Thesis submission to the University of London
for the degree of
Master of Philosophy
by
Helen Elizabeth Smith
Department of Biochemical Engineering
University College London
Torrington Place
London WCIE 7JE
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U642707
Published by ProQuest LLC(2015). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
I would like to thank Dr Edwin Davies, Dr Tom Seddan and Dr Glyn Hobbs for their constant support and belief in my ability whilst undertaking my work at UCL.
I would also like to thank professors Peter Dunnill and Mike Hoare for their assistance throughout the project duration.
My thanks also to Roy, Kamran, Raf, Irene, and Dave for their humour, and Leigh for the many cups of coffee and chats!
ABSTRACT
This project began as an extension of the work undertaken by Harrison, (1996) which
looked at the large-scale downstream processing of antibody fragments. After this preliminary work a change was made to the structure of research, whereby I began to look at the theoretical basis for the systematic selection of downstream processing techniques. This thesis examines the reports of protein recovery in the literature as an attempt to create a means of choosing the most appropriate process. The work described
focuses on the selection of downstream processing techniques for the purification of therapeutic proteins.
Analysing the data collected from 96 protein purification papers published in 1994, attempted to find a factor that could be used to combine yield and purification factor, thereby generating an Operational Effectiveness. This is described as the fraction of contaminant removed for a 95% yield. A review of the most frequently used chromatography steps and the sequence for their use was also undertaken. The order of effectiveness for the steps was given as affinity chromatography = hydrophobic interaction chromatography > ion exchange = gel filtration > precipitation.
CONTENTS
1. INTRODUCTION...8
1.1. Biochemical engineering challenges for protein purification... 8
1.1.1. Antibody engineering for commercial application...8
1.1.1.1. Project significance... 9
1.1.2. Antibody structure and features of antibody fragments... 10
1.1.2 .1. Structure and function of whole antibodies...10
1.1.2.2. Glycosylation...11
1.1.2.3. Antibody fragment production by enzymatic cleavage...12
1.1.2.4. Single chain antibody fragments...12
1.1.3. Applications of antibody fragments and engineered antibodies... 13
1.2. Features of biochemical materials as they affect separation... 14
1.2.1. Separation criteria... 17
1.2.1.1. Physicochemical properties... 17
1.2.2. Product location... 18
1.2.3. Escherichia coli as a host organism for recombinant protein...18
expression 1.2.4. Host organisms used for recombinant antibody fragment... 21
expression 1.3. Objectives of downstream processing... 24
1.3.1. Process selection... 25
1.3.2. Process interactions... 26
1.3.2.1. Regulatory considerations... 26
1.3.3. Scale of protein purification... 27
1.3 3 .1. Laboratory scale... 28
1.3.3.2. Considerations for process scale... 28
1.4. Primary protein recovery... 30
1.4.1. Product release techniques...31
1.4.1.1. Mechanical product release methods... 32
1.4.1.2. Non-mechanical disruption techniques... 33
1.4.2. Process design issues for periplasmic product release...34
1.4.3. Solid-liquid separation techniques...35
1.4.3.1. Gravity driven separation (Centrifugation)... 36
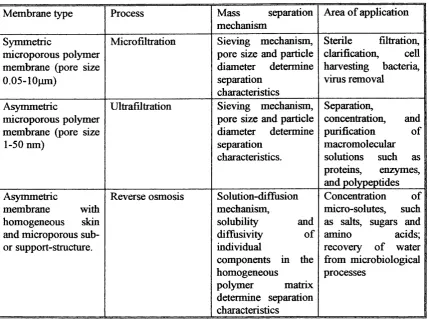
1.4.4. Membrane processes...37
1.4.4.1. Microfiltration... 38
1.5.1. Ion exchange... 41
1.5.2. Hydroxylapatite...41
1.5.3. Hydrophobic interaction chromatography... 41
1.5.4. Size exclusion chromatography... 42
1.5.5. Affinity purification... 42
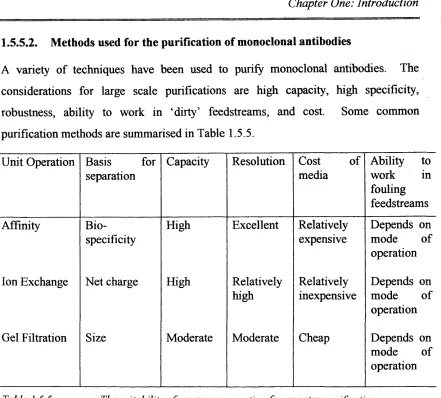
1.5 5 .1. Purification of antibodies... 43
1.5.5 .2. Methods used for the purification of monoclonal antibodies.45 1.6. Finishing operations...45
1.6.1. Concentration and drying techniques... 45
1.6.2. Drying...46
1.6.2.1. Formulation and freeze drying (lyophilisation)...46
1.6.2.2. Crystallisation...47
1.7. Objectives... 48
2. IDENTIFICATION OF THE MOST COMMON PROTEIN...49
PURIFICATION SEQUENCES FROM PUBLISHED LITERATURE 2.1. Introduction...50
2.1.1. Protein separation strategy...51
2.1.2. Operational interactions ... 53
2.2. Early analyses of operations used in the laboratory... 55
2.2.1. Choice of fractionation procedures... 56
2.3. Protein Fractionation... 59
2.3.1. Background theory...59
2.3.2. Richardson method for protein optimisation...61
2.4. Protein purification operations... 63
2.4.1. Determination of purification factor and initial purity...64
2.4.2. Discussion... 69
3 Effectiveness of protein purification operations...71
3.1. Introduction... ...71
3.1.1. Database...72
3 .1.1.1. Sequence of use...73
3 1.1.2. Protein product recovery... 73
3.2. Theoretical considerations of purification processes... 74
3.2.1. Pure product definition... 74
3.2.2. Performance factor description... 75
3.3. Data analysis... 77
3.3.1. Yield...78
3.3.2. Fractional purification factor...80
4. Therapeutic protein, monoclonal antibody and antibody... 85
fragment purification 4.1. Introduction... 85
4.2. Review of work undertaken for the production of monclonal... 88
antibodies and antibody fragments 4.3. Results...90
4.4. Case study of: Celltech Groups Pic... 94
4.5. Discussion... 97
5. ECONOMIC ASPECTS OF THERAPEUTIC PROTEIN PRODUCTION...98
5.1. Introduction... 98
5.2. Time to market...100
5.2.1. Quality... 101
5.3. Process costs...102
5.3.1. Cost factors in designing a process... 105
5.4. Theoretical bioprocess design for antibody fragment production...105
5.5.1. Process factors... 106
5.5.1.1. Process description...106
5.5.2. Basis of design...107
5.5.2.1. Unit operations...108
5.5. Estimation of world antibody market... 108
5.5.1. Economic evaluation for the production of monoclonal antibodies 110 5.5.2. Comparison of tPA and monoclonal antibody production costs I l l 5.6. Economic considerations of downstream processing...112
5.6.1. Costs of running large scale chromatographic separations...114
5.6.1.1. Model formulation... 115
5.7. Considerations for cost in the manufacture of therapeutic proteins...ll6 5.7.1. Cost calculation... 116
5.7.2. Typical costs of manufacturing product... 119
5.8. Discussion...119
6.0. PROCESS DESIGN AND GOOD MANUFACTURING... 121
PRACTICE ISSUES FOR PRODUCING THERAPEUTIC PROTEINS 6.1. Introduction... 121 6.2. A summary of current regulatory issues for modified monoclonai....l22
6.3. Suggested process flowsheet... 125
6.4 Puriflcation techniques... 127
6.4.1. Feed pre-treatment... 127
6.4.2. Initial purification... 127
6.4.3. Chromatographic purification... 129
6.4.4. Product impurities... 130
6.4.5. Prion protein contamination... 133
6.5. Process GMP... 133
6.5.1. Elements of the validation study... 134
6.6. Summary...135
7.0. Conclusions and Future work... 136
Conclusions... 136
Future Work... 137
Bibliography...138
Regulation reviews... 147
CHAPTER ONE
Introduction
1.1 Biochemical engineering challenges for protein purification
The increasing use of macromolecules, such as proteins and nucleic acids in the pharmaceutical, food and chemical industries has generated an increasing need for
practical and economical large-scale processing techniques. Biochemical engineering
is the discipline that underpins the large-scale processing of biological materials and the operation of biochemical reactors (Dunnill, 1987), which performs a similar role for biological materials as chemical engineering provides for non-biological
substances (Hoare and Dunnill, 1989). Until recently, few alternatives had existed for the production of therapeutic proteins or vaccines. This is of particular importance in
the production of therapeutic proteins, where, for such proteins to become
commercially viable, production must be achieved on the multi-ton scale and at a very low cost (Fulton, 1994).
1.1.1 Antibody engineering for commercial application
Recent advances in the field of genetic engineering have led to the development of
many potential microbial strains that can be used to produce pharmaceuticals, therapeutics and food products. Due to the increasing commercial demand for
antigen binding antibody fragments which now have industrial, environmental and
diagnostic applications, there is a need for a production process whereby these
proteins can be produced efficiently and at low cost. The relatively small size of the
antigen binding antibody fragments allows easier access to target sites of limited
access and makes these particular polypeptides attractive for the basis of medical
and Lefranc, 1990; Gavit et a l, 1992). The global market for antibodies was
predicted by CEST (UK Centre for Exploitation of Science and Technology) to be
worth around $6 billion dollars by the year 2000 (Hodgson, 1991).
The traditional production of monoclonal antibodies in mammalian cell culture, by Kohler and Milstein (1975) was developed using hybridoma technology. However,
this mechanism is too complex and uneconomical for use in very large scale
production. An alternative bacterial expression system using Escherichia coli has
been investigated, and the ease with which growth may occur by a straightforward
fermentation, has demonstrated an alternative method for the production of Fv and
Fab antibody fragments (Pluckthun and Skerra, 1989; Skerra, Pftizinger & Pluckthun, 1991, Berry and Pierce, 1993, Harrison et al, 1998).
1.1.1.1. Project significance
The increasing interest in the production of enzymes and recombinant proteins from
microorganisms has created many challenges in the area of biochemical engineering, not least that of the recovery of the desired product.
The contribution of this thesis is to examine the reports of protein recovery provided in the literature and to use this information to establish whether a more convenient
and, therefore, process efficient, generic, method is possible. The downstream processing of antibody fragments were chosen as a test case for the potential generic
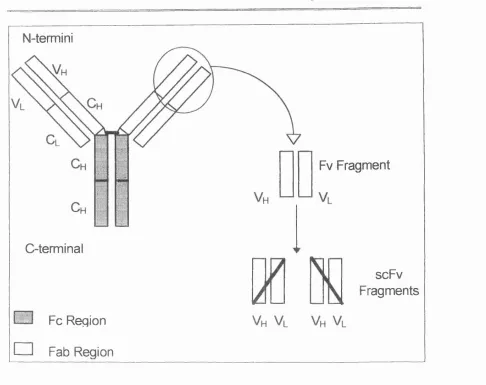
1.1.2 Antibody structure and features of antibody fragments 1.1.2.1 Structure and function of whole antibodies
Antibody molecules are composed of four polypeptide chains consisting of two heavy
chains and two light chains (Figure 1.1.2), with disulphide bonds and Van der Waals
forces holding these chains together in a Y-shaped structure. The structure can be
further divided into two different regions, constant (C) and variable (V). The amino
terminal end provides variability in both the heavy and light chains. These variable regions are known as Vh and Vl respectively, and are responsible for the recognition
and binding of antigen therefore forming the antigen binding site. The remainder of
the structure consists of constant domains and is, referred to as Ch or Cl. The
constant heavy domain is sub-divided into three structurally discrete regions Chi, Ch2,
Ch3. These domains mediate binding of the immunoglobulin to the host tissues,
N-termini
Fv Fragm ent
%
O term inal
scF v Fragm ents
LZj Fc Region
O Fab Region
Figure 1.1.2. Schem atic representation of an antibody a n d derived antibody fragments. A dapted from B etter an d Weikmannn, 1993.
The whole antibody molecule may be cleaved by papain on the C-terminal side of the disulphide bond at the hinge region resulting in two Fab fragments and an Fc fragment Fv fragments are the result o f digestion o f the Fab regions by pepsin, and as recombinant proteins the two domains Vjj and V, may be joined together in either order by a polypeptide linker to form single chain Fv (scFv) fragments. The most frequently utilised polypeptide linker has the amino acid sequence (Gly4Ser)3 (
Chester and Hawkins 1995; Huston et ai, 1988). This can be found in more detail later (Section 1.1.2.4.).
1.1.2.2 Glycosylation
Antibodies are natually glycosylated in mammalian cells (James, et al, 1995); Jenkins
contrast with the expression of many other recombinant proteins, the ability of the
host to perform such modifications does not appear to compromise the antigen
binding properties, however, the lack of glycosylation does affect some of the effector
functions. For industrial and most in vitro uses of antibodies, glycosylation is immaterial.
1.1.2.3 Antibody fragment production by enzymatic cleavage
Antibodies can be enzymatically cleaved at several strategic places to form antibody fragments. Cleavage of the antibody molecule with papain results in the production
of two identical Fab (fragment antigen binding) fragments, which comprise of the complete light chain and the Fd region of the heavy chain (Porter, 1959). The rest of
the molecule is known as the Fc (fragment crystallisable) fragment, which although it
does not contain the antigen-binding activity does, however, elicit an effector function in response to an activated immune system. Cleavage of whole antibodies with
pepsin occurs at the C-terminal side of the hinge region resulting in the production of F(ab') 2 fragments which contain both antigen binding sites (Petermann and
Pappenheimer, 1941). These fragments can easily be reduced to form Fab fragments.
1.1.2.4 Single chain antibody fragments
Genetic engineering has also enabled the production of single-chain Fv (scFv)
fragments, which consist of a single Vh and Vl connected by a short polypeptide
linker (Raag and Whitlow, 1995). The fragment has been engineered as an E. coli
plasmid vector and expressed as a single protein (Bird et al., 1988; Huston et al., 1988).
Single chain Fv antibody fragments (scFv) are novel recombinant proteins composed
of the Vh domain and the Vl domain linked together by a short polypeptide linker.
Recombinant DNA technology allows this to be engineered in E. coli plasmid vectors and expression as a single protein (Bird et a l, 1988). The inclusion of a polypeptide
linker in scFv fragments ensures the equal expression of both domains, which may
also aid the association of the immunoglobulin chains once translated, therefore,
increasing fragment stability (Glockshuber et a l, 1990).
the two domains together would comprise 15 peptide residues. Computer searches of
three dimensional peptide structure libraries identified suitable linkers that could join
Vh and Vl of the variable regions of three different monoclonal antibodies by linking
the C-terminus of one domain to the N-terminus of the other (Bird et ai, 1988). The
secondary structure must not be so complex that it is difficult to fold or interferes with antigen binding, however, if the linker is devoid of secondary structure it may be
susceptible to proteolysis. Linkers should therefore, be long enough to span the distance between the two variable domains, possess sufficient flexibility to allow Vh-
Vl association, and be sufficiently hydrophilic to enable a surface location on the
molecule (Harrison, 1996). The design of scFv and a number of linker polypeptides have been extensively reviewed, with (Gly4Ser) 3 being the most commonly used
(Huston et al., 1991), where serine residues confer hydrophilicity and glycine allows for flexibility.
1.1.3 Applications of antibody fragments and engineered antibodies
For more than a decade there have been attempts to use monoclonal antibodies (mAb) as therapy for diverse human diseases, including coronary artery disease, infection
control and detection, cardiac and renal failure, autoimmune diseases, and cancer diagnosis and therapy. An extensive review into the specific use of antibodies and
antibody fragments in medical applications was made by several authors, with the main areas involving the use of antibodies being for the carriage of radionuclides,
enzymes, genes, drugs or toxins to target cells (Lefranc and Lefranc, 1990; Jones, 1992; Chester and Hawkins, 1995).
The envisaged application of an antibody will ultimately determine the choice of
fragment required to elicit a response. Industrial and laboratory applications of
antibodies require high levels of binding and stability, whereas their use in medicine
will be dependent upon the need of the natural effector functions of the whole
antibody and whether a smaller fragment of the antibody could induce the same affect
(Pluckthun, 1991). The pharmokinetic stability of the antibody fragment is another
important feature, as disintegration of the fragment prior to reaching its target cell,
The application of antibodies for cancer diagnosis and therapy requires small antibody fragments, to allow optimal penetration, whilst also possessing a high
affinity and specificity for its antigen, without evoking an immune response during
repeated therapy. The small size of Fv and scFv fragments has, therefore, made them
attractive components for use in medical products. The reduced size of the fragments
allows both the rapid build-up and subsequent clearance at the target tissue (Colcher,
et al., 1990), and they can be easily attached to radiolabels or toxins to enable tumour
imaging and treatment (Bird and Walker, 1991).
Alternative applications of smaller antigen binding molecules such as the Fv and scFv fragments are in protein purification. The small size of these fragments allows
greater immobilisation capacity on porous supports, thereby increasing column
capacity for the target antigen (Berry and Pierce, 1993). Other industrial applications
include their increasing employment in biosensors and environmental techniques, and research has been initiated into their use as additives during food processing, and in
the cosmetics industry (Harris, 1991).
The presence of a linker molecule also generates further possible applications for antibody fragments. For example, the linker may be used to attach drugs or affinity
handles (Tai, et al, 1990) or for immobilization onto solid supports (Harrison, 1996).
1.2 Features of biochemical systems
In general, biological materials occur as components in a complex mixture of other
materials. This is true of both materials for which a production route has been
determined, such as fermentation and for materials which are to be extracted from naturally produced sources, such as blood plasma (Schawen and Melling, 1985; Bell
et al, 1983). Hence, the presentation of such materials in a useful form would
normally require a sequence of separation and purification operations.
There are two major differences between biological and non-biological products.
Firstly, many non-biological industrial products are already at a high concentration at
the start of their separation and purification, whereas, biological products are
generally dilute. Secondly, many non-biological products are produced in mixtures
amenable to rigorous chemical analysis whereas, biological products are generally
comprehensive analytical specification (Bowen, 1992), though this situation is
changing as a result of advances in mass spectrometry.
Fermentation broths are dilute aqueous solutions, with a continuously changing broth
composition and changing cellular properties during the culture time. It is therefore
important to cease the fermentation and begin the separation and purification at the
optimum time. For extracellular products it is important to know the rate at which the
desired product is produced so as to maximise yield and to separate the cells from the
broth before lysis increases separation difficulties.
Some of the most important physical properties of biological cells and their
dispersions from a separation viewpoint include, size, specific gravity, rheology,
stability. Typical sizes of materials obtained from a separation and purification
procedure are shown in Table 1.2.
Type Size pm
Cell debris 0.4
Bacterial cells 0.1-2.0
Yeast cells 3.0-10.0
Mammalian cells -40
Plant cells -100
Table 1.2 Table to show the typical size o f cells and cell debris. (Adapted from
Walsh and Headon, 1997).
Cell debris must be separated if cell lysis occurs during fermentation or if the cells are
deliberately disrupted to release an intracellular product. Such separations often
require the addition of flocculants (Habib et al, 1997) to aid product purification as
the size of debris and of microbial cells is commonly below the normal range for
conventional filtration processes or the debris fouls the finer filters or membranes
(Clarkson et al., 1993; Titchner-Hooker et al). Differences in specific gravity
between cells and the suspending broth determine the performance of sedimentation
and centrifugation. Such differences may in fact be small, for example, the typical
specific gravity of yeast cells being 1.07 compared to the typical specific gravity of a
1992). The administration of such cell flocculants before centrifugation have only
been shown to be of limited value due to the occlusion of broth between the floes of
individual cells (Clarkson et al., 1993; Titchner-Hooker et al).
Fermentation broths are also subject to a environmental changes in conditions when
they leave the controlled aseptic conditions of the fermenter. For example, actively
growing cells from an aerated culture can suddenly be deprived of oxygen and
experience a fall in substrate concentrations, which may ultimately produce a substantial change in the physical properties of the cells. The broth may also become
vulnerable to contamination by foreign organisms, which demand refrigeration of the
product. A further aspect of stability involves the resistance of cells to shear damage
which although not being a major problem with bacterial and yeast cells, can easily damage plant and animal cells (see section 1.4.5.).
Problems with product stability are also likely to arise with both high and low molecular weight products, with the main mechanisms identified for such product
loss being either chemical degradation or microbial degradation. In the case of chemical degradation of protein products, the product may only have a narrow
stability range to pH or temperature changes, outside of this range dénaturation and loss of function will occur (Fish and Lilly, 1984). The action of degradative enzymes,
such as proteases, can also facilitate the break down of active molecules (Veide et ai,
1984) and the action of enzymes can be accelerated by high temperatures. It is therefore often beneficial to process proteins and enzymes at low temperatures, and as
quickly as possible at those stages in the process where such enzymes may be present
(Kula et al, 1982). The action of degradative enzymes may only become apparent
with careful analysis. With therapeutic proteins there is the possibility of degradative
enzymes making small alterations to protein structure, cleaving the polypeptide chain
at a single position. While this may not alter the physiological action of the protein, it may cause substantial alteration to its antigenic properties (Bowen, 1992).
Fermentation media and downstream processes involving cells, cell debris, proteins or
other organic materials also provide a rich medium for the growth of contaminating
microorganisms (Ogez et a l, 1989). This is undesirable in terms of the prospective
microbial contamination of the product. Microbial product contamination can result
material to the separation process which it was not designed to separate or to the loss
of product by degradation (Gavit et al, 1992). The downstream processing scheme
must, therefore, be designed to exclude such contamination possibilities and be
configured to inhibit the growth of microorganisms (Ogez et a l, 1989).
1.2.1. Separation criteria
Biochemical systems share a number of common features which dominate any
discussion of techniques necessary for the downstream product recovery or processing. These include, dilute aqueous solutions, complex multicomponent
mixtures, poorly defined components, variable composition and product stability
(Liddell, 1994). Typical properties of a desired protein include if s primary amino
acid sequence; isoelectric point; solubility characteristics; stability characteristics
under the influence of pH and temperature; sedimentation and diffusion coefficients; and product location (Walsh and Headon, 1997).
1.2.1.1. Physicochemical properties
The key to efficient separations is to exploit the differences in the physicochemical
properties of the product and the other components present as effectively as possible (Bowen, 1992). These include size, electrical properties, hydrophobicity, solubility
and stability. Separation processes such as membrane filtration and gel filtration making use of differences in molecular weights. Many biological products including
amino acids, antibiotics and proteins bear a net electrical charge. Charge properties
depend on pH and the ionic composition of the process stream and may be used directly in ion exchange chromatography and electrophoretic separation. These
properties also have a profound influence on the performance of other separation
processes, such as membrane separations where precise control of solution conditions
is essential to process optimisation (Bowen, 1992).
Solubility is a result of a molecule’s charge and hydrophobic properties, but is often
characterised as a fundamental property. Product solubility can be utilised effectively
in controlled precipitation, for example, proteins are normally least soluble at the pH
Protein products are often unstable in an aqueous environment, especially those that
have originated from hydrophobic regions of the cell, whilst products, which have
been formulated into a dry state, will slowly lose activity. Protein dénaturation can be
reversed if only the hydrogen bonds, hydrophobic interactions or the disulphide bridges have been disrupted. However, irreversible changes may occur due to the
hydrolysis of bonds, oxidation and free radical attack and rupture of disulphide
bridges.
1.2.2. Product location
The location of the product has a substantial impact on the downstream processing procedure employed (Flaschel and Freihs, 1993). If the protein product is situated in
the cytoplasm, the concentrated cell mass must first be broken. Consequently, the
target protein must then be separated from a complex mixture of proteins, nucleic acids and eventually cellular compartments and debris (Hoare and Dunnill, 1989). A
gentler method of cell disruption may be applied if the target protein is secreted into
the periplasm of Gram-negative bacteria, with disruption being a superfluous task in the case of extracellular proteins (Flaschel and Freihs, 1993).
It is not straightforward to decide if it is the intracellular or extracellular location that provides a simpler means for product recovery (Foster, 1995). Where the product is released into the fermentation medium, substantial purification should be possible by
removal of the insoluble material such as cells and media components. However, purification may be less than anticipated due to the release of other protein
components into the medium by intact cells or on cell lysis (Fish and Lilly, 1984). In
cases where the product is retained within the cells, then simply recovering the cells
will give substantial concentration of the desired product. There is then the additional
step of breaking the cells open to release the product which will generate cell debris fragments and cause the release of other soluble material into the medium (Kula et
al., 1990).
1.2.3. Escherichia coli as a host organism for recombinant protein expression
E. coli is a genetically versatile organism in which recombinant DNA technology is
Heterologous gene expression in E. coli has frequently been used to produce large
quantities of protein, however, the exact category of the protein produced depends on
many factors, including both the nature of the protein to be expressed and the type of
expression system used. One of the major advantages of recombinant technology is
the ability to engineer cells to produce a product into a pre-defined location. Each
location has certain advantages and disadvantages in terms of bioprocessing.
Proteins expressed in E. coli may remain soluble in the cytoplasm (Schooner et al.,
1985), or become sequestered into inclusion bodies (Simons, et al., 1984), before
being transported across the cytoplasmic membrane. They may accumulate in the periplasmic space (Lunn, et al., 1990), or as in some cases, be secreted into the
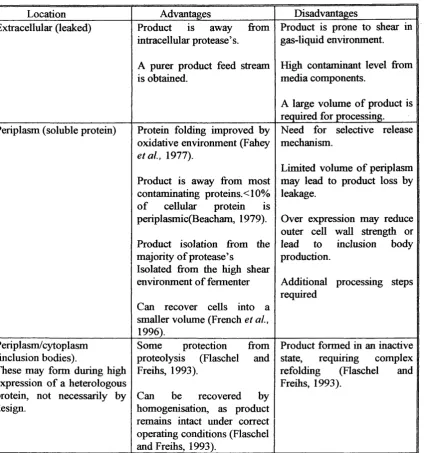
Location Advantages Disadvantages Extracellular (leaked) Product is away from
intracellular protease’s. A purer product feed stream is obtained.
Product is prone to shear in gas-Uquid environment. High contaminant level from media components.
A large volume of product is required for processing. Periplasm (soluble protein) Protein folding improved by
oxidative environment (Fahey
etal, 1977).
Product is away from most contaminating proteins. <10% o f cellular protein is periplasmic(Beacham, 1979). Product isolation from the majority of protease’s
Isolated from the high shear environment of fermenter Can recover cells into a smaller volume (French et al.,
1996).
Need for selective release mechanism.
Limited volume o f periplasm may lead to product loss by leakage.
Over expression may reduce outer cell wall strength or lead to inclusion body production.
Additional processing steps required
Periplasm/cytoplasm (inclusion bodies).
These may form during high expression of a heterologous protein, not necessarily by design.
Some protection from proteolysis (Flaschel and Freihs, 1993).
Can be recovered by homogenisation, as product remains intact under correct operating conditions (Flaschel and Freihs, 1993).__________
Product formed in an inactive state, requiring complex refolding (Flaschel and Freihs, 1993).
Table 1.2.3 Advantages associated with product re-location due to genetic engineering.
Genetic manipulation techniques have allowed for the introduction of plasmid DNA
encoding antibody genes under the control of a strong promoter, which will allow for
the generation of high antibody fragment titres at specific locations within the cell.
The rapid growth rate and comparatively simple method of cultivation render E. coli
an attractive host for large scale recombinant antibody production. The expression of
recombinant antibodies in E. coli is well documented (for reviews see Pluckthun and
Microbial research has centred upon using E. coli as the host, which commonly expresses the antibody fragments as inclusion bodies. However, this creates a number
of problems for large scale production due to the complexity of the protein renaturation process. Recent work performed has led to the control of antibody
fragment expression in E. coli (Harrison, 1995). Downstream processing with this
system is still laborious, and it may be more desirable to develop a system whereby a
high level of antibody fragments are secreted directly into the fermentation broth.
Scale-up of processes involving E. coli has also been extensively investigated. To
date the expression of Fab, Fv, scFv, single domains and various antibody based
molecules has been demonstrated in E. coli (Berry and Pierce, 1993, Harrison et al.
1998). However, the organism is known to have certain disadvantages, including its
improper protein folding and the inability to perform certain post-translational
modifications required for eukaryotic protein activation for example glycosylation
(Pluckthun, 1991). For the purpose of producing antibody fragments however, except for Fc antibody fragments glycosylation is immaterial, as it will not influence the
antigen binding ability of the fragment (Pluckthun, 1991).
1.2.4 Host organisms used for recombinant antibody fragment expression
Non-mammalian heterologous systems have been used as an alternative for antibody
fragment production, whereby the gene for an antibody fragment is engineered into the host microorganism and this is then grown up in a fermenter. Genetic
manipulation has enabled the expression of antibody fragments in a number of
microorganisms including Escherichia coli, Saccharomyces cerevisiae and
Aspergillus. However, expression in recombinant baculovirus infected insect cells
(zu Pulitz, et al, 1990) and transgenic plants (Owen, et al, 1992; Ma and Hein, 1995)
has also been performed. The choice of host will ultimately affect the isolation and
purification procedures required for recovery of the recombinant product.
Bacteria have great appeal as expression systems for antibody fragment production
because of their ease of growth. However, in spite of the success of using E. coli for
Filamentous fungi are attractive hosts for the production of foreign proteins because
of their high secretory capacity. However, relatively few cases have provided heterologous protein yields comparable with the g/L levels obtained for certain
homologous enzymes (Mackenzie, 1994). Yield improvements have been achieved
by changing the secretion signals attached to the target protein, by altering the growth
medium, and by mutating the host to produce a strain with a higher secretory capacity
(Dunn-Coleman, et a l.,\9 9 \).
The high rate of native protein secretion in Aspergillus spp. has also made them an
ideal candidate for the industrial production of heterologous proteins. Aspergillus spp
have numerous advantages as protein expression systems. These include; rapid growth on simple, inexpensive media; a high degree of genetic characterisation; and
good protein secretion properties (Mackenzie et ai, 1994).
Aspergillus niger has been used successfully to secrete hen egg white lysozyme (HEWL) at levels of approximately 10 mg/L (Archer et al, 1990). Studies performed
by Mackenzie and associates (1994), identified that growth temperature and medium
composition had the greatest effect on lysozyme levels. Yields of up to 30-60 mg/L were obtained in a rich medium, with the highest levels of secreted lysozyme being
obtained at 37 °C. Therefore, Aspergillus, offers promise as a host for the large scale production of secreted, heterologous proteins provided that expression and targeting
of the recombinant protein to the efficient secretory apparatus can be achieved (Archer et al, 1990).
The eukaryotic filamentous fungus Trichoderma reesei was found to be highly
efficient for the expression of recombinant Fab fragments (Nyyssonen et al, 1993). When the Fd chain was fused to part of the T. reesei cellulase cellobiohydrase I and
co-expressed with the light chain, gene titres of 150 mg/mL of Fab were achieved in
fermenter cultures grown on cellulase inducing medium. Fab fragments were properly
assembled and secreted into the culture medium, where cleavage of the fusion-protein
was mediated by a T. reesei extracellular protease. The antibody fragments described
here are the first multi-chain molecules produced in filamentous fungi. The yields in
Trichoderma are comparable to those of hybridoma cell lines and far exceed those
obtained in most other microbial production systems. Single chain antibody
Saccharomyces cerevisiae was first used for the expression of whole antibodies
(Wood et al, 1985). Both heavy and light chains could be secreted into the culture
medium but intracellular localisation was prominent. In cells expressing plasmids for
both chains functional antibodies were detected and the heavy chain could be
glycosylated. Horwtiz et al, (1988) achieved extracellular concentrations of the light
chain at 100 ng/mL, and the heavy chain at 50-80 ng/mL from S. cerevisiae
harbouring plasmids for both proteins. Alternative yeast host organisms for antibody fragment expression include Schizosaccharomyces pombe (Davis et al, 1991) and
Pichia pastor is (Bidder et al, 1995). P. pastor is was shown to be highly suitable for
scFv expression on a small scale and titres of 100 mg/L were achieved. This organism combines the fast growth of bacteria with eukaryotic secretion machinery
and induction could be efficiently managed by the addition of methanol.
Another example of a system which combines eukaryotic properties with the ease
associated with handling microbial systems to produce both functional and
glycosylated antibodies is the use of recombinant baculovirus infected insect cells. When both light and heavy chains were expressed together in suspension culture, the
system was found to produce up to 30 mg/L whole antibody molecules (zu Pulitz, et al, 1990).
The Streptomyces spp. are filamentous Gram positive soil bacteria, which are widely
known as producers of commercially important antibiotics. They produce a variety of extracellular proteins and have been considered as hosts for the production of
heterologous proteins. The secretion system has been applied to a few foreign
proteins including interferon-alpha-1 (Noack, et al., 1988) and interleukin-2 (Bender
et al., 1990). More recently, a study has been performed to establish the secretory
production of the Fv domain of HyHELlO (Smith-Gill, et al., 1987) in S. lividans.
This antibody is one of a series of anti-lysozyme monoclonal antibodies whose
epitope has been found to overlap with the substrate binding site of hen egg-white
lysozyme (HEWL) (Padlan, et al., 1989). High level expression of foreign genes in
Streptomyces promises to be a useful strategy when coupled to signal sequences and
promoters, with site directed extracellular secretion of Fv in S. lividans producing
yields of 1 mg/L (Ueda, et al., 1993). Over expression of heterologous genes in E coli
In contrast, inclusion bodies have not yet been observed in Streptomyces. Therefore,
high level expression of foreign genes in Streptomyces promises to be a useful
strategy especially when coupled with improvements, in for example, promoters and
signal sequences.
Lastly, the expression of antibody fragments in transgenic plants offers an attractive
and economical alternative approach for production at an increased scale. All that is
required for production is a field, water, pesticide and fertiliser (Owen et ai, 1992),
although containment is likely to demand the use of greenhouses. The first
demonstration of antibody expression in plants involved Agrobacterium tumefaciens
mediated transformation of tobacco plants with genes encoding either complete light
or heavy chains (Hiatt et al, 1989). Crossing these plants resulted in progeny which could express both chains simultaneously resulting in the accumulation of functional
antibodies in the leaves, with the antibody comprising 1.3% of total leaf protein.
Expression of antibody proteins in plants has since been extended to the production of antibody fragments such as Fab and scFv targeted to specific locations in a number of
plants (for review see Conrad and Fielder, 1994).
However, given the absence of plant-derived mAbs from clinical trials for human or
animal application, none of the issues related specifically to plant-derived antibodies have been formally addressed at the regulatory level and, therefore, few have been
dealt with rigorously at the experimental level. Other concerns may be focused on the
glycosylation pattern, which in the context of the antibodies that are required for
some therapies, may provide a new antigenic challenge to the human immune system
(Ma and Hein, 1995, Carol Potera, 1999).
Information concerning the production of antibodies from mammalian systems can be
found in a number of research papers, including Kohler and Milstein (1975),
Pluckthun and Skerra, (1989) and Skerra, Pftizinger and Pluckthun, (1991).
1.3 Objectives of downstream processing
The increased production of biological materials and the increasing application of
biotechnology within the pharmaceutical industry has dictated the introduction of
large-scale processing techniques into the bio-industry for the recovery of biological
defined as “Downstream Processing”, a term which encompasses the recovery and
purification operations that follow biochemical reactions such as, fermentation; plant and tissue culture; and whole foods production (Knight, 1989). These techniques play
a central role in the commercial success of new protein-based diagnostics and
therapeutics, whereby, the purification steps taken provide a definition of the often
complex product (Knight, 1989). The challenges that were raised with respect to the
purification of enzymes have since expanded to encompass a variety of new protein
products including those of the monoclonal antibodies, antibody fragments and
complex therapeutic molecules.
A number of review articles, including those of Fish and Lilly (1984), Bonneijea et al.
(1986), and Hoare and Dunnill (1987), have been written to consider the interactions between the unit operations involved in the industrial production and subsequent
recovery of any protein product. The ultimate challenge is to select the best
combination of substrate, enzyme or organism, bioreactor, and separation for a given product. Ultimately it is the économes of the process which determines it’s choice.
1.3.1 Process selection
The success of a process is dependant on the careful design and subsequent
optimisation of the purification scheme. The cost of obtaining the product at the
desired purity and scale is ultimately dependant upon the choice and sequence of purification steps. Process optimisation is another crucial element of putting a
recovery process in place because it has a pronounced effect upon overall production
cost and success rate. For example, in optimising chromatographic operations, it is
advisable to both clarify the feedstocks and develop regeneration procedures that
allow the reuse of packing materials (Eriksson and Sandahl, 1991). This reduces both
down-time and labour required for column repacking and significantly reduces raw
material and quality control costs. Many of the principles apply to the optimisation of
filtration operations (see section 1.4.4), including regeneration procedures that allow
membrane reuse and determination of operating ranges of pressure and
1.3.2 Process interactions
Sofer and Briton (1983) have reviewed the principles for incorporating
chromatographic steps into protein purification schemes. Their recommendations
included using a high capacity technique, such as ion-exchange as the initial
chromatographic step to facilitate later purification steps by the reduction of process
volumes. They also recommend the use of a highly specific technique, such as
affinity chromatography at an early point in the recovery process to facilitate the rapid
separation of the product from any contaminants, which would otherwise degrade it over time. However, affinity separation is an expensive operation and fouling is very
serious early in the process. Sofer and Briton (1983) also suggested ways of linking
multiple chromatographic operations in order to eliminate intervening buffer
exchange steps, whilst addressing some of the considerations for the scale-up of
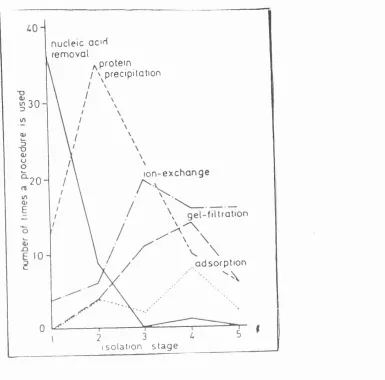
column chromatography. A more recent study was undertaken by Bonneijea et al.
(1986), and involved the analysis of 100 published protein purification articles. The analysis highlighted several clear preferences regarding the use and order of various purification methods. For example, homogenisation (Sidiqui et al, 1997) and
precipitation were the most common initial steps, followed by ion-exchange in an
attempt to partially purify and reduce process volumes.
1.3.2.1 Regulatory Considerations
The steps required for any process ultimately depend on the quality criteria and
quantity of the final product required by the manufacturer, factors which are
dependent on the intended product application (Ostlund, 1986). There are a number
of considerations that need to be taken into account for the large-scale processing of
monoclonal antibodies, for example, the criteria derived for the production of monoclonal antibodies destined for in vivo use are likely to be very stringent due to
the products from hybridoma cells being considered as recombinant DNA technology
products (Ostlund, 1986). Both the US Food and Drug Administration (FDA) and the
National Institute for Biological Standards and Control in the UK have produced draft
guidelines for the in vivo use of monoclonal antibodies. These guidelines provide
details of the criteria that the regulatory bodies expect producers of monoclonal
antibody products to consider in the product development and license applications.
reaction in a patient, that is, protein, viral and nucleic acid contaminations (Edmond
et ai, 1986; Hoare and Dunnill, 1989). As monoclonal antibodies are the products of
malignant cells, there is a risk of transferring viruses and nucleic acids associated
with malignancy through the product. The importance of bacterial toxins such as
pyrogens is also well recognised, and these must be removed during the purification
process. If the final product is destined for in vitro use, the process should also be operated under aseptic conditions (Ostlund, 1986).
In developing tests for in-process quality assurance, a manufacturer must consider
that the product has to conform to regulatory standards. In-process controls must
demonstrate that the product is pure, safe and effective for its intended application
(Edmond et al, 1986). Monoclonal antibody products destined for use in vitro use do
not need to meet such stringent criteria and the degree of purity required will be
determined by its intended application. A more in depth analysis of the good manufacturing practice and validation requirements can be found in Chapter 6.
1.3.3 Scale of Protein Purification
Protein purification protocols are initially designed at a laboratory scale, so it is
important to recogmse that the scale at which commercial production of proteins from
microorganisms occurs varies greatly with the product and may, therefore, influence the techniques that can be used (Fish and Lilly, 1984).
On the basis of several assumptions concerning cellular yields and protein contents,
Pickett and Haddock (1983) concluded that for many proteins produced by
‘genetically-engineered’ strains, the fermentation volume requirements are likely to be small. The scale of production of the material will, to a certain degree, control the
processes that, can be used, and although the scale of operation across which unit
operations can be used is becoming increasingly blurred, there are still processes that
are more suited to certain scales rather than others (Liddell, 1994). All of the
methods used in primary separation, including centrifugation and filtration, can be
1.3.3.1 Laboratory Scale
Protein Purification protocols are initially designed at laboratory level and scale-up
studies are then undertaken in order to produce sufficient quantities of the protein to
meet market demands as economically as possible (Dwyer, 1984). In general, the
cost associated in producing a unit quantity of any protein will decline with increasing
the scale of production (Table 1.3.3.1).
Most chemicals and raw materials can be purchased at reduced cost in bulk
Many overhead costs remain independent of production scale
Labour costs (per unit of product produced) decrease sharply with increased production scale
Table 1.3.3.1: Reasons for the decline in production cost per unit quantity ofproduct
with the increased production scale (Adaptedfrom Walsh and Headon, 1997).
Many of the techniques used in a laboratory scale purification are not amenable to scale-up. For example, the application of sonication for bacterial cell wall disruption,
whilst feasible on a small scale, is inefficient when applied to large scale procedures. Likewise the use of lysozyme to degrade bacterial cell walls is a commonly used
laboratory scale technique, however, the large scale application of such a procedure
would be uneconomic (Ogez et al, 1989). Other routinely used laboratory scale protein purification techniques must be modified before they can be successfully
employed on a large scale, with for example, laboratory scale centrifugation,
generally being performed in a batch centrifuge, whereas continuous flow centrifuges are employed in industrial-scale purification systems (Christi and Moo-Young, 1986).
1.3.3.2 Considerations for Process Scale
Rational design of protein purification, with all stages being amenable to direct scale-
up, is especially desirable when working with therapeutic proteins (Walsh and
Headon, 1997). Such proteins are initially produced in small quantities which are
then subjected to animal trials. If encouraging results are obtained, limited clinical
studies may then be initiated and whilst such trials may require limited protein, the
trials are in fact expensive to run and could take several years to complete (Gosse, et
al, 1996). Successful clinical trials will trigger the scale-up of production in line
changes made to the original purification process would invalidate the earlier clinical
studies. Therefore it is important to ensure that the purification system initially
developed in the laboratory can be scaled up without difficulty. The three stages
involved in the scale-up of protein purification are outlined in Figure 1.3.3.3.
However, in recent years, under the Comparability Protocol, it is possible for some
changes to be made to the production process, provided that the product can be well
specified or characterised to an acceptable new level.
The design of such a protein purification procedure should contain the minimum
number of steps required to yield a protein product within the designated product
specifications (Fish an Lilly, 1984). Although the scale and level of purification
required can vary from product to product, most purification systems share some common attributes, these being an extraction step as the first step in the purification
protocol, followed by the preliminary treatment of the crude extract to clarify and concentrate the crude material.
The engineering of E. coli to secrete functional chimeric antibody fragments which
can be purified directly from the fermentation medium has a number of potentials for large-scale production, notably the avoidance of excessive dénaturation and
renaturation steps required for the purification of intracellularly produced proteins (Gavit et ai, 1992). Gavit et ai (1992) developed a process to prepare biologically
active chimeric ING-2 Fab secreted from E. coli using the same vectors and secretion
system described by Better et al, (1973). The process designed by Better et a l
(1973) allowed a greater than 95% purity to be achieved, for endotoxin levels to be
reduced to less than 0.05 EU/mg Fab, and for DNA concentrations to be reduced to less than 1 pg/mg Fab (Gavit et a l, 1992).
The purification scheme for ING-2 employed by Gavit et al (1992) consisted of a
series of ion-exchange chromatography steps interspersed with membrane
diafiltration or ultrafiltration, with the effectiveness of each step in the removal of
contaminating proteins, endotoxins, DNA and salts being evaluated. Choosing a
secretion system precludes the need for cell lysis and the problems associated with
1.4. Primary protein recovery
The selection of purification steps depends on the nature of the end product, its
concentration, the side products present, the stability of the biological material, and
the necessary degree of purification required. Biochemical engineers are concerned
with the safe, efficient and economic processing of a wide range of materials of
biological origin, with their interest spanning operational scales of a few grams in the
case of some highly potent human proteins, to multi-tonne products such as foodstuffs
(Hoare & Titchner-Hooker, 1990).
There is now increasing pressure to develop new methods of recovering cells and cellular products from fermentation material (Strathmann, 1985; Cooney, 1990). Any
separation technique that is developed for the recovery of a product must be designed
so that the operating conditions cause minimal damage to the product or cells being
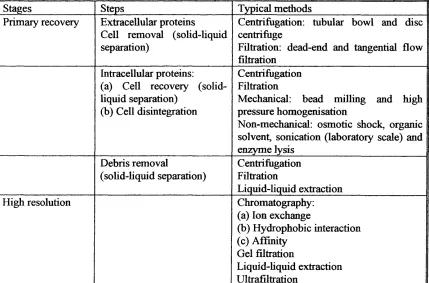
recovered. Typical methods involved in protein downstream processing are outlined in Table. 1.4.
Stages Steps Typical methods Primary recovery Extracellular proteins
Cell removal (solid-liquid separation)
Centrifugation: tubular bowl and disc centrifuge
Filtration: dead-end and tangential flow filtration
Intracellular proteins; (a) Cell recovery (solid-liquid separation)
(b) Cell disintegration
Centrifugation Filtration
Mechanical: bead milling and high pressure homogenisation
Non-mechanical: osmotic shock, organic solvent, sonication (laboratory scale) and enzyme lysis Debris removal (sohd-liquid separation) Centrifugation Filtration Liquid-liquid extraction High resolution Chromatography:
(a) Ion exchange
(b) Hydrophobic interaction (c) Affinity
Gel filtration
Liquid-hquid extraction Ultrafiltration
1.4.1. Product release techniques
The use of genetic engineering to target the production of a heterologous protein to
the E. coli periplasm provides an opportunity for product recovery without
introducing additional intracellular or extracellular contaminants (Chaib et al, 1995;
Biedermann and Jepsen, 1989). The opposite approach is taken by Christi and Moo-
Young (1986). Here they suggest that an intracellular product increases the process
expense as additional cell concentration and process release steps are required. They
have, therefore, recommended that product release into the fermentation medium is
an advantage, but this will ultimately demand the production of large product
concentrations
Steps involved in product release have been examined by Kula and Shutte (1987), and
Christi and Moo-Young (1986), who identified the need to employ a sufficiently
gentle release method to avoid product contamination through the release of cytoplasmic material or nucleic acids. A summary of the available product release
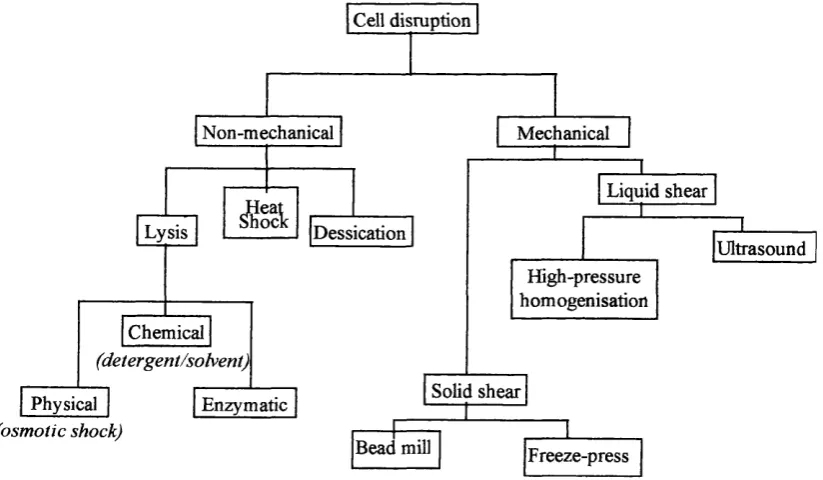
techniques is shown in Figure 1.4.1.
IPessication
(detergent/solvent)
(osmotic shock) Lysis
Physical
Chemical
Enzymatic Solid shear
Ultrasound
Bead mill
Liquid shear Cell disruption
Mechanical
Freeze-press Non-mechanical
High-pressure homogenisation
Figure 1.4.1 Product release techniques. (Adapted from Christi and Moo-Young, 1986).
At present, non-mechanical disruption methods such as osmotic shock, chemical and
due to the specificity of these techniques for single organisms or products, and
difficulties with scale up (Shutte and Kula, 1990). Mechanical methods may involve
liquid or shear operations with the most commonly used industrial scale
homogenisation being, high pressure homogenisation, microfluidisation and bead
milling.
1.4.1.1, Mechanical product release methods
The effectiveness of a mechanical release mechanism is dependent upon the cell wall
strength, which to some extent depends on the culture conditions. During starvation
or limited growth, physiological signals may trigger enforced cell wall development.
Therefore, the mechanical stability of the microorganisms is not a constant, but
dependent on the species involved, growth conditions and biomass history (Kula and
Schutte, 1987).
Mechanical methods for cell disintegration are currently preferred for large scale
work. The two approaches generally taken being high pressure homogenisation or high speed agitator bead mills (Kula and Schutte, 1987). The passage of some cells
through these apparatus will cause cell breakage, thereby releasing cytoplasmic
contaminants and protease’s into the product stream (Veide et al, 1984).
The use of protease inhibitors is accompanied with further process and personnel
problems. The addition of the inhibitor before product release is initiated will
increase the risk of operator exposure, whereas addition after completion of product release will probably be too late to prevent product degradation (Fischer, 1996).
Further disadvantages are associated with high pressure homogenisation, including
the release of heat during the procedure (Takesawa et a l 1990). Heat release is due
to adiabatic compression, where typically 2 °C are released for every 10 MPa compression (Christi and Moo-Young, 1986). The effects of heating can be
significant, especially when the feed has a high solids content. Therefore, for multi
pass cell treatment interstage cooling is required to minimise the rate of proteolysis
and to avoid thermal deactivation of the product.
The other mechanical product release methods, ultrasonication and the freeze press
are only suitable for work on a laboratory scale. Ultrasonication requires a large
free radical will also destoys proteins. Alternatively, the freeze press requires passing
frozen cells through flow restrictions, which when considering the impact that ffeeze-
thaw actions can have on protein configurations renders the technique inappropriate
(Fischer, 1996).
1.4.1.2 Non-mechanical disruption techniques
Selective release of the periplasmic product has been achieved using solvents and
other chemicals to permeabilise the outer cell membrane. Triton X-100 and
guanidine hydrochloride were used by Naglak and Wang (1990) to permeabilise the
outer membrane of E. coli. They identified that selective release of the periplasmic
contents was possible using guanidine hydrochloride alone, and that the addition of
Triton X-100 led also to the release of the cell’s cytoplasmic contents.
Chaib et al. (1995), compared the chemical treatment of E. coli with guanidine hydrochloride and an osmotic shock procedure for the selective release of cystatin C.
Both approaches were considered options for scale-up by these researchers. A high
dilution product stream resulted from the osmotic shock of E. coli which may not be attractive if protein purification is to be considered. The use of guanidine
hydrochloride provided a more selective product release with a 85-90 % purity being
achieved in a small liquid volume.
A key consideration involving the use of chemicals to aid product release is their
impact on the desired product. Guanidine hydrochloride is a chaotropic agent which
has the ability to denature proteins at high concentration, and can solubilise outer
membrane proteins from E. coli affecting the release of periplasmic contents.
Guanidine hydrochloride was found to cause partial unfolding of cystatin C, rendering
it more susceptible to attack by proteinases or oxidases (Chaib, 1995). Studies
performed with P-lactamase producing recombinant E. coli revealed that 94%
recovery of this periplasmic enzyme was achieved using 0.2M guanidine
hydrochloride over a period of 2-5 hours (Naglak and Wang, 1990). Due to the low
levels of protein released by this method the enzyme specific activity was high.
Osmotic shock was initially developed as a small scale technique. It does, however,
provide a useful alternative to chemical release techniques, as the osmotic shock
(Fischer, 1996). The technique employed by Chaib et a l (1995) is typical. The cells
were recovered by centrifugation and then re-suspended in 25 % (w/v) sucrose
solution containing 100 mM Na2EDTA and 200 mM Tris-HCl at pH 9. After
incubation at 4 the suspension was centrifuged again, and the cells re-suspended in
cold 10 mM Tris buffer, with the periplasmic contents being released into the buffer
in order to balance the osmotic pressure. Biedermann and Jepsen (1989), use a
similar protocol but employ microfiltration for the solid-liquid separations . The
membrane based approach provides a contained method for the periplasmic protein
release, and as long as fouling is not a problem, will lead to a product stream
containing few solids (Levesley and Hoare, 1999).
Biedermann and Jepsen (1989), found that the addition of lysozyme during osmotic
shock resulted in a higher degree of periplasmic release. Similar results have been reported by French et al. (1996) and is thought to be due to degradation of the
peptidoglycan matrix in the periplasm. Chisti and Moo-Young (1986), suggest that
the use of lysozyme will lead to an increased selective recovery of the periplasmic proteins, as the enzyme is targeted towards the periplasmic peptidoglycan. The use of
lysozyme was previously thought too expensive (Naglak and Wang, 1990), but as
research advances other fermentative sources of lysozyme may become available; e.g., T4 lysozyme has been produced in E. coli, and protein engineering has been used
(insertion of poly-histidine tails) to allow the recovery and recycling of the enzyme
using Immobilised Metal Ion Affinity Chromatography (Sloane et al. 1996, Willoughby et al., 1999). O’Brien et al. (1996) report the successful application of
this lysozyme in the selective release of recombinant a-amylase from E. coli.
Product release of a-amylase from E. coli may also be possible using a heat shock,
heating the broth to 80 °C and holding this temperature for 10 minutes (Lee et al.
1992). This process is unlikely to be selective to periplasmic proteins, and is also
likely to lead to aggregation and inactivation of proteins present, rendering the
processing of the stream produced difficult (Fischer, 1996).
1.4.2. Process design issues for periplasmic product release
Due to the variety of proteins which may be produced from microorganisms there is