic colorectal cancer develop clinical resistance
and eventually stop responding to therapy. This resistance has been well-documented
with 5-fluorouracil-based regimens.
5-fluorouracil (5-FU) is the backbone of treatment for advanced and metastatic
colorectal cancer, as nearly all patients will receive a 5-FU-containing regimen (Figure
1.1).13,14 The prominence of 5-FU in metastatic colorectal cancer (mCRC) treatment is
largely a function of its consistent efficacy throughout five decades of use.15 However,
one of the biggest challenges for the management of mCRC is 5-FU treatment failure,
especially in patients who initially respond but become resistant. Indeed, the five-year
survival of those with mCRC is less than ten percent.12 Deaths due to
chemotherapy-resistant mCRC disproportionately account for its very high mortality rate.16 Given that
5-FU is the third most commonly used anti-neoplastic agent, its resistance represents a
major clinical problem.
Targets Associated With Variable 5-FU Resistance
Understanding the mechanism of action for 5-FU may offer insight into
mechanisms of its resistance. 5-FU is metabolized in vivo by thymidine phosphorylase
ultimately into fluorodeoxyuridine monophosphate (FdUMP, Figure 1.2), which binds
TS. TS is the rate-limiting enzyme in pyrimidine nucleotide synthesis, and it inhibits
deoxythymidine monophosphate (dTMP) production.17Since dTMP is essential for DNA
biosynthesis and repair, its depletion results in 5-FU’s cytotoxicity.18Dihydropyrimidine
dehydrogenase (DPD)-mediated metabolism of 5-FU into dihydrofluorouracil (DHFU) is
the rate-limiting step of 5-FU catabolism; up to 80% of 5-FU is metabolized by DPD in
the liver.19
Due to the importance of 5-FU to the treatment of colorectal and other
gastrointestinal cancers, there have been numerous investigations into 5-FU resistance.
The primary approach of these previous studies has been to evaluate genes, mRNA, and
proteins thought to be important to 5-FU resistance (Figure 1.3).20-23
tumor site, altered 5-FU distribution secondary to the cancer, and increased 5-FU
elimination. Altered 5-FU metabolism, such as decreased catabolic activation and
increased inactivation of metabolites by a metabolizing enzyme (i.e. TP), can also cause
resistance.21 Additionally, pharmacodynamic contributors to 5-FU resistance are
important. Target-associated resistance includes increased levels of 5-FU’s target enzyme
(thymidylate synthase, TS) or its substrate (dUMP) and thymidylate synthase gene
(TYMS) amplification.24 Wild-type p53 also inhibits TYMS promoter activity, so
mutated p53 represents a potential mechanism of target-associated resistance.25 In
aggregate, over 20 enzymatic targets comprise the 5-FU pharmacokinetic and
pharmacodynamic pathway. Their true predictive value with respect to 5-FU response has
yet to be thoroughly elucidated.
This review will summarize major targets with known involvement in the 5FU
pharmacodynamic and pharmacokinetic pathway (Figure 1.2). Variation at either the
gene, mRNA, or protein level for these targets is may potentially be associated with
resistance to 5-FU. This chapter will review their biologic roles, their pharmacologic
relevance with respect to 5-FU, available data on their involvement in 5-FU resistance,
and their therapeutic utility as predictive markers for 5-FU efficacy. Based on this
discussion, a targeted approach will be presented as a new tool for circumventing some of
the technical challenges associated with traditional investigations of 5FU resistance.
Thymidylate Synthase
Thymidylate synthase (TS) is a critical enzyme for DNA synthesis. It catalyzes
the methylation of 2V-deoxyuridine-5V-monophosphate (dUMP) to
5V-monophosphate (dTMP) thereby mediating the rate-limiting step of pyrimidine
biosynthesis.26 Accordingly, it is an important molecular target for many chemotherapy
agents including 5-FU. Inhibition of TS by FdUMP, the active metabolite of 5-FU, is
considered the primary mechanism of action of 5-FU. Numerous studies have evaluated
polymorphisms and copy number aberrations in TS associated with variable response to
5-FU.Overexpression of TS has been linked to resistance to 5-FU. Notably, a
polymorphic tandem repeat in the promoter region of the thymidylate synthase enhancer
region influences TS expression and segregates 5-FU responders from non-responders.27
Multiple studies show that three copies of the TS (TSER*3) tandem repeat gives greater
TS expression than two copies (TSER*2).28-30Patients with stage 3 and 4 CRC who have
TSER*2/TSER*2 or TSER*2/TSER*3 genotypes have improved response and survival
from 5-FU compared to TSER*3 homozygous patients.24,27
TS levels have also been directly associated with 5-FU response in both advanced
CRC and mCRC patients. Advanced CRC and mCRC patients with low TS levels have
2.2 and 3.3 greater odds, respectively, of responding to 5-FU compared to patients with
high TS levels.31 Additionally, data from multiple studies corroborate that tumors
expressing high TS levels are associated with poorer survival than tumors expressing low
TS levels.32,33 Additionally, high thymidylate synthase gene copy number has been
implicated in poor survival in mCRC patients receiving 5-FU.34 Collectively, this data
suggests that high TS levels, irrespective of cause (i.e. copy number, tandem repeat in
However, there are complicating factors that have limited the utility of TS as a
biomarker including: the lack of standardized methodologies for assaying TS or TYMS
and negative and even contradictory data. Additional studies are needed with consistent
methodologies and 5-FU regimens that incorporate multiple TS variants and TS levels to
delineate the true predictive value of TS. Non-TS variants also appear important in
explaining 5-FU resistance and their integration into a model with TS would likely
contribute to the overall predictive value.
Thymidine Phosphorylase
Thymidine phosphorylase (TP) catalyzes the reversible interconversion of
thymine and thymidine, which are pyrimidine and nucleoside bases that are critical for
DNA synthesis.35,36 Additionally, since TP catalyzes a deoxyribosyl transfer reaction
responsible for the biologic activation of 5-FU, it has also been investigated as a cause of
5-FU resistance in a variety of clinical settings.21,37 However, the data is highly
discordant. In CRC patients receiving 5-FU, tumor TP mRNA and protein levels have
direct, no, or inverse associations with patient outcome, such as survival.38-40Presumably,
low TP levels would be associated with poorer response or survival in patients receiving
5-FU since TP is responsible for activating the prodrug (5-FU). Nonetheless, in addition
to bioactivation, TP also has dual and contradictory functionality as a proangiogenic
factor.41 Thus, the malignant phenotype of pronounced angiogenesis conferred by high
TP may counterbalance or override any clinical utility of excess 5-FU activation. The
predictive utility of TP levels alas remains convoluted.
Dihydropyrimidine Dehydrogenase
Dihydropyrimidine dehydrogenase (DPD) is an enzyme important for metabolic
degradation of nucleotides, a process important for cellular detoxification, and the
production of free pyrimidines that can be recycled for DNA and RNA synthesis. This
activity is also important for 5-FU metabolism, as DPD mediates the initial and
rate-limiting step of 5-FU catabolism.42 DPD has been evaluated for its impact on 5-FU
resistance. High DPD levels would be expected to correlate with poor 5-FU outcome
since over 80% of 5-FU is catabolized by DPD.43 However, similar to the TP data, the
results are very ambiguous. Studies of CRC patients receiving 5-FU have shown direct,
no, and inverse associations between tumor DPD mRNA and protein levels and 5-FU
outcome.44-50 Perhaps these discrepant results may be related to the reduced DPD
expression and activity in colorectal tumors compared to normal mucosa.51-54 The
discordance may also be attributable to the non-standardized methods for measuring DPD
levels. The lower DPD expression in some colorectal tumors may make a clear
association between 5-FU resistance and DPD levels difficult.
Deoxyuridine Triphosphate
Deoxyuridine triphosphatase (DUT) is an enzyme that regulates intracellular
2V-deoxyuridine-5V-triphosphate (dUTP) levels. DUT catalyzes the conversion dUTP to
dUMP, a critical substrate for TS. Thus, DUT is believed to indirectly modulate the
effectiveness of 5-FU. Inhibition of TS by 5-FU results in accumulation of dUTP, which
serves as a stimulus for uracil misincorporation into DNA and ultimately cellular death.55
However, lack of accumulation of dUTP due to excess DUT would prevent uracil
levels have also been investigated for 5-FU resistance. Ladner and colleagues found that
response was exclusively seen in CRC patients who had low DUT (p=0.005).55Time to
progression was also significantly longer (p=0.017) in patients with low DUT; however,
there was no difference in overall survival (p=0.09). Knockdown of DUT with siRNA
caused two of three cancer cell lines to become more sensitive to TS-inhibitor FUdR.56
However, other in vitro studies indicated no significant association between altered
cellular 5-FU resistance phenotype and DUT mRNA expression.57DUT appears to be an
important target for 5-FU resistance, but its true predictive value has yet to be defined.
Uridine Cytidine Kinase 2
Uridine cytidine kinase (UCK) is an enzyme that catalyzes the phosphorylation of
dUMP to deoxyuridine diphosphate (dUDP), the initial step in the production of the
pyrimidine nucleoside triphosphates required for RNA and DNA synthesis. Since dUMP
is the critical substrate for TS, it has been hypothesized that UCK ultimately affects
5-FU-mediated cytotoxicity.58 Although UCK exists in two isoforms, UCK1 and UCK2,
only UCK2 protein and mRNA levels correlate with UCK enzymatic activity.58
Additionally, studies in rat pheochromocytoma cells and normal rat tissues suggest that
the UCK pathway is a preferred pathway for 5-FU activation.59 It was believed that
overexpression of UCK2 would result in 5-FU sensitization; however, no difference in
cytotoxicity existed in cells overexpressing UCK2 compared to cells with normal UCK2
levels.59 Both colon tumor and CRC cell lines exhibited higher levels of UCK2 than
normal tissue, other tumors, and other cell lines investigated.58The clinical relevance of
UCK2 levels with respect to 5-FU activity is currently unknown, especially in lieu of the
fact that 5-FU can be activated and mediate its cytotoxicity independent of UCK2.60
Ribonucleotide Reductase
Biologically, ribonucleotide reductase is an enzyme that produces
deoxyoligonucleotides from ribonucleotides, a step that is essential for DNA synthesis
and repair.61,62Human ribonucleotide reductase exists in two subunits RRM1 (regulatory)
and RRM2 (catalytic).61 Both subunits are necessary for enzymatic activity and are
encoded by different genes on separate chromosomes. Since RRM1 and RRM2 are both
critical for DNA synthesis, they have been evaluated by multiple studies as therapeutic
targets in various cancers. Importantly ribonucleotide reductase reduces FUDP, a 5-FU
intermediate metabolite, to FdUDP. FdUDP is subsequently dephosphorylated into
FdUMP, the active 5-FU metabolite inhibiting TS mediating 5-FU’s cytotoxicity. This
pathway operates independent of TP and may be associated with 5-FU resistance. Recent
data suggests that RRM1 and RRM2 may have conflicting roles in tumor malignancy.
Whereas overexpression of RRM1 has significant inhibited tumor growth and incidence
of metastases via PTEN (Phosphatase and Tensin Homolog), overexpression of RRM2 is
associated with increased tumor invasiveness and metastases via various oncogenes. 63-67
Additionally, RRM2 protein level is significantly associated with the incidence of
metastasis in colon cancer.68The pharmacologic significance of RRM1 and RRM2 with
respect to 5-FU resistance in mCRC is presently unknown.
Non-metastatic cells
Non-metastatic cells (NME) is an enzyme encoded by the first discovered
metastasis suppressor; presently there are over 20 known metastasis suppressor genes.69,70
NME has many biological functions including: histidine kinase activity, binding of other
Both NME1 and NME2 genes encode the hexamer nucleoside diphosphate kinase
responsible for NME kinase function. Through phosphorylation, NME catalyzes
conversion of UDP and dUDP to UTP and dUTP, respectively. Notably dUTP is a
substrate for DUT and is ultimately used in DNA synthesis. NME’s kinase activity is also
responsible for activating 5-FU metabolites FUDP and FdUDP to FUTP and FdUTP,
respectively, which is important for 5-FU cytotoxicity. Due to its biologic and
pharmacologic roles, NME has also been investigated for its role in 5-FU resistance.
Many studies have been conflicting: while some studies have found that NME1
expression (mRNA and protein) correlates with development of distant metastases, other
studies have not. 72-76 Loss of heterozygosity (LOH) of the NME1 gene occurs in up to
52% of colorectal liver metastases.77-79 However, some studies were unable to detect
LOH in NME1.80It is unknown whether NME expression and LOH has predictive utility
in mCRC patients treated with 5-FU.
Conclusions
5-Fluorouracil, a nucleotide analog, has successfully been used to treat a broad
range of malignancies including: colorectal, head and neck, and pancreatic cancers. There
are many pharmacogenomic targets within the 5-FU pharmacokinetic and
pharmacodynamic pathway that are related to its response and resistance. However, the
impact of their altered expression on 5-FU resistance in mCRC patients remains
unknown. For example, TYMS (gene) amplification has been linked to 5-FU resistance in
mCRC patients; however, data on the impact of altered protein expression of pathway
targets in this patient group is unavailable, unconvincing or conflicting. Additionally, the
impact of 5-FU exposure on the expression of the remainder of the 5-FU pathway
proteins remains unknown. Future studies are needed to investigate the expression of
these targets in 5-FU sensitive and resistant mCRC patients to determine whether altered
expression can explain 5-FU resistance.
Many studies unfortunately focus on single gene candidates. Despite gains in
knowledge of pharmacokinetic and pharmacodynamic contributors to 5-FU resistance,
the mechanistic determinants to 5-FU resistance remain unclear. With 24 proteins
comprising the 5-FU pharmacokinetic and pharmacodynamic pathway known to date, it
is quite improbable that 5-FU resistance would be wholly attributable to one target.
Indeed, it is becoming apparent that the cause of 5-FU resistance is likely multifactorial.21
Future studies must elucidate the complex interplay of pharmacokinetic and
pharmacodynamic contributors to 5-FU resistance.
Another challenge in establishing clear associations between altered protein
expression of 5-FU pathway genes and 5-FU resistance is that the qualitative means of
detecting expression is subject to inter-evaluator variability. Qualitative methods of
protein expression also suffer from lack of sensitivity, which can make finding
associations between altered target expression and 5-FU resistance particularly difficult if
the effect or sample size is small or modest. The nominal observations of traditional
pathologist-based scoring of protein expression are unable to detect subtle differences in
staining intensity. Thus, there is much disparity between the interpretations of observers
with pathologist-based scoring. Additionally, traditional immunohistochemistry means of
themselves to high-throughput, quantitative, sensitive, and unbiased tools for determining
protein expression.
Introduction to Dissertation
5-FU is the cornerstone of treatment for colorectal and other gastrointestinal
cancers, so its resistance represents a major clinical problem. Placing the 40-50%
response rate of 5-FU based therapy into context means that not only are there 50-60% of
patients who do not respond but also that the majority of those who do respond
eventually relapse. Additionally, CRC accounts for approximately 10% of cancer-related
deaths domestically. Colorectal cancer most commonly metastasizes to the liver; these
metastases are surgically removed whenever feasible. Even with surgical and medical
intervention, mCRC is disproportionately deadly with less than a 10% 5-year survival
rate.81The focus of this dissertation project is to identify molecular determinants of 5-FU
resistance in metastatic colorectal cancer. Consequently, this dissertation is separated into
two sections. Part one centers on the protein expression results determined by automated
quantitative analysis (AQUA) in mCRC patients sensitive and resistant to 5-FU. Part two
is concerned with the mechanism for differential expression amongst 5-FU sensitive and
resistant mCRC patients.
By carefully phenotyping patients who have consented to have their liver
metastases banked at the UNC Tissue Procurement Facility, patients were stratified based
on their 5-FU exposure. A comprehensive review of the clinical phenotyping database of
mCRC patients and how it was used to explore 5-FU resistance is provided in Chapter 2.
The purpose of Chapter 3 is to identify 5-FU pathway targets with altered expression
associated with 5-FU resistance in metastatic colorectal cancer patients using AQUA.
Since protein expression will be quantified for multiple targets, this approach overcomes
the aforementioned challenges of the single target-approach and the technical limitations
of qualitative protein expression determination. Chapter 4 will communicate the
mechanisms by which the 5-FU pathway targets associated with 5-FU exposure cause
resistance. Lastly, the results of this research effort will be placed into context of the
existing literature, and recommendations for a future research agenda that will advance
Figure 1.1
Figure 1.15-Fluorouracil Structure
Figure 1.2
Figure 1.3
Figure 1.35-Fluorouracil Pharmacokinentic and Pharmacodynamic Pathway. T.E. Klein, J.T. Chang, M.K. Cho, K.L. et al."Integrating Genotype and Phenotype Information: An Overview of the PharmGKB Project" (220k PDF),The Pharmacogenomics Journal(2001) 1, 167-170.
References
1. Yang R, Niepel M, Mitchison T, Sorger P. Dissecting variability in responses to cancer chemotherapy through systems pharmacology. Clin Pharmacol Ther. Jul 2010;88(1):34-38.
2. Giacchetti S, Perpoint B, Zidani R, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil-leucovorin as first-line treatment of metastatic colorectal cancer.J Clin Oncol.Jan 2000;18(1):136-147.
3. Douillard J, Cunningham D, Roth A, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial.Lancet.Mar 2000;355(9209):1041-1047.
4. Wolpin BM, Mayer RJ. Systemic treatment of colorectal cancer.
Gastroenterology.May 2008;134(5):1296-1310.
5. Evans W, Relling M. Pharmacogenomics: translating functional genomics into rational therapeutics.Science.Oct 1999;286(5439):487-491.
6. Evans W, Johnson J. Pharmacogenomics: the inherited basis for interindividual differences in drug response.Annu Rev Genomics Hum Genet.2001;2:9-39.
7. McLeod H, Evans W. Pharmacogenomics: unlocking the human genome for better drug therapy.Annu Rev Pharmacol Toxicol.2001;41:101-121.
8. Evans W, McLeod H. Pharmacogenomics--drug disposition, drug targets, and side effects.N Engl J Med.Feb 2003;348(6):538-549.
9. Kalow W, Tang B, Endrenyi L. Hypothesis: comparisons of inter- and intra-individual variations can substitute for twin studies in drug research.
Pharmacogenetics.Aug 1998;8(4):283-289.
10. Marsh S, McLeod H. Cancer pharmacogenetics. Br J Cancer. Jan 2004;90(1):8-11.
11. Watters J, McLeod H. Cancer pharmacogenomics: current and future applications.
Biochim Biophys Acta.Mar 2003;1603(2):99-111.
12. American Cancer Society. Cancer Facts & Figures2010.
13. de Gramont A, Vignoud J, Tournigand C, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer.Eur J Cancer.Feb 1997;33(2):214-219.
15. Moertel C. Chemotherapy for colorectal cancer. N Engl J Med. Apr 1994;330(16):1136-1142.
16. Society AC. Cancer Facts & Figures2010.
17. Sobrero A, Guglielmi A, Grossi F, Puglisi F, Aschele C. Mechanism of action of fluoropyrimidines: relevance to the new developments in colorectal cancer chemotherapy.Semin Oncol.Oct 2000;27(5 Suppl 10):72-77.
18. Parker W, Cheng Y. Metabolism and mechanism of action of 5-fluorouracil.
Pharmacol Ther.1990;48(3):381-395.
19. He Y, Wei W, Zhang X, et al. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in Chinese cancer patients. J Clin Pharm Ther. Jun 2008;33(3):307-314.
20. Ahnen D, Feigl P, Quan G, et al. Ki-ras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study.
Cancer Res.Mar 1998;58(6):1149-1158.
21. Peters G, Backus H, Freemantle S, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta. Jul 2002;1587(2-3):194-205.
22. Etienne M, Chazal M, Laurent-Puig P, et al. Prognostic value of tumoral thymidylate synthase and p53 in metastatic colorectal cancer patients receiving fluorouracil-based chemotherapy: phenotypic and genotypic analyses. J Clin Oncol.Jun 2002;20(12):2832-2843.
23. Liang J, Huang K, Cheng Y, et al. P53 overexpression predicts poor chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV colorectal cancers after palliative bowel resection. Int J Cancer. Feb 2002;97(4):451-457.
24. Villafranca E, Okruzhnov Y, Dominguez M, et al. Polymorphisms of the repeated sequences in the enhancer region of the thymidylate synthase gene promoter may predict downstaging after preoperative chemoradiation in rectal cancer. J Clin Oncol.Mar 2001;19(6):1779-1786.
25. Lee Y, Chen Y, Chang L, Johnson L. Inhibition of mouse thymidylate synthase promoter activity by the wild-type p53 tumor suppressor protein. Exp Cell Res.
Aug 1997;234(2):270-276.
26. Carreras C, Santi D. The catalytic mechanism and structure of thymidylate synthase.Annu Rev Biochem.1995;64:721-762.
27. Marsh S, McLeod H. Thymidylate synthase pharmacogenetics in colorectal cancer.Clin Colorectal Cancer.Nov 2001;1(3):175-178; discussion 179-181.
28. Kawakami K, Omura K, Kanehira E, Watanabe Y. Polymorphic tandem repeats in the thymidylate synthase gene is associated with its protein expression in human gastrointestinal cancers.Anticancer Res.1999 Jul-Aug 1999;19(4B):3249-3252.
29. Kawakami K, Salonga D, Park J, et al. Different lengths of a polymorphic repeat sequence in the thymidylate synthase gene affect translational efficiency but not its gene expression.Clin Cancer Res.Dec 2001;7(12):4096-4101.
30. Horie N, Aiba H, Oguro K, Hojo H, Takeishi K. Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5'-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct. Jun 1995;20(3):191-197.
31. Qiu L, Tang Q, Bai J, et al. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine-based chemotherapy: evidence from 24 studies. Int J Cancer. Nov 2008;123(10):2384-2389.
32. Aschele C, Debernardis D, Casazza S, et al. Immunohistochemical quantitation of thymidylate synthase expression in colorectal cancer metastases predicts for clinical outcome to fluorouracil-based chemotherapy. J Clin Oncol. Jun 1999;17(6):1760-1770.
33. Popat S, Matakidou A, Houlston R. Thymidylate synthase expression and prognosis in colorectal cancer: a systematic review and meta-analysis. J Clin Oncol.Feb 2004;22(3):529-536.
34. Wang TL, Diaz LA, Romans K, et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proceedings of the National Academy of Sciences of the United States of America.2004;101(9):3089-3094.
35. FRIEDKIN M, ROBERTS D. The enzymatic synthesis of nucleosides. I.
Thymidine phosphorylase in mammalian tissue. J Biol Chem. Mar
1954;207(1):245-256.
36. Iltzsch M, el Kouni M, Cha S. Kinetic studies of thymidine phosphorylase from mouse liver.Biochemistry.Nov 1985;24(24):6799-6807.
37. Zimmerman M, Seidenberg J. Deoxyribosyl Transfer. I. Thymidine
Phosphorylase and Nucleoside Deoxyribosyltransferase in Normal and Malignant Tissues.J Biol Chem.Aug 1964;239:2618-2621.
39. Ichikawa W, Uetake H, Shirota Y, et al. Both gene expression for orotate phosphoribosyltransferase and its ratio to dihydropyrimidine dehydrogenase influence outcome following fluoropyrimidine-based chemotherapy for metastatic colorectal cancer.Br J Cancer.Oct 2003;89(8):1486-1492.
40. Hasegawa S, Seike K, Koda K, et al. Thymidine phosphorylase expression and efficacy of adjuvant doxifluridine in advanced colorectal cancer patients. Oncol Rep.Apr 2005;13(4):621-626.
41. Nakayama Y, Inoue Y, Nagashima N, et al. Expression levels of thymidine phosphorylase (TP) and dihydropyrimidine dehydrogenase (DPD) in patients with gastrointestinal cancer.Anticancer Res.2005 Nov-Dec 2005;25(6A):3755-3761.
42. Diasio R, Lu Z. Dihydropyrimidine dehydrogenase activity and fluorouracil chemotherapy.J Clin Oncol.Nov 1994;12(11):2239-2242.
43. Heggie G, Sommadossi J, Cross D, Huster W, Diasio R. Clinical
pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile.
Cancer Res.Apr 1987;47(8):2203-2206.
44. Ichikawa W, Uetake H, Shirota Y, et al. Combination of dihydropyrimidine dehydrogenase and thymidylate synthase gene expressions in primary tumors as predictive parameters for the efficacy of fluoropyrimidine-based chemotherapy for metastatic colorectal cancer.Clin Cancer Res.Feb 2003;9(2):786-791.
45. Salonga D, Danenberg K, Johnson M, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase.Clin Cancer Res.Apr 2000;6(4):1322-1327.
46. Kornmann M, Link K, Galuba I, et al. Association of time to recurrence with thymidylate synthase and dihydropyrimidine dehydrogenase mRNA expression in stage II and III colorectal cancer. J Gastrointest Surg. 2002 May-Jun 2002;6(3):331-337.
47. Kornmann M, Schwabe W, Sander S, et al. Thymidylate synthase and
dihydropyrimidine dehydrogenase mRNA expression levels: predictors for survival in colorectal cancer patients receiving adjuvant 5-fluorouracil. Clin Cancer Res.Sep 2003;9(11):4116-4124.
48. Ikeguchi M, Makino M, Kaibara N. Thymidine phosphorylase and
dihydropyrimidine dehydrogenase activity in colorectal carcinoma and patients prognosis.Langenbecks Arch Surg.Oct 2002;387(5-6):240-245.
49. Ciaparrone M, Quirino M, Schinzari G, et al. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil.
Oncology.2006;70(5):366-377.
50. Koopman M, Venderbosch S, van Tinteren H, et al. Predictive and prognostic markers for the outcome of chemotherapy in advanced colorectal cancer, a retrospective analysis of the phase III randomised CAIRO study. Eur J Cancer.
Jul 2009;45(11):1999-2006.
51. McLeod H, Sludden J, Murray G, et al. Characterization of dihydropyrimidine dehydrogenase in human colorectal tumours.Br J Cancer.1998;77(3):461-465.
52. Johnston S, Ridge S, Cassidy J, McLeod H. Regulation of dihydropyrimidine dehydrogenase in colorectal cancer.Clin Cancer Res.Sep 1999;5(9):2566-2570.
53. Uetake H, Ichikawa W, Takechi T, Fukushima M, Nihei Z, Sugihara K. Relationship between intratumoral dihydropyrimidine dehydrogenase activity and gene expression in human colorectal cancer. Clin Cancer Res. Oct 1999;5(10):2836-2839.
54. Yamashita K, Mikami Y, Ikeda M, et al. Gender differences in the
dihydropyrimidine dehydrogenase expression of colorectal cancers. Cancer Lett.
Dec 2002;188(1-2):231-236.
55. Ladner R, Lynch F, Groshen S, et al. dUTP nucleotidohydrolase isoform expression in normal and neoplastic tissues: association with survival and response to 5-fluorouracil in colorectal cancer. Cancer Res. Jul 2000;60(13):3493-3503.
56. Koehler S, Ladner R. Small interfering RNA-mediated suppression of dUTPase sensitizes cancer cell lines to thymidylate synthase inhibition. Mol Pharmacol.
Sep 2004;66(3):620-626.
57. Schmidt W, Kalipciyan M, Dornstauder E, et al. Dissecting progressive stages of 5-fluorouracil resistance in vitro using RNA expression profiling. Int J Cancer.
Nov 2004;112(2):200-212.
58. Shimamoto Y, Koizumi K, Okabe H, et al. Sensitivity of human cancer cells to the new anticancer ribo-nucleoside TAS-106 is correlated with expression of uridine-cytidine kinase 2.Jpn J Cancer Res.Jul 2002;93(7):825-833.
59. Mascia L, Ipata P. Activation pathways of 5-fluorouracil in rat organs and in PC12 cells.Biochem Pharmacol.Jul 2001;62(2):213-218.
60. Van Rompay A, Johansson M, Karlsson A. Substrate specificity and
phosphorylation of antiviral and anticancer nucleoside analogues by human deoxyribonucleoside kinases and ribonucleoside kinases. Pharmacol Ther. Nov 2003;100(2):119-139.
62. Nordlund P, Reichard P. Ribonucleotide reductases. Annu Rev Biochem.
2006;75:681-706.
63. Gautam A, Li Z, Bepler G. RRM1-induced metastasis suppression through PTEN-regulated pathways.Oncogene.Apr 2003;22(14):2135-2142.
64. Bepler G, Sharma S, Cantor A, et al. RRM1 and PTEN as prognostic parameters for overall and disease-free survival in patients with non-small-cell lung cancer.J Clin Oncol.May 2004;22(10):1878-1885.
65. Zhou B, Tsai P, Ker R, et al. Overexpression of transfected human ribonucleotide reductase M2 subunit in human cancer cells enhances their invasive potential.
Clin Exp Metastasis.Jan 1998;16(1):43-49.
66. Duxbury M, Ito H, Zinner M, Ashley S, Whang E. RNA interference targeting the M2 subunit of ribonucleotide reductase enhances pancreatic adenocarcinoma chemosensitivity to gemcitabine.Oncogene.Feb 2004;23(8):1539-1548.
67. Liu X, Zhou B, Xue L, et al. Metastasis-suppressing potential of ribonucleotide reductase small subunit p53R2 in human cancer cells. Clin Cancer Res. Nov 2006;12(21):6337-6344.
68. Liu X, Zhou B, Xue L, et al. Ribonucleotide reductase subunits M2 and p53R2 are potential biomarkers for metastasis of colon cancer. Clin Colorectal Cancer.
Jan 2007;6(5):374-381.
69. Steeg P, Horak C, Miller K. Clinical-translational approaches to the Nm23-H1 metastasis suppressor.Clin Cancer Res.Aug 2008;14(16):5006-5012.
70. Steeg P, Bevilacqua G, Kopper L, et al. Evidence for a novel gene associated with low tumor metastatic potential.J Natl Cancer Inst.Apr 1988;80(3):200-204.
71. Besant P, Attwood P. Mammalian histidine kinases. Biochim Biophys Acta. Dec 2005;1754(1-2):281-290.
72. Ayhan A, Yasui W, Yokozaki H, Kitadai Y, Tahara E. Reduced expression of nm23 protein is associated with advanced tumor stage and distant metastases in human colorectal carcinomas. Virchows Arch B Cell Pathol Incl Mol Pathol.
1993;63(4):213-218.
73. Myeroff L, Markowitz S. Increased nm23-H1 and nm23-H2 messenger RNA expression and absence of mutations in colon carcinomas of low and high metastatic potential.J Natl Cancer Inst.Jan 1993;85(2):147-152.
74. Heide I, Thiede C, Poppe K, de Kant E, Huhn D, Rochlitz C. Expression and mutational analysis of Nm23-H1 in liver metastases of colorectal cancer. Br J Cancer.Dec 1994;70(6):1267-1271.
75. Yamaguchi A, Urano T, Fushida S, et al. Inverse association of nm23-H1 expression by colorectal cancer with liver metastasis. Br J Cancer. Nov 1993;68(5):1020-1024.
76. Ichikawa W. Positive relationship between expression of CD44 and hepatic metastases in colorectal cancer.Pathobiology.1994;62(4):172-179.
77. Cohn K, Wang F, Desoto-LaPaix F, et al. Association of nm23-H1 allelic deletions with distant metastases in colorectal carcinoma. Lancet. Sep 1991;338(8769):722-724.
78. Leone A, McBride O, Weston A, et al. Somatic allelic deletion of nm23 in human cancer.Cancer Res.May 1991;51(9):2490-2493.
79. Wang L, Patel U, Ghosh L, Chen H, Banerjee S. Mutation in the nm23 gene is associated with metastasis in colorectal cancer. Cancer Res.Feb 1993;53(4):717-720.
80. Iacopetta B, DiGrandi S, Dix B, Haig C, Soong R, House A. Loss of heterozygosity of tumour suppressor gene loci in human colorectal carcinoma.
Eur J Cancer.1994;30A(5):664-670.
Chapter 2
Abstract
Introduction: Colorectal cancer is the third most common non-cutaneous
malignancy in the United States and the third most frequent cause of cancer-related
deaths. The presence of distant metastases is particularly problematic, as the 5-year
survival is 5-8%, despite 5-FU-based therapy. The primary objectives of this
comprehensive review are to detail how data were collected to develop a clinical database
resource that was used in subsequent studies investigating 5-FU resistance and to
characterize the mCRC patients comprising this clinical cohort.
Methods: Electronic and paper patient records from North Carolina Memorial
Hospital and community hospitals within the region were reviewed to obtain pertinent
dates, chemotherapy regimens, treatment outcomes, and baseline disease characteristics.
The Social Security Death Index was also queried to obtain survival data when
unavailable from patient charts. Patients were only included if they had mCRC, had their
colorectal liver metastases banked at North Carolina Memorial Hospital, and could be
phenotyped with respect 5-FU exposure.
Results:Chart reviews for 140 patients were performed; one-hundred and twenty
one of these patients had mCRC and could be phenotyped for 5-FU exposure.
Sixty-seven of the 121 (55%) were exposed to 5-FU, and the remaining patients (54/121) were
unexposed. There were no significant differences with respect to baseline characteristics
with the exception of age at diagnosis of liver metastases (p=0.01). From surgical
resection of colorectal liver metastases, the median overall survival was 885 days and
Conclusions: Patients were well-characterized with respect to 5-FU exposure.
The 5-FU exposed and unexposed groups did not significantly differ with respect to
demographical data. This patient cohort is well-suited to investigate clinical resistance.
Introduction
Colorectal cancer (CRC) is the third most common non-cutaneous malignancy in
the United States and the third most frequent cause of cancer-related deaths.1In 2010, an estimated 142,570 cases of CRC will be diagnosed and 51,370 people will die from this
disease.1 Despite the substantial progress that has been made in recent years, CRC
remains a major clinical problem. The most critical prognostic factor for CRC is
pathologic stage. The American Joint Committee on Cancer (AJCC) is the most common
staging criteria used.2,3 There is an inverse relationship between CRC patients’ AJCC
stage and their expected prognosis. As AJCC staging increases from stage 1 (early,
localized primary tumor) to stage 4 (distant metastases), the 5-year survival decreases
from 90% to less than 10%. Note that 30-40% of patients with localized CRC eventually
develop distant metastases and nearly 20% of patients present initially with metastatic
disease. Surgical resection of metastases in patients with a few metastases confined to a
specific organ has been shown to be curative in some patients.4However, the majority of mCRC patients succumb to the disease.
Perhaps, the most insidious problem for mCRC patients is chemotherapy
resistance, particularly 5-FU resistance since it is part of the therapeutic backbone of
mCRC treatment. Although patients will initially respond to 5-FU based therapy, most
develop recurrences. These patients will typically receive a new regimen that contains
5-FU, and while most won’t respond, the few who do respond do so in an unpredictable
manner. The lack of validated predictive markers for 5-FU response results in patients
being exposed to unnecessary toxicity and a regimen with marginal chances of success.
FU resistance because they result in altered expression and copy number aberrations in
5-FU pathway proteins, mRNA, and genes. However, the results from clinical studies
evaluating this resistance have been conflicting, and the studies are often underpowered
and have limited external validity.5-8
It is critical that future studies not ignore the enriched phenotyping strategy.
Clearly, the aforementioned data suggests that not all mCRC patients have the same
response to 5-FU. The enriched strategy attributes differences in patient outcomes to
underlying molecular differences. These molecular differences can be identified by
comparing patients most likely to possess high concentrations of variation associated
with resistance to those predisposed to be sensitive. An underlying presumption is the
tumors remaining despite adequate 5-FU-based treatment are clinically-resistant to 5-FU.
Giving such patients more 5-FU-based regimens would be expected to provide limited
therapeutic utility while simultaneously exposing them to increased chemotherapy-related
toxicity. Differences in gene copy number and expression are expected between 5-FU
exposed and unexposed patients, as they represent two distinct mCRC patient
populations: resistant and sensitive, respectively. Moreover, stratifying patients based on
their 5-FU exposure capitalizes on the fact that exposed tumors are enriched with
variation resulting in resistance that is not present in unexposed samples. When such
variation has previously been identified through enrichment, it has been unambiguously
implicated in 5-FU resistance.9,10 As such, exposed tumors can be further exploited as a valuable discovery tool for investigating 5-FU resistance (Figure 2.1). Functional
validation of these targets with knockdown studies would be expected to modulate the
5-FU resistance phenotype in CRC cells. By methodically characterizing mCRC patients
with respect to clinical resistance (5-FU exposure), these molecular etiologies underlying
5-FU resistance can be systematically dissected. Thus, while it is expected that exposed
and unexposed patients will not significantly differ with regards to their baseline disease
characteristics and demographics, the fundamental molecular heterogeneity between
these two populations as a consequence of clinical resistance to 5-FU (exposure) serves
as the basis for stratification and is the focus of this dissertation. Since confounding
variables cannot be controlled for in this dataset, survival data will not be compared
between exposed and unexposed. Accordingly, this chapter details how mCRC patients
were phenotyped based on their 5-FU exposure and other pertinent clinical information
and how these data were pooled to create the clinical database used for investigating
Methods
Data Sources
Patient medical records were retrieved from colorectal cancer patients who had
undergone surgical resection of their liver metastases at North Carolina Memorial
Hospital between 1998 and February 2009. Paper and electronic charts from North
Carolina Memorial Hospital and community hospitals within the region were reviewed.
Survival data was also obtained from the Social Security Death Index when survival was
unspecified in patient medical records. Examination of clinical records was approved by
an institutional review board and was performed in adherence to Health Insurance
Portability and Accountability Act regulations. A waiver of informed consent was
granted.
Eligibility Criteria
Patients were included in this analysis if they met the following criteria: (1) had
pathological diagnosis of metastatic colorectal cancer; (2) had colorectal liver metastases
banked at the University of North Carolina Translational Pathology Laboratory (formally
known as the Tissue Procurement Facility); (3) their exposure or lack of exposure to
5-FU for the immediate 6 months preceding resection of their liver metastases could be
verified. Patients with ambiguous 5-FU exposure phenotype were excluded, such as
patients where it is unclear whether they received 5-FU in the six months before their
liver resection. All exposed patients received at least 4 weeks of 5-FU-based
chemotherapy.
Data Extraction
The following data were extracted for each patient: first and last name, date of
birth, gender, race, ethnicity, diagnosis data of primary and metastatic lesions, disease
stage as determined per AJCC criteria, number and location of metastases, surgeon who
performed resection of liver metastases, date of resection of liver metastases,
radiofrequency ablation therapy, chemotherapy regimen within six months prior to
resection of liver metastases, post surgery chemotherapy (regimen not documented), date
of last follow up or death, date of recurrence or last pronouncement of no evidence of
disease, height, weight, dosage reduction (when applicable), and referring physician. The
data extraction was performed primarily by Roshawn Watson with some assistance from
Michael Hudson and Joan Van Ord. Disagreements about data were resolved by
consensus between the author, Christine Walko, and Bert O’Neil.
Statistical Analyses
All univariate statistical analyses were performed with SAS software, version 9
(SAS Corp, Cary, NC). P<0.05 was considered statistically significant. Overall survival
was calculated from both a) the date of the surgical excision of the metastases and b)
from the diagnosis date of metastatic colorectal cancer to the date of death or last
follow-up. Survival was computed by the Kaplan–Meier method. Disease-free survival was
determined from the date of the surgical excision of the metastasis to the date of
recurrence or last declaration of no evidence of disease. Data were censored when
Results
Demographics
The study population comprised of a total of 140 patients with colorectal liver
metastases (Table 2.1). Nineteen patients eventually were excluded because they had
inadequate documentation of presurgical chemotherapy, unconfirmed histology,
noncolorectal primary tumors, or inadequate tissue available. The remaining 121 mCRC
patients consisted of sixty-seven 5-FU exposed and fifty-four unexposed. Their ages at
the diagnosis of their liver metastases ranged between thirty-eight and eighty-six (mean
age of sixty-one). This cohort was comprised of sixty-one male patients and sixty female
patients. The racial distribution of patients in this trial was: White, n = 90; Black, n =27;
and Asian/Other, n=4. Other patient characteristics are listed in Table 2.1. The mean age
at diagnosis in the unexposed group was 6 years greater than in the 5-FU exposed group
(p=0.01); no other statistically significant differences in demographic data were observed
between the two groups.
Survival
The median follow-up period of 434 days (95%confidence interval [CI], 335 to
533 days) for the 121 patients analyzed in this study. The median overall survival time
was 885 days from surgical resection of liver metastases and 1308 days from diagnosis of
liver metastases (Figures 2.2 and 2.3, respectively). The median disease-free survival
time was 294 days from surgical resection of liver metastases (Figure 2.4).
Chemotherapy
Sixty-seven mCRC patients received 5-FU containing regimens within the six
months preceding the resection of their liver metastases. The other components of the
regimens were also documented (see Table 2.1). Thirty-one (46%) patients received
oxaliplatin as part of their regimen. Twenty-four (36%) patients received bevacizumab.
Fourteen (21%) patients received irinotecan. One (1.4%) patient received cetuximab.
Another patient was prescribed cetuximab but immediately discontinued usage after
experiencing anaphylaxis during the administration of initial dose. Clearly, these
chemotherapy regimens reflect the current diversity and complexity in the treatment of
Discussion
Drug resistance is a primary reason for treatment failure and death in cancer
patients. The causes of drug resistance in malignancies are relatively unknown, despite
numerous investigations into the putative mechanisms. Recent data suggests that by
comparing molecular differences between 5-FU exposed and unexposed patients, the
underlying molecular contributors to 5-FU can be elucidated.9 The presumptive hypothesis is that the residual tumor post-5-FU exposure is enriched with somatic
changes causing the resistance whereas the clinically-sensitive tumors would respond to
an adequate course of 5-FU. A relevant in vivo example is the genetic alterations that
occur in the BCR/ABL gene in chronic myelogenous leukemia patients during Gleevac
treatment. These alterations unequivocally suggest that the BCR/ABL gene plays a
significant role in Gleevac resistance.11,12Similarly, changes in 5-FU targets that are only
evident tumors already clinically resistant (residual tumor) to the 5-FU are
unambiguously associated with 5-FU resistance.
Using this model as the basis investigate drug resistance, a clinical cohort was
developed consisting of both 5-FU exposed and unexposed patients. The results
demonstrate that the exposed and unexposed patients do not significantly differ in
baseline disease characteristics and demographics with one minor exception: the mean
age of the unexposed patients was 6 years greater than exposed patients. The implications
are that differences between the exposed and unexposed will be attributable to 5-FU
exposure, clinical resistance to 5-FU, and will have relevance to the clinical management
of mCRC.
A strength of this design is that it allows for clinical resistance to be evaluated
using in vivosamples from well-characterized patients. Additionally, the cohort consists
of patients from both community and university settings, which means data from this
cohort reflects real-world 5-FU resistance in mCRC patients. The fact that all included
patients had resectable disease is also notable. Historically, only 10-20% of patients with
colorectal liver metastases are resection candidates, and this cohort is among the largest
studies investigating 5-FU resistance in this patient cohort.
There are also some important considerations when evaluating data from this
cohort. First, by excluding non-resection candidates, the study design screens out patients
with the most aggressive disease. Resection candidates typically have higher performance
status and more contained liver disease than non-resection candidates. In contrast to the
overall 5-year survival of 5-8% seen in mCRC patients, survival in patients amendable to
resection is significantly higher at 40-50%.13-17 Nonetheless, resection candidates have significant resistance considering that they are still prone to numerous recurrences and
only 50-60% survive at 5 years. Second, chemotherapy prior to six months before
resection is not documented, so some patients in unexposed group likely received 5-FU
before this point. There was no precedent dictating when data collection started, so by
consensus of medical oncologist and gastroenterologists, six months preceding resections
was agreed to be reasonable. Third, patients who were exposed to 5-FU were often
treated with other agents, so their influence cannot be separated from 5-FU. The patient
cohort examined in the Wang et al study, were treated with only 5-FU, as this was the
and survival, so 5-FU monotherapy is no longer gold standard. This is a common
consideration in any contemporary in vivo study investigating 5-FU resistance and
certainly not limited to this analysis.
It is tempting to directly contrast survival of the unexposed and exposed patients;
however, there are several limitations to the validity of such a comparison. First, survival
would be influenced by post-surgical access to care and patient preferences. Since both of
these were not assessed, they would be important potential confounders to a survival
analysis. Second, the drugs included in the post-surgical adjuvant therapy, duration of
therapy, and additional procedures were also not included in the database, yet they can all
influence survival. Lastly, the presence of extrahepatic disease in mCRC patients has
repeatedly been shown to decrease survival, so a meaningful survival analysis should also
be adjusted to reflect these data.18-23 Acquisition of the aforementioned data was beyond the scope and resources for this project. Thus, a comparison of survival based on 5-FU
phenotype, without controlling for these confounders, would make interpretation
ambiguous and was never planned.
The primary conclusions drawn from this review are that the exposed and
unexposed patient groups do not significantly differ with respect to baseline disease
characteristics and demographics. Also, the included patients are a distinct subset of
mCRC patients who were eligible for resection. In aggregate, the data collected on this
clinical cohort will be critical to determining molecular contributors to 5-FU resistance in
mCRC patients. Based on the available data and samples, the database has lent itself to
numerous types of analyses including expression analysis (Chapters 3 and 4), copy
number analysis (Chapter 4). Overall, stratifying patients on the basis of 5-FU exposure
Figure 2.1
Figure 2.1Model for an ‘enriched’ approach for genomic discovery. The hypothesis is that residual tumor after chemotherapy treatment is clinically resistant and thereby harbors functionally important somatic alterations.
Figure 2.2
Overall Survival from Surgery
0 500 1000 1500 2000 2500
0 50
100 KM stat
Exp.
Days Survived
P
er
ce
n
t
S
u
rv
iv
al
Figure 2.3
Diseaese-Free Survival from
Diagnosis
0 1000 2000 3000
0 50 100 150
KM stat Exp.
Days Survived
Figure 2.3Kaplan-Meier curve depicting the disease-free survival of the entire clinical cohort from diagnosis of liver metastases.
Figure 2.4
Disease-Free Survival from
Surgery
0 500 1000 1500 2000 2500
0 50 100 150
KM stat Exp.
Days Survived
P
er
ce
n
t
S
u
rv
iv
al
Table 2.1
Characteristic Unexposed 5-FU Exposed P Gender
Male 31 (57%) 30 (45%)
Female 23 (43%) 37 (55%) NS
Race/Ethnicity
White 38 (70%) 52 (78%)
Black 13 (24%) 14 (21%)
Asian/Other 3 (6%) 1 (2%)
Hispanic 0 1 (2%) NS
Age (mean) at diagnosis1 65 (range 42 – 86) 59 (range 38– 85) 0.01
Tumor
1 5 2
2 8 5
3 22 42
4 3 2 NS
Nodes
0 28 26
1 15 19
2 9 16 NS
Metastases2
0 32 29
1 18 35 NS
Initial No. of Metastases
1 28 26
2 11 12
>2 7 15
Range 1-6 1-14
Median 1 1.5 NS
Neoadjuvant chemotherapy (%)3
5-Flourouracil-containing regimen 67 (100) 5-Fluorouracil-Oxaliplatin regimen 31 (46) 5-Fluorouracil-Irinotecan regimen 14 (21) 5-Fluorouracil-Bevacizumab regimen 24 (36)
1Diagnosis refers to diagnosis of liver metastasis
2TNM criteria was taken at diagnosis of colorectal cancer not diagnosis of liver
metastases
3Regimens included more than one agent
Table 2.1Clinical cohort demographics and baseline disease characteristics.
References
1. American Cancer Society. Cancer Facts & Figures2010.
2. Greene F, Stewart A, Norton H. A new TNM staging strategy for node-positive (stage III) colon cancer: an analysis of 50,042 patients. Ann Surg. Oct 2002;236(4):416-421; discussion 421.
3. Greene F, Stewart A, Norton H. New tumor-node-metastasis staging strategy for node-positive (stage III) rectal cancer: an analysis. J Clin Oncol. May 2004;22(10):1778-1784.
4. Tomlinson J, Jarnagin W, DeMatteo R, et al. Actual 10-year survival after resection of colorectal liver metastases defines cure. J Clin Oncol. Oct 2007;25(29):4575-4580.
5. Peters G, Backus H, Freemantle S, et al. Induction of thymidylate synthase as a 5-fluorouracil resistance mechanism. Biochim Biophys Acta. Jul 2002;1587(2-3):194-205.
6. Ciccolini J, Evrard A, Cuq P. Thymidine phosphorylase and fluoropyrimidines efficacy: a Jekyll and Hyde story. Curr Med Chem Anticancer Agents. Mar 2004;4(2):71-81.
7. van Kuilenburg A. Dihydropyrimidine dehydrogenase and the efficacy and
toxicity of 5-fluorouracil.Eur J Cancer.May 2004;40(7):939-950.
8. Uetake H, Ichikawa W, Takechi T, Fukushima M, Nihei Z, Sugihara K.
Relationship between intratumoral dihydropyrimidine dehydrogenase activity and
gene expression in human colorectal cancer. Clin Cancer Res. Oct
1999;5(10):2836-2839.
9. Wang TL, Diaz LA, Romans K, et al. Digital karyotyping identifies thymidylate synthase amplification as a mechanism of resistance to 5-fluorouracil in metastatic colorectal cancer patients. Proceedings of the National Academy of Sciences of the United States of America.2004;101(9):3089-3094.
10. Watson R, Muhale F, Thorne L, et al. Amplification of thymidylate synthetase in metastatic colorectal cancer patients pretreated with 5-fluorouracil-based chemotherapy.Eur J Cancer.Aug 2010.
11. Gorre M, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. Aug 2001;293(5531):876-880.