GTPases-activated Signalling Cascades in the
Developing Neuron
Monica Alexandra Domingues Serrador da Mota
A thesis submitted for the Degree of Doctor of Philosophy
Eisai London Research Laboratories,
University College London
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U642241
Published by ProQuest LLC(2015). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
I w ould like to express m y gratitude to m y supervisor. C hantai Bazenet, for her expert guidance, help and encouragem ent th roughout the course of this work.
My special thanks to Joanne Taylor for her advice and critical reading of this thesis.
I w ould also like to thank m y office m ates. Bina Shah and Jacqueline Catchick and everyone at Eisai for creating a friendly and enjoyable e n v iro n m e n t in w hich to work.
Ab s t r a c t... 2
Ac k n o w l e d g m e n t s... 3
Ta b l eo f Co n t e n t s... 4
Ta b l eo f Fig u r e s...9
Ab b r e v ia t io n s...11
1. In t r o d u c t i o n... 1 2 1.1 Apoptosis... 12
1.1.1 History of ceU death...12
1.1.2 Apoptosis and necrosis... 13
1.2 The role of apoptosis...16
1.2.1 Physiological apoptosis... 16
1.2.2 Pathological apoptosis... 17
1.3 Apoptosis in the nervous system... 18
1.3.1 Developmental apoptosis... 18
1.3.2 Pathological apoptosis... 19
1.4 Regulation of cell death... 21
1.4.1 Balance between life and death...21
1.4.1.1 Survival pathways... 21
1.4.1.2 Apoptotic pathways... 24
1.4.2 Signal transduction players of apoptosis... 26
1.4.2.1 Programmed cell death in the developing nematode worm...26
1.4.2.2 The Bcl-2 fam ily... 27
1.4.2.3 Caspases...30
1.4.2.4 Involvement of the JNK pathway in transcription-dependent death... 33
1.4.2.5 The JNK family of kinases...34
1.4.2.6 Substrates of the JNKs...36
1.4.2.7 Targets of c-Jun...38
1.5 Upstream activators of the JNK pathway...39
1.5.1 The p75 neurotrophin receptor... 40
1.5.2 The Rho family of GTPases...43
1.5.2.1 Primary structure... 43
1.5.2.3 Biological functions... 50
1.5.2.3.1 Actin Rearrangements... 50
1.5.2.3.2 JNK and p38 MAP kinase pathways and transcriptional activation... 51
1.5.2.3.3 Regulation of apoptosis...52
1.5.2.3.4 Role in disease... 53
1.5.3 MARK cascades...53
1.5.3.1 PA K s... 55
1.5.3.1.1 Structure...55
1.5.3.1.2 Regulation... 56
1.5.3.1.3 Biological functions... 57
1.5.3.1.4 Role in disease... 59
1.5.3.2 A S K l... 60
1.5.3.2.1 Structure...60
1.5.3.2.2 Biological functions and regulation...60
1.5.3.2.3 Role in disease... 63
1.5.3.3 MLK3... 63
1.5.3.3.1 Structure...63
1.5.3.3.2 Function and Regulation... 64
1.6 Sympathetic and sympathetic-hke neurons as a model of neuronal cell death... 65
1.6.1 Superior cervical ganglion neurons... 65
1.6.2 PC12 cells...67
1.7 Thesis aim s... 68
2. Ma t e r ia l sa n d Me i h o d s... 6 9 2.1 Materials... 69
2.1.1 Chemicals, equipment and suppliers...69
2.1.2 Antibodies... 73
2.1.3 Bacterial strains... 75
2.1.4 Plasmids... 75
2.1.5 Stock solutions, media and buffers...75
2.2 Methods... 81
2.2.1 Molecular b iology...81
2.2.1.1 General...81
2.2.1.1.1 Phenokchloroform extraction... 81
2.2.1.1.2 Ethanol or Isopropanol precipitation... 81
2.2.1.5 Ligation of D N A ...82
2.2.1.6 Preparation of competent E. coli...83
2.2.1.7 Transformation of competent bacteria... 83
2.2.1.8 Screening for the presence of recombinant plasmid... 83
2.2.1.9 Plasmid preparation...84
2.2.1.9.1 Small scale (Miniprep)...84
2.2.1.9.2 Large scale (Maxiprep)...84
2.2.1.10 Preparation of total RNA from SCG neurons...86
2.2.1.11 Reverse transcriptase-polymerase chain reaction (RT-PCR)... 86
2.2.1.12 DNA sequencing... 86
2.2.2 CeU Biology...87
2.2.2.1 CeU Culture... 87
2.2.2.1.1 Primary superior cervical neurons... 87
2.2.2.1.2 PC12 cells... 88
2.2.2.1.3 Jurkat ceUs... 88
2.2.2.2 Microinjection...88
2.2.2.3 ViabiUty assays... 89
2.2.2.3.1 Survival assay... 89
2.2.2.3.2 TUNEL (TdT-mediated dUTP nick end) labeUing... 89
2.2.2.4 Immunocytochemistry... 89
2.2.3 Biochemistry...90
2.2.3.1 Preparation of ceU lysate...90
2.2.3.2 Protein estimation...91
2.2.3.3 Immunoprécipitation... 91
2.2.3.4 In vitro kinase assay...91
2.2.3.5 Polyacrylamide gel electrophoresis of proteins...92
2.2.3.6 Immunoblotting of proteins...92
3. Th eim p o r t a n c eo f Cd c4 2 i nth ein d u c t io no fa p o p t o s iso fs y m p a t h e h cn e u r o n s ... 9 3 3.1 Introduction...93
3.2 Immunocytochemical analysis of injected ceUs... 95
3.3 Cdc42 and R ad induce apoptosis in SCG neurons... 97
3.4 Cdc42 and R ad are required for NGF withdrawal-induced apoptosis... 100
3.5 Cdc42 lies upstream of R a d ... 101
3.9 The broad spectrum caspase inhibitor zVAD-fmk inhibits Cdc42-induced apoptosis in SCG
neurons...109
3.10 Discussion... I l l 4 . Mu t a t io n a l a n a l y s iso f Cd c4 2 ...1 1 5 4.1 Introduction... 115
4.2 Effect of the various Cdc42 mutants on the survival of SCG neurons in the presence of N G F.. 117
4.3 Activation of JNK by Cdc42 effector mutants...119
4.4 Discussion...121
5. Th eroleo f P A Ksi nn e u r o n a la p o p t o s is...1 2 3 5.1 Introduction... 123
5.2 PAKl and PAK2 are expressed in rat sympathetic neurons...124
5.3 PAKs decrease the viability of primary rat sympathetic neurons...125
5.4 PAKs are not required for NGF withdrawal- and Cdc42-induced death of SCG neurons... 128
5.5 Differential activation of JNK or of the c-jun promoter by the PAK isoform s... 130
5.6 PAKl activity does not increase upon NGF withdrawal... 134
5.7 Analysis of PAK2 activation... 135
5.8 Discussion...138
6. Th ef u n c t io n o f A S K l i nt h ein d u c t io n o fa p o p t o s iso fs y m p a t h e h cn e u r o n s...1 4 0 6.1 Introduction... 140
6.2 Activated ASKl induces apoptosis...141
6.3 ASKl-induced apoptosis in SCG neurons is caspase dependent... 142
6.4 ASKl is an important component of NGF withdrawal-induced death... 144
6.5 ASKl is crucial for the activation of the JNK pathway after NGF withdrawal in SCG neurons. 147 6.6 ASKl is required for Cdc42-induced death... 152
6.7 Discussion...155
7. Ev id e n c efo ra ro leo f M L K 3 inn e u r o n a la p o p t o s i s... 1 5 7 7.1 Introduction... 157
7.2 MLK3 is expressed in sympathetic neurons... 158
7.3 Characterisation of the MLK3 mutants and expression in SCG neurons...160
7.4 MLK3 induces neuronal apoptosis...164
7.5 MLK3 catalytic activity increases following NGF withdrawal... 167
7.6 MLK3 activity is required for NGF withdrawal-induced death of sympathetic neurons... 169
7.10 Discussion... 181
8 . Di s c u s s i o n...1 8 5
PUBLICATIONS ASSOCIATED WITH THIS INVESTIGATION... 1 9 4
Figure 1.1 Survival pathways... 23
Figure 1.2 Apoptotic pathways by death factors...25
Figure 1.3 C. elegans cell death pathway... 27
Figure 1.4 The role of cytochrome c release in apoptosis... 30
Figure 1.5 The JNK pathway and apoptosis... 39
Figure 1.6 General structure of the Rho GTPases... 44
Figure 1.7 The GTPase molecular switch...45
Figure 1.8 Mammalian targets of Rho, Rac and Cdc42... 49
Figure 1.9 General structure of PAKl-3... 56
Figure 1.10 General structure of A SK l...60
Figure 1.11 General structure of MLK3...64
Figure 3.1 Expression of RhoA, Ras, Rac and Cdc42 in sympathetic neurons...96
Figure 3.2 Activated Ras and RhoA do not induce apoptosis in SCG neurons...98
Figure 3.3 Activated Cdc42 and Racl induce apoptosis in SCG neurons...99
Figure 3.4 Dominant negative mutants of Cdc42 and Racl protect sympathetic neurons against NGF withdrawal-induced death... 100
Figure 3.5 Cdc42 is upstream of Racl... 102
Figure 3.6 Activation of Cdc42 results in an increase in the level of c-Jun protein and its phosphorylation... 103
Figure 3.7 Cdc42-induced apoptosis requires AP-1 activity...106
Figure 3.8 Cdc42-induced death is not mediated by SEKl... 108
Figure 3.9 zVAD-fmk can protect sympathetic neurons from V12Cdc42-induced death... 110
Figure 4.1 Cdc42 mutants decrease the survival of SCG neurons in the presence of NGF... 117
Figure 4.2 Effect of various Cdc42 mutants on the nuclear morphology of SCG neurons... 118
Figure 4.3 Increase in the level of phosphorylated c-Jun induced by Cdc42 mutants...120
Figure 5.2 Constitutively activated PAK induces cell death in the presence of NGF...126
Figure 5.3 Effect of dominant negative mutants of PAKl and PAK2 on the survival of SCG neurons in the absence of NGF...128
Figure 5.4 V12Cdc42-induced death of SCG neurons is not rescued by a dominant negative mutant of PAKl... 129
Figure 5.5 Differential effect of PAKl and PAK2 on c-Jun phosphorylation...131
Figure 5.6 Differential effect of PAKl and PAK2 on c-jun activation... 132
Figure 5.7 PAKl kinase activity in PC12 cells deprived of NGF...134
Figure 5.8 Proteolytic cleavage of PAK2... 136
Figure 5.9 The caspase inhibitor zVAD-fmk does not rescue SCG neurons from PAK2-mduced death...137
Figure 6.1 Induction of neuronal cell death by ASKl-AN in SCG neurons...142
Figure 6.2 The caspase inhibitor zVAD-fmk protects SCG neurons from ASKl-induced death 143 Figure 6.3 ASKl-KR prevents NGF withdrawal-induced cell death in SCG neurons... 145
Figure 6.4 ASKl-dependent activation of the JNK pathway... 149
Figure 6.5 FLAGA169 blocks ASKl-induced apoptosis... 151
Figure 6.6 Effect of a dominant-negative ASKl on Cdc42-induced cell death... 153
Figure 7.1 MLK3 is expressed in sympathetic neurons... 159
Figure 7.2 Characterisation of the MLK3 mutants and expression in SCG neurons...162
Figure 7.3 MLK3 induces neuronal apoptosis... 165
Figure 7.4 MLK3 kinase activity is increased following NGF withdrawal in PC12 cells and sympathetic neurons... 168
Figure 7.5 MLK3 is required for NGF-withdrawal-induced apoptosis...169
Figure 7.6 MLK3 is required for Cdc42-induced death...173
Figure 7.7 MLK3 activates the JNK pathway in neurons...177
Figure 7.8 Relationship between MLK3 and A SK l...179
Ab b r e v ia t io n s
AD A lzheim er's disease AP-1 activator protein-1
Apaf-1 apoptotic protease activating factor ASK apoptosis signal-regulating kinase ATP activating transcription factor ATP adenosine 5'-triphosphate
BDNF b rain derived neurotrophic factor CAT chloram phenicol acetyl transferase CRIB Cdc42/Rac interactive binding DTK dual leucine zipper bearing kinase D N A deoxyribonucleic acid
DRG dorsal root ganglion
ERK extracellular signal-regulated kinase GAP GTPase activating protein
GDI guanine nucleotide dissociation inhibitor GEE guanine nucleotide exchange factor
GP-IgG guinea pig IgG
GTP guanosine 5'-triphosphate IL in te rle u k in
JIP JNK interacting protein JNK c-Jun N H2-term inal kinase
MAPK m itogen-activated protein kinase MEKKl M APK/ERK kinase kinase 1 MLK m ixed lineage kinase
N F-kB nuclear factor kB NG F nerve grow th factor
p75NTR p75 neurotrophin receptor PAK p21-activated kinase
PCD program m ed cell death
PI3K phosphatidylinositol-3-kinase R N A ribonucleic acid
ROS reactive oxygen species
RT-PCR reverse transcriptase-polym erase chain reaction SAPK stress-activated protein kinase
SCG superior cervical ganglion SEKl SAPK/ERK kinase 1 SRE serum response elem ent T N F -a tu m o u r necrosis factor a T rk tyrosine receptor kinase
TUNEL term inal deoxynucleotidyl transferase-m ediated dUTP nick-end labelling
U V u ltra-v io let
1. Introduction
1.1 Apoptosis
1.1.1 History of cell death
Cell death has been observed and docum ented by scientists for decades. Indeed, in 1914 Ludwig G raper published a paper entitled "A new point of v iew regarding the elim ination of cells" (Graper, 1914), w hich p rovided the first step tow ards understanding the m echanism s of cell death. H ow ever, the im portance of this w ork was lost during the w ar years and it w as not until 1951 th at G lüksm ann rediscovered the significance of cell death in d ev elo p m en t (Glücksmann, 1951). For instance, he recognised that cell death helps to shape th e form of organs and to elim inate structures no longer useful. In the follow ing years it became a well accepted fact that cells m ust be lost continuously fro m norm al tissues to balance cell division and that this loss of cells accom panied atrophy an d physiological involution of tissues and organs, b u t its m orphological characteristics had not yet been clearly defined. At the tim e, the process of cell death best characterised was know n as coagulative necrosis and it w as the result of noxious stim uli or of irreversible disturbances of cellular hom eostasis.
It was only in 1971 that John Kerr described other structural changes w ith in dying cells that did not conform w ith the known concepts b u t that were consistent w ith an active, "inherently controlled phenom enon" (Kerr, 1971). Kerr used electron microscopy to look at lightly injured or m ildly hypoxic livers. He fo u n d that, rather than swelling and rupturing, cells shrank, lost contact to neighbouring cells, condensed their nuclear chrom atin and w ere phagocytosed and degraded. Kerr called this process "shrinkage necrosis" and later renam ed it as "apoptosis" (apoptosis in Greek is used to describe the "dropping off" or the "falling off" of petals from flowers or leaves from trees).
times and places in the developing embryo and in m etam o rp h o sin g insects, to highlight the fact that it is an intrinsically program m ed event in the developm ental plan of an organism (Lockshin and Beaulaton, 1974). How ever, it was soon realised that these cell deaths could be m odified by e n v iro n m e n ta l factors and w ere not always unavoidable. N ow PCD im plies a pre-existing genetic control and the differential expression of genes th at m ay regulate or be regulated by the activation and execution of cell death. This program m e appears to be present in all the nucleated m am m alian cells tested so far as staurosporine (a broad-spectrum kinase inhibitor) could induce apoptosis in the presence of cycloheximide (a protein synthesis inhibitor) (Weil et al., 1996). This result was also observed in other organism s such as the fly Drosophila m elanogaster and the nem atode w orm Caenorhabditis elegans (C. elegans), suggesting th at it m ight be present in every cell of all m ulticellular organism s as originally suggested by U m ansky (Umansky, 1982; Steller, 1995; Shaham and H orvitz, 1996). These observations suggest that com ponents of the cell death program m e are in place and ready to go in the cytoplasm of all anim al cells. The only exception found so far is the anucleated h u m an red blood cell, w hich did not undergo apoptosis w hen treated w ith staurosporine in the presence or absence of cyclohexim ide (Weil et al., 1996). This study dem onstrates that anucleated erythrocytes do n o t seem to have the m olecular m achinery required for PCD and suggests that the nucleus m ight be responsible for their resistance. Indeed, in m any cases of cell death, both in v itro and in v iv o , a requirem ent for p ro tein synthesis has been observed. For instance, the death of lym phocytes treated w ith glucocorticoids can be blocked by the inhibition of protein synthesis (Cohen and Duke, 1984), as is th e developm ental n euronal death in the chick em bryo (O ppenheim et al., 1990). Taken together, these results suggest that in m ost anim al cells there is a p re existing cytoplasmic death apparatus w hich m ay or m ay n o t require p ro tein synthesis depending on the death stim ulus a n d /o r cell type.
1.1.2 Apoptosis and necrosis
nuclear chrom atin, blabbing of the cell surfaces, a decrease in the rates of R N A (ribonucleic acid) and protein synthesis, and DNA (deoxyribonucleic acid) fragm entation [detected by DNA laddering and TUNEL (term inal deoxynucleotidyl transferase-m ediated dUTP nick-end labelling) staining]. T he cells condense, lose cell-cell contact and fragm ent into m em brane-bound apoptotic bodies that are rapidly phagocytosed. Because the contents of the apoptotic bodies are n o t released, there is no damage caused to the neighbouring tissues and a n inflam m atory response is not illicited. W ithin the phagocytic cells there is a further degradation of the fragm ents by lysosomes. The apoptotic process is highly d ependent on energy, and affects characteristically single cells. A typical feature th at is com m only described as a hallm ark of apoptosis is the activation of nucleases that degrade chrom osom al DNA. DNA dam age represents an irreversible step in the death process. H ow ever DNA fragm entation is n o t observed in all cases of apoptosis (Cohen et al., 1992; O berham m er et al., 1993; Tomei et al., 1993). For instance, DNA fragm entation is n o t detected in cultured hepatocytes treated w ith tu m our grow th factor P 1 (TGF-pi) (O berham m er et al., 1993).
nucleases w hich cleave the DNA into fragm ents of various sizes (Hawkins et ah, 1972; A fanas'ev et ah, 1986). In vivo, in contrast to apoptosis, necrosis u sually involves a large zone of cells such as in the liver follow ing damage by hepatotoxins or in the brain, heart and kidney follow ing ischaemia. As the cells die and burst, an acute inflam m atory response is triggered at the periphery of th e necrotic zone [for review see (Trump and Berezesky, 1994)].
In the last years variations in the characteristics of apoptosis have been observed. For instance, another type of PCD, in addition to apoptosis, is the so- called lysosom al cell death or autophagy. It is characterised by the prim ary expansion of lysosomes and other vacuoles w hich rem ove specific cell organelles and condense the cytoplasm and a late nuclear condensation and fragm entation (also detected by TUNEL staining) (Dunn, 1990a; D unn, 1990b). As in apoptosis there is no m em brane disruption and no induction of an inflam m atory response. This type of PCD has been observed in longer living cells w ith m assive cytoplasm such as rat m am m ary gland cells and M anduca labial gland cells (Lockshin and Zakeri, 1994; Zakeri et al., 1996). More recently, autophagy has been observed as a n integral p a rt of the inductive phase of apoptosis in sym pathetic n eurons deprived of NGF (nerve grow th factor), dem onstrating that apoptosis and autophagy do n o t necessarily occur exclusive of one another and m ay overlap (Xue et ah, 1999). H ow ever it is not yet clear w hat the biochemical relationship betw een autophagy and apoptosis is.
In addition to these variations w ithin PCD, it has been observed that m an y cells m ay exhibit characteristics of both apoptotic and necrotic death, and therefore the distinction betw een these two types of cell death has b lurred (Clarke, 1998). For instance, in a m odel of excitotoxically lesioned new born rat, kainic acid induced DNA laddering and death of neurons exhibiting a variety of m orphologies, ranging from necrosis to apoptosis. Now adays, it is conventionally believed th at both types of cell death are two extremes in a co n tin u u m (Portera-Cailliau et al.,
external stim uli that does not involve the activation of a p rogram m ed m echanism (necrosis).
1.2 The role of apoptosis
1.2.1 Physiological apoptosis
Cell death by apoptosis is of central im portance for both developm ent and hom eostasis of m ulticellular organism s. Recently, Jacobson et al. (Jacobson et al., 1997) defined 5 functions of apoptosis during anim al developm ent:
(1) Sculpting of the body. For instance, inter digital cell death is necessary to create separate articulated digits (Saunders, 1966). Apoptosis has also been show n to be involved in the developm ent of sexual dim orphism . In m am m als, the fem ale M ullerian and m ale W olffian ducts are form ed in both sexes d u rin g developm ent, b u t are later deleted d uring sexual differentiation (G lücksm ann, 1951; Saunders, 1966).
(2) Rem oval of unw anted structures. For example, d u rin g m etam orphosis of am phibians, regression of the tadpole tail occurs by apoptosis (Yoshizato, 1989). Similarly, in some m oths, the deletion of larval intersegm ental muscles and abdom inal prolegs are also a result of apoptosis (Schwartz et al., 1990; Weeks et al., 1992). Likewise, structures from ancestral species, such as pronephric tubules (which form kidneys in fish and am phibian larvae), th at are no longer required in mam m als, are deleted by apoptosis (Glücksmann, 1951; Saunders, 1966).
(3) Control of cell num bers. This is of crucial im portance for the developm ent of the nervous system w here both neurons and oligodendrocytes are produced in excess and then elim inated by apoptosis (see section 1.3).
(4) Rem oval of abnorm al, misplaced or harm ful cells. For instance, im m ature B and T lym phocytes die by apoptosis because they express inappropriate antigen receptors, are self reactive or fail to detect a foreign antigen (Golstein et al., 1991; Motyka and Reynolds, 1991; Cohen et ah, 1992; Rothenberg, 1992).
In developed organism s, apoptosis is also observed in the rem oval of harm ful cells. For instance, in the im m une system, self-reactive cells continue to be deleted, as are cells that are virally infected, as a protection m echanism to stop the infection spreading to neighbouring cells (Levine et aL, 1993). Also, as soon as neu tro p h ils are no longer required they are elim inated by apoptosis to avoid inflam m ation (Savill et al., 1993). A poptosis is also observed in cells w hich h a v e D NA dam age and in tissues undergoing reversible expansion, like the h o rm o n e- dependent cells of the lactating breast (Bardon et al., 1987). M oreover, apoptosis is an im p o rtan t m echanism in the elim ination of cells th at are carrying oncogenic m utations and that could potentially lead to cancer (Korsmeyer, 1992). All of these functions suggest that apoptosis is of prim e im portance in the hom eostasis of organism s.
1.2.2 Pathological apoptosis
Because the control of cell num ber is the result of a balance betw een cell proliferation and cell death, deregulation of the death m echanism s can cause either uncontrolled grow th or excessive death. Indeed, deregulation of apoptotic functions has been linked to the aetiology of m any diseases.
infection w ith adenovirus is e lb (Rao et al., 1992), w hich codes for a protein th at is structurally hom ologous to the anti-apoptotic Bcl-2.
A lternatively, deregulation of apoptosis can cause excessive cell death an d lead to a variety of diseases. For instance, in h um ans, reduction in lym phocyte num bers can be caused by the h u m an im m unodeficiency virus, leading to th e acquired im m unodeficiency syndrom e (AIDS) (M eyaard et al., 1992). A loss or decrease of blood flow (ischaemia) can lead to m yocardial infarction or stroke an d there is evidence th at the dam age occurring peripherally to the central ischaem ic zone is due to delayed apoptosis (Thompson, 1995). A poptosis also plays a role in the loss of specific neuronal populations w hich is the hallm ark of m an y neurodegenerative diseases (section 1.3).
1.3 A poptosis in the nervous system
1.3.1 D evelopm ental apoptosis
The m ost general function of vertebrate neuronal apoptosis is illustrated by the neu ro tro p h ic theory. This theory grew from pioneering studies by Rita Levi- M ontalcini, Viktor H am burger and Stanley Cohen on developing NGF- dependent sym pathetic and sensory neurons (Cohen et aL, 1954; H am burger et al., 1981; Levi-M ontalcini, 1987). The neurotrophic theory is based on the assum ptions th at the survival of developing vertebrate n eu ro n s depends o n particular target-derived trophic factors (or neurotrophins) and that the n e u ro n s are produced in excess, so that only a proportion gets enough trophic support from their target cells for survival. The advantages of this neurotrophic system is th at (1) inappropriate neuronal projections are elim inated as these neurons do not receive adequate neurotrophic factor; (2) the chance th at all target cells become innervated increases; and (3) the num ber of target cells m atches the num ber of neurons (M artin et al., 1988). Therefore, this neurotrophic m echanism ensures th at target cells become innervated in a precise and sufficient m anner [reviewed by (Pettm ann and H enderson, 1998)]. However, the regulation of neuronal cell death is m uch m ore complex than envisaged by the neu ro tro p h ic theory as developing neurons do not depend exclusively on the neurotrophins produced by their target cells (Korsching, 1993). M any require pre-synaptic in p u t for su rv iv al, such as dorsal root ganglion (DRG) neurons [reviewed in (O ppenheim , 1991; Clarke, 1992)] and some require horm ones, like hippocam pal neu ro n s (Sloviter et al., 1989; McEwen and Gould, 1990).
1.3.2 Pathological apoptosis
trophic factor, as illustrated by the fact that following global transient ischaem ia, delayed death of the neurons in the CA l region of the hippocam pus can be prevented b y injection of NGF into the ventricle (Shigeno et ah, 1991). H ow ever it is not always clear w hat the exact contribution of n euronal apoptosis tow ards disease progression is. Most cases of neurodegenerative diseases are sporadic and therefore can arise from a variety of different factors. Hence, studies of hereditary forms of neurodegenerative diseases have provided a lot of insight into th e m echanism s of neuronal cell death. For instance, m utations in the ^-am yloid and presenilin-1 and -2 genes have been linked to A lzheim er's disease (Yamatsuji et ah, 1996; Guo et ah, 1998; M attson et ah, 1998), m utations in the C u /Z n superoxide dism utase (SOD-1) gene are the cause of certain fam ilial am yotrophic lateral sclerosis (Kunst et ah, 1997) and polyglutam ine expansion of certain genes such as h u n tin g tin cause H u n tin g to n 's disease (M artindale et ah, 1998). F u rth erm o re, these proteins such as P-amyloid, presenilin-2 and h u n tin g tin , as well as dow nregulation of SOD-1 activity, have been show n to induce neuronal apoptosis in vitro (Forloni et ah, 1993; R othstein et ah, 1994; C otm an and A nderson, 1995; M ark et ah, 1995; W olozin et ah, 1996) and in v iv o (huntingtin) (Zeitlin et ah, 1995).
p rev en t these diseases. Taken together, these observations suggest that apoptosis plays a p a rt in neurodegeneration although its req u irem en t m ay vary fro m disease to disease.
1.4 Regulation of cell death
1.4.1 Balance between life and death
Tissue m odelling d u rin g developm ent and tissue hom eostasis during adult life is regulated by a dynam ic equilibrium betw een su rv iv a l/g ro w th and apoptosis. Survival and apoptosis are closely associated and a disturbance of th e balance betw een these two processes often leads to pathological situations as described above. Some of the m olecular m echanism s controlling cell survival and apoptosis are discussed below.
1.4.1.1 Survival pathways
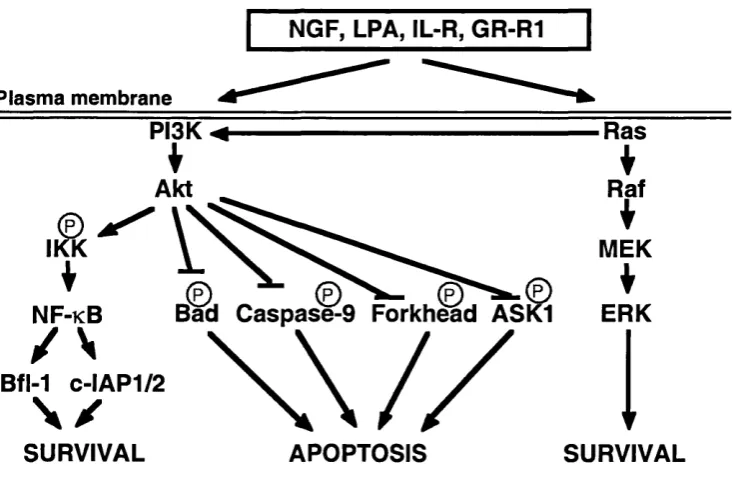
Raff and co-workers (Raff, 1992) suggested that cellular suicide is regulated by a default death pathw ay present in every single cell and only those cells th at m anage to inactivate this pathw ay have the privilege to survive. It is a well- k now n fact that extracellular survival factors play a role in m aintaining cell viability (Eastman, 1995). Therefore, if the cell death m achinery is constitutively expressed, then one of the functions of survival factors m u st be to suppress those death pathw ays. The ability of these factors to prom ote survival have been attributed, at least in part, to the phosphatidylinositol-3-kinase (PI3K)/Akt kinase cascade.
NGF, LPA, IL-R, GR-R1
Plasma membrane
PI3K-#-Akt
IKK
NF-kB Bad Caspase-9 Forkhead ASK1
/ \
Bfl-1 C-IAP1/2
w / /
-Ras
I
Raf
I
MEK
ERK
SURVIVAL APOPTOSIS SURVIVAL
Figure 1.1 Survival pathw ays.
Diagram illustrating putative mechanisms by which extracellular factors are able to promote cell survival.
arabinoside-induced death (A nderson and Tolkovsky, 1999; Xue et aL, 2000). The requirem ent for ERK activity in cell survival appears therefore to vary depending on the cell type as well as on the type of apoptotic insult. The possibility that there are other pathw ays to be discovered that could be m ore or less im p o rtan t depending on the cell type still remains.
1.4.1.2 A poptotic pathw ays
(receptor-interacting protein) and TRAF2 (TNFR-associated factor 2), w hich induce pathways that lead to the activation of NF-kB and JNK/AP-1 (c-Jun N H2-
terminal kinase/activator protein-1) (Chinnaiyan et al., 1996; Hsu et al., 1996). The induction of JNK activity has been linked with IN F 's ability to induce apoptosis (Ichijo et al., 1997; Nishitoh et al., 1998; Hoeflich et al., 1999). As m entioned above, NF-KB can induce the expression of anti-apoptotic Bfl-1 and the caspase inhibitors c-IAPl and C-IAP2, thereby suggesting a control system by which apoptosis induced by IN F is usually inhibited (Wang et al., 1998a). Figure 1.2 illustrates some of the mechanisms by which death factors may mediate apoptosis.
Another member of the IN F receptor superfamily is the p75 n e u ro tro p h in receptor (p75NTR), which plays a role in the induction of apoptosis in the nervous system (see section 1.5.1).
■as Ligand NF
NF-R1
■as
Plasma membrane
FADD D FADD
APOPTOSIS
TRADb n .TRADD
Ü I
T R A F 2 J Jr i p
I (kinase) caspase-8 caspase-8
N F -K B
a c t i v a t i o n ASK1
SURVIVAL
JNK
caspase-3
APOPTOSIS
APOPTOSIS
Figure 1.2 Apoptotic pathways by death factors.
Binding of ligand induces receptor trimérisation which recruits caspase-8 via the adaptors FADD
and/or TRADD. Oligomerisation of caspase-8 may result in self activation of proteolytic a ctiv ity
and trigger the activation of downstream caspases. In another pathway, mediated only by TNF
binding, RIP binds to TRADD and transduces an apoptotic signal through the death domain. In
addition, TRAF2 may also recruit ASKl which activates the JNK pathway. Furthermore, RIP
1.4.2 Signal transduction players of apoptosis
1.4.2.1 Program m ed cell death in the developing n em atode w orm
Egl-1 --- 1 Ced-9---1 Ced-4 ---- ► Ced-3 ► DEATH
Figure 1.3 C. elegans cell death pathway.
Egl-1 inhibits Ced-9, which itself inhibits Ced-4, which activates Ced-3.
C loning of the above-m entioned genes led to the identification of m am m alian hom ologues w hich perform ed sim ilar functions in the regulation of apoptosis. For instance, ced-3 possesses 29% hom ology w ith the h u m a n interleukin ip -converting enzym e (ICE) (Yuan et al., 1993) w hich is a m em ber of the caspase family of proteases (Alnemri et a l, 1996). CED-4 shares hom ology w ith the recently identified Apaf-1 (Apoptotic protease activating factor-1) (Zou et al., 1997). The sequence of ced-9 revealed 24% hom ology w ith the m am m alian bcl-2 oncogene, w hich also negatively regulates cell death (H engartner and H orvitz, 1994); w hilst egl-1 has hom ology to pro-apoptotic m em bers of the Bcl-2 fam ily (Conradt and H orvitz, 1998). The role of these in the regulation of apoptosis w ill be explained in the sections below.
1.4.2.2 The Bcl-2 fam ily
A nti-apoptotic Bcl-2 is the founding m em ber of an expanding gene fam ily that includes the anti-apoptotic Bc1-Xl, Bcl-w, Bfl-1 and M cl-l, as well as pro- apoptotic m em bers such as Bax (Bcl-2 associated pro tein X), Bak, Bcl-Xs, Bad, Bik, Bid, Bim, Bok, M td, Krk and H rk [(Hsu et a l, 1997a; Inohara et al., 1998; O 'C o n n o r et al., 1998) also review ed in (Reed, 1997)].
Bcl-2 has been show n to delay m ultiple cell death pathw ays in m any cell types, including those m ediated by grow th factor deprivation (neuronal, haem opoietic, lym phoid and fibroblastic cells), tu m o u r necrosis factor a (TNF-a), neu rotrophin w ithdraw al, UV and y-radiation, heat shock, reactive oxygen species (ROS), calcium ionophores, glutam ate excititoxicity, p53 and caspase activation [reviewed in (Reed, 1994; White, 1996)]. By contrast, pro-apoptotic m em bers of th e Bcl-2 fam ily appear to prom ote apoptosis (Sakakura et al., 1996; M cCurrach et al.,
The overall sequence hom ology between different Bcl-2 fam ily m em bers is low. It is concentrated around four specific regions called the Bcl-2 hom ology dom ains (BH 1-4), which correspond to a-helical segm ents w hich m ediate p ro tein interactions [for review see (Adams and Cory, 1998; A ntonsson and M artin o u , 2000)]. Bcl-2 fam ily m em bers can interact in different ways to form hom o- or heterodim ers and their relative abundance m ight play a m ajor role in determ ining the response to a death signal (Oltvai and Korsmeyer, 1994; Sedlak et aL, 1995). For instance, the BH l and BH2 dom ains have been show n to be required for Bcl-2 and Bc1-Xl to bind Bax and block apoptosis (Borner et ah, 1994). M any of
the anti-apoptotic m em bers show hom ology on all four dom ains (e.g. Bcl-2, Bcl- Xl, Bcl-w). In contrast, all pro-apoptotic m embers, except for Bcl-Xg, lack the BH4 dom ain w hich is crucial for the anti-apoptotic function. This is illustrated by th e fact that m utants of Bcl-2 that lacked the BH4 dom ain behaved like killer p ro tein s (Cheng et al., 1997). The BH3 dom ain, on the other hand, is a crucial death dom ain in all pro-apoptotic m em bers (Zha et al., 1997) and several BH3-only proteins exist (e.g. Bid, Bad, Bim, Hrk).
The biochem ical m echanism s by which Bcl-2 fam ily m em bers regulate cell death are complex and not yet completely clear. The m ajority of anti-apoptotic m em bers are integral m em brane proteins w hich are localised in the m itochondria, endoplasm ic reticulum (ER) or nuclear m em branes (Hockenbery et al., 1990; Krajewski et al., 1993; de Jong et al., 1994; Z hu et al., 1996), w hereas m ost pro-apoptotic m em bers are found in the cytosol or cytoskeleton before a death signal (Hsu et al., 1997b; Gross et al., 1998; Puthalakath et al., 1999). Follow ing a death stim ulus, cytosolic m onom eric Bax undergoes a conform ational change that allows it to hom odim erise and to translocate to the m itochondrial o u ter m em brane (W olter et al., 1997; Gross et al., 1998). The presence of anti-apoptotic proteins such as Bcl-2 or Bc1-Xl can prevent Bax activation in response to a death
(Datta et al., 1997; del Peso et al., 1997; H arada et al., 1999), resulting in its sequestration to the cytosol by 14-3-3 proteins (Zha et al., 1996). Recently, Bad has also been show n to be phosphorylated by A kt at serine 155 and its m u ta tio n enhanced the pro-apoptotic activity of Bad (Virdee et al., 2000). Serine 155 locates w ith in the BH3 dom ain and it has been suggested that its phosphorylation m ay in d u ce a conform ational change w hich favours the dissociation of Bad from anti- apoptotic Bcl-2 fam ily m em bers (Virdee et al., 2000), thereby neutralising Bad's pro-apoptotic activity. Phosphorylation of Bcl-2 has also been show n to affect its anti-apoptotic activity (H aidar et al., 1995; Ito et al., 1997; Poom m ipanit et a l, 1999). A good exam ple of activation by cleavage is the proteolysis of cytosolic Bid by caspase-8 u p o n Fas or TNF-a treatm ent (Li et al., 1998; Gross et al., 1999; H an et al., 1999). The truncated carboxy term inus of Bid translocates to the m ito ch o n d ria, w here it seems to be required for the release of cytochrom e c (Gross et al., 1999), w hich then complexes w ith cytosolic caspase-9 and Apaf-1 (the m am m alian CED-4 hom ologue) (Liu et al., 1996b; Zou et al., 1997; Z ou et al., 1999) to cleave and activate the effector caspase-3 (Li et al., 1997) w hich ultim ately lead to cell death (Figure 1.4 and see section 1.4.2.3). There is also evidence th at the expression of som e Bcl-2 fam ily m em bers is transcriptionally regulated. For instance, the pro- apoptotic H rk and Bim are upregulated in response to a death signal (Inohara et al., 1997; W hitfield et al., in press).
release of cytochrom e c from the m itochondria by interacting w ith anti-apoptotic Bcl-2 or Bc1-Xl (Yang et ah, 1995). Furthermore, Bid seems to m ediate the release of cytochrome c by inducing a conform ational change in Bax. Following a conform ational change, translocation to the m itochondria and oligom erisation, Bax is thought to insert into the m itochondrial outer m em brane and trigger the release of cytochrom e c (Eskes et al., 1998; Jurgensm eier et al., 1998), probably through the opening of a specific channel such as the FTP (perm eability tran sitio n pore) (Marzo et al., 1998), VDAC (voltage-dependent anion channel) (Shimizu et ah, 1999), or even the Bax pore itself. The release could be blocked by overexpression of Bcl-2 (Kluck et ah, 1997; Yang et ah, 1997b), suggesting that anti- apoptotic Bcl-2 fam ily m em bers m ay exert their functions by blocking the release of cytochrome c.
Bid Bax Bad
\
Apaf-1C y tc -H Caspase-9 dATP
Bcl-2
Ll j
--- ►APOPTOSISBcl-xL
Figure 1.4 The role of cytochrome c release in apoptosis.
Various cell death stim uli cause cytochrome c to be released from the m itochondria into the cytosol.
Cytosolic cytochrome c forms a com plex w ith caspase-9, Apaf-1 and in the presence of dATP,
pro-caspase-9 is activated and leads to the activation of caspase-3 and apoptosis.
1.4.2.3 Caspases
com m on to all of these proteases, w ith the four am ino acids im m ediately N - term inal to the cleavage site defining the specificity of the substrate [reviewed in (N icholson and Thornberry, 1997)]. The CED-3 hom ologues are now know n as caspases (cysteine p ro teases that cleave their substrates after an aspartate residue) and to date, 14 m em bers have been identified (caspase-1 to 14) [for review see (Shearw in-W hyatt and Kum ar, 1999)].
Of the caspases know n to date, not all seem to play a role in apoptosis. Caspase-1, -4, -5 and -11 appear to be prim arily involved in cytokine processing, w hereas caspase-2, -3, -7, -8, -9 and -10 have been show n to play a direct role in apoptosis. A lot of the evidence supporting this has come from studies o n knockout mice. For instance, caspase-1 and -11 knockout mice have defects in IL-i p and IL -la productIL-ion (KuIL-ida et al., 1995; Li et al., 1995; W ang et al., 1998b). Caspase-3 and -9 knockout mice die soon after b irth and show p ro fo u n d developm ental defects such as a higher brain m ass due to lack of neuronal cell death, thereby dem onstrating the im portance of these caspases in n euron cell d eath (Kuida et ah, 1996; H akem et ah, 1998; Kuida et ah, 1998; Woo et ah, 1998).
be activated by class I caspases, they are term ed dow nstream , effector or class II caspases. These effector caspases have been show n to be responsible for th e cleavage of m any proteins involved in the process of cell death [see below and for review see (Nicholson and Thornberry, 1997; Cryns and Yuan, 1998)]._
In addition to being activated by death pathw ays, caspases can also be a target of regulation by com ponents of survival pathw ays. For instance, caspase-9 phosphorylation by Akt renders it inactive probably by blocking the intrinsic catalytic activity of caspase-9 (Cardone et ah, 1998).
1.4.2.4 Involvement of the JNK pathway in transcription-dependent death
The m ost com pelling evidence for the role of transcription in apoptosis came from studies perform ed on neurons. In different types of prim ary n e u ro n s cultured in v itro , inhibitors of transcription or tran slatio n block cell death induced by survival factor w ithdraw al (Martin et aL, 1988; Scott and Davies, 1990; D'Mello et aL, 1993; Milligan et aL, 1994). These observations suggested that the rem oval of survival signals m ay activate regulatory pathw ays w hich lead to th e activation of specific genes w hose products prom ote cell death (Johnson and Deckwerth, 1993). Some of the signalling pathw ays th at lead to apoptosis h a v e begun to be defined and m any molecules that either induce or block apoptosis have been identified. The c-Jun NHz-term inal kinase (JNK; also referred to as stress-activated protein kinase or SAPK) pathw ay has been show n to play a n im portant role in both the induction of apoptosis and cell survival in vivo and in a variety of cell systems [for review see (Leppa and Bohm ann, 1999)].
In addition to an increase in c-Jun expression, c-Jun phosphorylation by JNK has been show n to be required for the m ediation of apoptosis in m an y neuronal cell types. An increase in JNK activity has been observed soon after NGF w ithdraw al from SCG neurons (Virdee et aL, 1997; Filers et aL, 1998) and in differentiated PC12 cells, NGF w ithdraw al induced an activation of JNK and of p38/H O G l m itogen activated protein kinase w hereas the ERK pathw ay was inhibited (Xia et aL, 1995). Similarly, JNK activation has been observed in embryonic m otoneurons after rem oval of their survival factor (M aroney et aL, 1998). In vivo evidence for the importance of JNK in neuronal apoptosis has been obtained from jn k knockout mice. Indeed, knockout of the brain-specific jn k S gene, b u t not jn k l or jn k 2 , protected hippocam pal n eu ro n s from kainate-induced apoptosis (Yang et aL, 1997a; Dong et aL, 1998; Yang et aL, 1998). In addition, the hippocam pi of mice that have a c-jun locus carrying m utations in the phosphorylation sites (S er63^A la and Ser73-^Ala) are also protected from kainic acid-induced apoptosis (Behrens et aL, 1999). On the w hole, these findings suggest that pathw ays regulating the level of c-Jun protein an d its phosphorylation are im portant in neuronal cell death. Consequently, u p stream regulators of these pathw ays m ight be involved in m ediating neuronal cell death (section 1.5).
1.4.2.5 The JNK family of kinases
instance, JN K l was thought to preferentially bind and phosphorylate c-Jun w hereas JNK2 w ould preferentially bind and phosphorylate ATF-2 (activating transcription factor 2) (Kallunki et aL, 1994; Sluss et aL, 1994). H ow ever, it is n o w clear that those differences reflected the particular splice variants exam ined. Indeed, distinct tissues express different am ounts of the various spliced JNK isoforms an d the specific splice variant that binds and phosphorylates a particular substrate can be expressed by either the j n k l or the j n k l genes (Gupta et aL, 1996). Studies on j n k knockout mice have provided som e clues tow ards the understanding of the function of these kinases. Mice lacking JN Kl or JNK2 seem to be m orphologically norm al. However, they are im m unodeficient because of defects in T cell function (Constant et aL, 2000). In ad d itio n , jnk3'^' mice are developm entally norm al but are defective in the apoptotic response to excitotoxins (Yang et aL, 1997a). The jn k l/jn k 3 and jn k 2 /jn k 3 deficient mice h a v e also no phenotypic abnorm alities. However, j n k l / j n k l knockout m ice are embryonically lethal and have defects in neuronal apoptosis and exencephaly (Kuan et aL, 1999; Sabapathy et aL, 1999). Prim ary m u rin e fibroblasts fro m j n k l / j n k l knockout embryos totally lack JNK illustrating the specific n e u ro n a l expression pattern of the jn k3 gene (Tournier et aL, 2000). A ltogether, the studies of jn k knockout mice do confirm that there is a high degree of c o m p lem en tatio n betw een the j n k genes and that the tissue-specific defects in signal tran sd u ctio n m ay reflect a differential expression profile of the various JNK isoforms. This m ay complicate the analysis of jn k knockout mice and dem onstrate the need for studies of anim als that are deficient in all JNK isoforms (Tournier et aL, 2000).
The JNK pathw ay is activated by treatm ent of cells w ith p ro -in flam m ato ry cytokines such as TNF and IL-ip, by exposure of cells to en v iro n m en tal stresses such as UV light. X-rays, hydrogen peroxide (H2O2), heat, osm otic shock an d
1999; W id m an n et al., 1999). MAPKKK phosphorylates and hence activates MAPKK w hich in tu rn phosphorylates and activates MAPK. The JNKs (MAPK) are activated by dual phosphorylation w ithin the protein kinase subdom ain VIII, more specifically on Thr-183 and Tyr-185 in JNKl and JNK2 (Derijard et aL, 1994; W hitm arsh and Davis, 1996) and Thr-221 and Tyr-223 in JNK3 (Lisnock et aL, 2000). Following phosphorylation, cytosolic JNKs translocate to the nucleus w here they can phosphorylate transcription factors. This phosphorylation has been show n to be m ediated by the MAPKKs MKK4 (Derijard et aL, 1994; Sanchez et aL, 1994; Lin et aL, 1995) and MKK7 (Tournier et aL, 1997). Distinct MAPKs are activated by different kinase m odules which respond to specific stim uli (Davis, 1994; Derijard et aL, 1994; W hitm arsh and Davis, 1996). Interestingly, a new group of proteins term ed JNK interacting proteins (JIPs) has been show n to interact w ith m em bers of the MLK (mixed lineage kinase) family of MAPKKKs such as MLK3 (mixed lineage kinase 3) and DTK (dual leucine zipper bearing kinase), two kinases upstream of JNKK, w ith MKK7 and w ith JNK thereby linking these kinase-signalling com ponents (Dickens et aL, 1997; W hitm arsh et aL, 1998; Yasuda et aL, 1999). The JIPs are therefore thought to act as m olecular scaffolds th at organise the JNK signal transduction pathw ay in response to specific stim u li (W hitm arsh et aL, 1998). The translocation of the activated JNKs into the n u cleu s seems to be controlled by JIPs (Dickens et aL, 1997) w hich m aintain the JNKs in the cytoplasm by acting as an anchor.
1.4.2.6 Substrates of the JNKs
(Curran and Franza, 1988). JNK binds ATF-2 and c-Jun at an N -term inal region and phosphorylates Thr69 and ThrZl in ATF-2 and Ser63 and Ser73 in c-Jun, w hich lie w ithin the activation dom ain of these transcription factors (Pulverer et al., 1991; Derijard et ah, 1994; Gupta et al., 1995; Karin and H unter, 1995; Livingstone et ah, 1995; van Dam et ah, 1995). Phosphorylation of ATF-2 and c-Jun leads to increased transcriptional activity, including A P -l's ow n transcriptional activity through the induced expression of c-Fos an d c-Jun (W hitm arsh an d Davis, 1996). c-Fos expression is regulated by activation of the serum response elem ent (SRE) in the c-fos prom oter by Elk-1 (Cavigelli et ah, 1995; Gille et ah, 1995; W hitm arsh et ah, 1995). The increase in c-Jun expression is m ediated by tw o m echanism s: an increase in AP-1 activity which then turns on the c-jun p ro m o ter by binding to its AP-l-like site (Whitmarsh and Davis, 1996); and p h o sp h o ry latio n of c-Jun w hich causes a decrease in ubiquitin-induced degradation of c-Jun, thereby increasing the half life of c-Jun (Fuchs et ah, 1996; M usti et ah, 1997). T he above studies clearly show that the JNK pathw ay can regulate the AP-1 transcriptional activity in response to different stimuli.
dem onstrate a w ay in w hich JNK m ay m odulate the regulation of im portant cell d eath and survival proteins.
1.4.2.7 Targets of c-Jun
One of the transcriptional targets of c-Jun includes the Fas ligand gene w hich has consensus AP-1 binding sites in its prom oter (Yang et aL, 1997a; Kasibhatla et aL, 1998). Recent findings have dem onstrated th at the u p reg u latio n of the expression of the Fas ligand gene is a com m on characteristic of trophic factor deprivation-induced neuronal cell death (H erdegen et aL, 1998; Le- N iculescu et aL, 1999; Martin-Villalba et aL, 1999; Raoul et aL, 1999). These studies suggest a m odel in which grow th factor w ithdraw al induces the transcription of a p o ten t death protein, such as Fas ligand, thereby am plifying the initial death stim ulus. H ow ever, no induction in the expression of Fas ligand was observed in sym pathetic n eurons deprived of NGF (Cesare Spadoni, u n p u b lish ed observations), suggesting that the m echanism s of cell death m ight differ depending on the neuronal type.
TNF, IL-ip, UV, X-rays, H2O2, osmotic shock, growth factor withdrawal
Plasma membrane
JNKKI
JNI
nucleus
JNK
p53 ATF-2 c-Jun Elk-1
Bax FasL Bim c-Jun >
APOPTOSIS
Figure 1.5 The JNK pathw ay and apoptosis.
The JNK cascade can be activated by cytokines, environmental stresses, osmotic shock and
withdraw al of grow th factors. Following phosphorylation, JNK translocates to the nucleus where i t
phosphorylates p53, ATF-2, c-Jun and Elk-1. p53 and c-Jun can activate target genes that promote
apoptosis.
1.5 U pstream activators of the JNK pathw ay
activation of the JNK pathw ay in some cellular system s (see section 1.5.3). T he upstream activators of the JNK pathw ay that were m ost relevant for the w ork presented in this thesis are discussed in more detail in the sections below.
1.5.1 T he p75 n eu ro tro p h in receptor
to m ediate apoptosis and th at this m echanism is essential for naturally occurring neuronal death. M oreover, p75NTR knockout mice have increased num bers of sym pathetic an d forebrain neurons (Van der Zee et aL, 1996; Bamji et aL, 1998) but also reduced n um bers of DRG neurons (Lee et aL, 1992) therefore displaying a phenotype of both survival and neuronal cell death. This dual nature of the p75NTR can be explained in the follow ing way: the decreased num bers of DRG neurons is a resu lt of the inability to form high-affinity NGF receptors at the period of target in n erv atio n w hich naturally occurs before birth (Lee et aL, 1992; M urray et aL, 1999), w hereas because the period of in n erv atio n for sym pathetic neurons norm ally occurs after birth, these neurons undergo a delayed cell death (Van der Zee et aL, 1996; Bamji et aL, 1998). These studies suggest that th e developm ent an d survival of the neurons is based u p o n the functional interplay of the signals generated by Trk and p75NTR. p75NTR expression is high d u rin g developm ent an d is dow nregulated during postnatal developm ent (Yan and Johnson, 1987). H ow ever, p75NTR is rapidly induced follow ing ischaemia or nerve lesion (Moix et aL, 1991; Lee et aL, 1995). Interestingly, in aged rat brain and A lzheim er's patients, high levels of p75NTR are observed in the basal forebrain and hippocam pus, correlating w ith the regions w here extensive cell death is observed (Kerwin et aL, 1993; W iley et aL, 1995). Taken together, these results suggest that p75NTR m ight be involved not only in developm ental neuronal cell death b u t also in the m echanism s of pathological neuronal loss.
(C asadem unt et aL, 1999), dem onstrating the im portance of NRIF in developm ental cell death. NADE is an adaptor molecule th at specifically binds th e DD of p75NTR (Mukai et aL, 2000). Co-expression of NADE w ith p75NTR induced cell death in 293 HEK cells, PC12 cells (p75NTR++% TrkA+) PC12 nnrS cells (p75NTR^\ TrkA ) and oligodendrocytes (p75NTR^^\ TrkA ) in response to NGF, suggesting that NADE plays an im portant role in NG F-induced death by transm itting the signal dow nstream of p75NTR. O verexpression of NRAGE allowed N G F-dependent apoptosis w ithin sym pathetic n e u ro n precursor cells (Salehi et aL, 2000). This seems to occur because NRAGE competes w ith TrkA for the same p75NTR binding site. Finally, p75NTR has also been show n to bind an d activate the small GTP-binding protein RhoA and n eu ro tro p h in binding abolished RhoA activation (Yamashita et aL, 1999). This study provided the first evidence that p75NTR can m odulate the activity of cytoskeletal proteins in a ligand-dependent m anner and it suggests a way in w hich the m orphological changes th at occur during apoptosis m ight be regulated.
Mechanisms of p75NTR induction of apoptosis - a putative role for JNK
because p75NTR binds each of the neurotrophins w ith sim ilar affinity, the specificity of the high-affinity neurotrophin receptor is d ependent on Trk (Chao and H em pstead, 1995). Because sympathetic n eurons do not express TrkB (the specific receptor for BDNF), BDNF's pro-apoptotic response seems to be m ediated by p75NTR.
1.5.2 The Rho family of GTPases
The Rho-subfam ily of GTPases form a subgroup of the larger Ras superfam ily of small GTP-binding proteins. The Ras superfam ily comprises o v er 50 m em bers w hich have been classified into 5 sub-families: Ras, Rho, Rab, Arf and Ran (Hall, 1990; Bourne et aL, 1991; W agner and W illiam s, 1994). These proteins share m any com m on features including sequence hom ology, sim ilar m olecular weights (20-25 kDa) and the ability to bind guanine nucleotides and hydrolyse GTP. The Rho family of proteins is highly conserved in eukaryotes and in m am m als and it includes Rho (A, B, C, D, E, F, G), Rac (1, 2, 3), Cdc42 (two splice variants), TCIO and TTF (Ridley, 1996). The best characterised m em bers are R hoA , Racl and Cdc42.
1.5.2.1 Primary structure
The m ain conserved regions w ithin the Rho proteins are: (1) a GTP binding and hydrolysis region (split into four or five separate dom ains th at come together in the protein's tertiary structure), (2) an effector dom ain, (3) a Rho insert d o m ain and (4) a C -term inus CAAL motif needed for p roper m em brane localisation (Figure 1.6).
(1) The GTP binding and hydrolysis dom ains have been deduced from structural sim ilarities to other GTPases and from the analysis of activated and d o m in a n t negative m utations and as the nam e indicates are im p o rtan t for the binding an d the hydrolysis of GTP [for review see (Halliday, 1983; Schweins and W ittin g h o fer, 1994)].
different effectors or regulators of Cdc42, such as guanine nucleotide exchange factors (GEFs), may bind to different subdomains of the effector domain, possibly even simultaneously. In addition, the binding of one effector to this dom ain could interfere with the binding of other proteins to this domain, providing another way of regulating the Rho-GTPases (see below). The predom inant binding partner for the effector domain in Cdc42 and Rac has been shown to be the CRIB (Cdc42/Rac interactive binding) domain which is found in many of their downstream effectors [see below and (Burbelo et aL, 1995)]. However, not all proteins that interact with Cdc42 and Rac have a CRIB domain, suggesting that there might be other mechanisms by which proteins interact with these GTPases. (3) It is the Rho insert domain that makes Rho GTPases unique within the Ras superfamily. This dom ain contains 13 extra amino acids and has been shown to play a role in Cdc42 interactions with its effector IQGAP and BNIP-2 (McCallum et aL, 1996; Wu et aL, 1997; Low et aL, 2000) and its GDIs (guanine nucleotide dissociation inhibitor) (Wu et aL, 1997).
A ctivation: D om inant n egative: A ctivation: G->V12 T->N17 G->L61
prenylated cleaved I CXXL G1 Effector Domain F->A3H G2 G3
— RTTÔ
Myper-G4 Insert Domain G5 variable region Y->C40 M embrane Localisation Domain
Figure 1.6 General structure of the Rho GTPases.
Regions G1 to G5 are highly conserved and essential for GTP binding and hydrolysis. The amino acids
that are commonly mutated and that were the subject of this investigation are noted. All Rho
GTPases are geranylgeranylated and methylated except RhoB, which is further modified by a
palmitoyl group and RhoE which is famesylated.
of these proteins is not required for their GTPase activity, it is crucial for their plasma membrane localisation and their biological functions (Glomset and Farnsworth, 1994).
The greatest differences between the Rho-GTPases are found in the C-term inus hypervariable region.
1.5.2.2 Regulation and mediation of the activity of the Rho GTPases
Rho GTPases, like Ras proteins, function as molecular switches w hich regulate the transmission of an upstream signal to dow nstream effectors. These proteins are thought to interact with their downstream effectors only when bound to GTP and this interaction is terminated following a conformational change induced by the hydrolysis of GTP. The GTP hydrolysis per se is an irreversible process, however the reversibility of the conformational change can be achieved by the exchange of GDP for GTP. It is these two opposing reactions that make this cycling mechanism possible (Figure 1.7).
GTP (^GE^ pGP
EFFECTOR
Rho
G TPase R h d S
GTPai
GDP
GDI
Figure 1.7 The GTPase molecular switch.
Rho GTPases are inactive when bound to GDP, however exchange of GDP for GTP by a GET converts
the GTPase into an active conformation which allows it to interact with a downstream effector. This
interaction is terminated following a conformational change induced by the hydrolysis of GTP which
is catalysed by a GAP. The intrinsic or catalysed GTP hydrolysis activity ensures that the GTPase
1.5.2.2.1 R egulators
The interconversion betw een the GTP and the GDP-bound form s is m ediated by three types of regulatory proteins: the GTPase activating p ro tein s (GAPs), the guanine nucleotide exchange factors (GEFs) (W hitehead et aL, 1997) and the guanine dissociation inhibitors (GDIs) (Fukum oto et aL, 1990).
The GAPs catalyse the GTP hydrolysis (Boguski and McCormick, 1993). To date, m ore than 25 m em bers of Rho-GAPs have been identified from yeast to h u m an and they include p50 Rho-GAP, Bcr, Abr, N -chim aerin, P-chim aerin, pl90GAP, RalBPl and Graf, to m ention only a few (D iekm ann et aL, 1991; Settlem an et aL, 1992; Barfod et aL, 1993; Leung et aL, 1993; Tan et aL, 1993; Lancaster et aL, 1994; Cantor et aL, 1995; H ildebrand et aL, 1996). The substrate specificity of the Rho GAPs towards m em bers of the Rho subfam ily varies w ith each GAP protein as well as their tissue distribution. For instance p50 Rho-GAP is expressed ubiquitously and Cdc42 is its preferred GTPase, w hereas N -chim aerin is only expressed in the brain and is specific for Rac (Diekm ann et aL, 1991; Barfod et aL, 1993; Lancaster et aL, 1994).
FD Gl are GEFs for Cdc42 b u t Dbl stim ulated PA Kl activation to a higher degree than it could stim ulate JNK activation, w hereas FGDl stim ulated JNK activation b u t n o t PA K l (Zhou et aL, 1998). H ow ever, despite this w ealth of inform ation o n Rho GEFs, the processes that regulate them and those th at they regulate have n o t yet been elucidated.
The guanine dissociation inhibitors (GDIs) bind to the GDP-bound form of Rho GTPases and stabilise the GDP-conformation by inhibiting the dissociation of GDP (Fukum oto et aL, 1990; Ueda et aL, 1990). H ow ever, later studies have sh o w n that Rho GDIs could also associate w ith GTP-bound Rho, Rac and Cdc42 and inhibit the intrinsic GA P-stim ulated GTPase activity by stabilisation of Mg""^ in th e nucleotide binding pocket (H art et aL, 1992; C huang et aL, 1993; Scheffzek et aL, 2000). In addition to this, Rho GDIs play an im p o rtan t role in the subcellular localisation of the Rho GTPases. Indeed, the Rho proteins are found in the cytoplasm w hen coupled to GDIs, but u p o n activation they are released from the GDI com plex and are translocated to the m em branes (Takai et aL, 1995). To date, three types of GDIs have been identified: (1) Rho GDI (Rho GDIa), the first identified, is ubiquitously expressed (Fukum oto et aL, 1990; Ueda et aL, 1990); (2) D4-GDI (Rho GDIp) predom inantly found in haem atopoietic tissues (Lelias et aL, 1993; Scherle et aL, 1993); and (3) Rho GDIy, m ainly expressed in the brain and pancreas (Adra et aL, 1997). There is still little know ledge of the physiological functions of Rho GDIs. They have been show n to inhibit some d o w n stream functions of Rho (N ishiyam a et aL, 1994; Coso et aL, 1995) and m ore recently, D4- GDI w as found to be a substrate for caspase-3 (Na et aL, 1996). H ow ever, not all Rho-GDIs are cleaved by caspase-3 (Essmann et aL, 2000). The cleavage of Rho-GDI (or perh ap s even other GDIs) by caspases d uring apoptosis m ay therefore change the activity an d the signalling by Rho-family GTP binding proteins.
1.5.2.2.2 D ow nstream effectors
Yeast tw o-hybrid system screens, combined w ith a ligand overlay assay w ith [^^S]GTPyS-Rho (W atanabe et al., 1996), led to the identification of a num ber of Rho targets including PKN (Amano et aL, 1996), rh o p h ilin (W atanabe et aL, 1996) and rhotekin (Reid et aL, 1996). A lthough both rh o p h ilin and rhotekin share the sam e binding m otif for Rho as PKN, they do not have a catalytic d o m ain suggesting th at they will have different activities. M ore recently, a kinase called pl60ROCK (Rho-associated coiled-coil containing kinase) was isolated (Ishizaki et aL, 1996). Isoenzym es of pl60ROCK such as ROKa and Rho kinase have also been identified as w ell as the m yosin-binding subunit (MBS) of m yosin pho sp h ate (Leung et aL, 1995; K im ura et aL, 1996; M atsui et aL, 1996). In addition to these proteins w hich w ere show n to bind GTP-Rho, other m olecules w ere found to be activated by GTP-Rho in cell lysates. They include PI3K, PI-4-phosphate 5-kinase and phospholipase D (Zhang et aL, 1993; Chong et aL, 1994; M alcolm et aL, 1994; K uribara et aL, 1995).