International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
897
Automatic Parallelization for Parallel Architectures Using
Smith Waterman Algorithm
Ananth Prabhu G
1, Dr. Ganesh Aithal
2 1Assistant Professor, Dept of CS&E, Sahyadri College of Engineering & Management, Mangalore. 2Professor, HOD CS&E, P.A. College of Engineering, Mangalore.
Abstract- Gene sequencing problems are one of the
predominant issues for researchers to come up with an optimized system model that could facilitate the optimum processing and efficiency without introducing overheads in terms of memory and time. This research is oriented towards developing such kind of system while taking into consideration of the dynamic programming approach called a Smith Waterman algorithm in its enhanced form decorated with other supporting and optimized techniques. This paper provides an introduction oriented knowledge transfer so as to provide a brief introduction of research domains, research gap and motivations, objective formulated and proposed systems to accomplish ultimate objectives.
Keywords- Smith-Waterman (SW), Multiple-Instruction,
Multiple-Data (MIMD), Single Instruction, Multiple Data (SIMD), Smith-Waterman algorithm (SW), Diagonal Parallel Sequencing and Alignment Approach (DPSAA),
I. INTRODUCTION
Bioinformatics and Computational Biology (BCB) is a relatively new multidisciplinary field which brings together many aspects of the fields of biology, computer science, statistics, and engineering. Bioinformatics extracts useful information from biological data and makes these more intuitive and understandable by applying principles of
information sciences, while computational biology
harnesses computational approaches and technologies to answer biological questions conveniently. In the present days scenario the approaches of genomics have played a vital role in optimizing parallel processing systems[1].
Genomics is an emerging field, constantly presenting many new challenges to researchers in both biological and computational aspects of applications. Genomics applications can be very computationally intensive, due to the magnitude of the data sets involved, such as the three billion base pair human genome. Some genomics applications include protein folding, RNA folding, de novo assembly, and sequencing analysis using hidden Markov models.
Many of these applications involve either searching a database for a target sequence, as done with hidden Markov models, or performing computations to characterize the behavior of a particular sequence, as performed by both protein and RNA folding. Genomics applications therefore require immense quantities of computation, and they also tend to require the movement and storage of very large data sets. These data sets are either in the form of a database to be searched, as in sequence searching with hidden Markov models, or in the form of a sequence to be computed and operated upon, such as the sequence of amino acids of a protein being folded. These two challenges tend to lead to long execution times for software programs, which can result in a tool being unusable for researchers[2].
While considering the potential application of genomics approach the short read genome mapping cannot be ignored. Short read mapping attempts to determine a sample DNA sequence by locating the origin of millions of short length reads in a known reference sequence. Genome mapping is performed in this manner, because machines that sequence, these short reads are able to do so in a very parallel manner, producing reads at a much faster rate than traditional sequencing technology. The increasing growth and complexity of high-performance computing as well as the stellar data growth in the Bioinformatics field stand as posts guiding this research work[1].
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
898 Once the target protein/DNA/RNA is reassembled, the string can be used for analysis. One type of analysis is sequence alignment. It compares the new query string to already known and recorded sequences [1]. Comparing (aligning) sequences is an attempt to determine common ancestry or common functionality [2]. This analysis uses the fact that evolution is a conservative process [3].
Over the last few years, the newly developed biological techniques became largely intertwined with Bioinformatics. The generated data is accessible for computational analysis; the computation is even required for the interpretation of the data. With protein-protein interaction, microarray and 2D gel databases with mass spectrometry now becoming available as a primary resource of the most relevant information, tomorrow's biological research will have a chance to be by a much greater extend initiated by computational analysis. The formalization of experimental evidence for transmembrane protein topology may take a role as an extension to the actual sequence annotation. This is a powerful tool, making sequence alignment the most common operation used in computational molecular biology [1]. Now that much of the actual process of sequencing is automated, a huge amount of quantitative information is being generated. As a result, the gene and protein databases such as GenBank and Swiss-Prot are nearly doubling in size each year. New databases of sequences are growing as well. In order to use sequence alignment as a sorting tool and obtain qualitative results from the exponentially growing databases, it is more important than ever to have effective, efficient sequence alignment analysis algorithms.
Advances in sequencing technology have significantly increased data generation, requiring similar computational
advances for Bioinformatics analysis. Computer
architectures based on reconfigurable computing can reduce application run times from hours to minutes, while addressing problems unapproachable with commodity servers. The increased capability also improves research quality by allowing more accurate, previously impractical approaches. This paper discusses the approach used a search and alignment program using the Smith-Waterman algorithm. The proposed system is optimized for architecture to dramatically reduce the time to perform large numbers of local alignments. A query file consisting of one or more nucleotide or protein sequences can be compared to a database using linear or tabular scoring and affine gap penalties.
II. PAIRWISE SEQUENCE ALIGNMENT
Pairwise sequence alignment is a one-to-one analysis between two of the sequences (strings). It takes as input a query string and a second sequence, outputting an alignment of the base pairs (characters) of both strings. A strong alignment between two sequences indicates sequence similarity. The similarity between a novel sequence and a studied sequence or gene reveals clues about the evolution, structure, and function of the novel sequence via the characterized sequence or gene. In the future, sequence alignment could be used to establish an individual's likelihood for a given disease, phenotype, trait, or medication resistance. The goal of sequence alignment is to align the bases (characters) between the strings. This alignment is the best estimate of the actual evolutionary history of Best here refers to the best alignment according to a specific evolutionary model used. When trying to determine the common functionality or properties that have been conserved over time between two sequences (sometimes genes), sequence alignment assumes that the two sample donors are homologous, descended from a common ancestor.
Regardless of the homology assumption, this is still a very relevant type of analysis. For instance, sequences of homologous genes in mice and humans are 85% similar on average [4], allowing for valid sequence analysis. An example of an exact alignment of two strings, S1 and S2, can consist of substitution mutations, deletion gaps, and insertion gaps known as indels. The terms are defined with regard to transforming string S1 into string S2: a substitution is a letter in S1 being replaced by a letter of S2, a mutation is when S1
≠
S2 a deletion gap character appears in S1 but does not appear in S2, and for an insertion gap, the letters of S2 do not exist S1 [4]. The following example contains thirteen matches, an insertion gap of length one, a deletion gap of length two, and one mismatch.AGCTA-CGTACACTACC AGCTATCGTAC--TAGC
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
899 A variation by Smith and Waterman allows for local alignment [6]. A minor adjustment by Gotoh [7] greatly improved the running time from 0(m2n) to 0(mn) where m and n are the sequence sizes being compared. It is this algorithm that is often referred to as the Smith-Waterman algorithm [8] [9] [10]. Both compare two sequences against each other. If the two string sizes are of size m and n respectively, then the running time is proportional to the product of their size, or0(mn). When the two strings are of equal size, the resulting algorithm can be considered an 0(n2) algorithm.
These dynamic programming algorithms are rigorous in that they will always find the single best alignment. The drawback to these powerful methods is that they are time consuming and that they only return a single result. In this context, heuristic algorithms have gained popularity for performing local sequence alignment quickly while revealing multiple regions of local similarity.
In this research work, then we has developed a highly efficient scheme for parallelization of gene sequencing while optimizing the existing approach of Smith-Waterman (SW) algorithm. A brief of this approach has been discussed in the following section. A complete research oriented discussion for pairwise sequence alignment has been discussed in Chapter-4 of the presented thesis.
III. SMITH-WATERMAN SEQUENCE ALIGNMENT
One of the predominant significance of Smith-Waterman algorithm (SW) is that this technique facilitates local sequencing and alignment facility which is a dynamic programming application that differs from the Needleman-Wunsch (NW) algorithm. Local alignment does not require entire sequences to be positioned against one another. Instead it tries to find local regions of similarity, or sub-sequence homology, aligning those highly conserved regions between the two sequences. Since it is not concerned with an alignment that stretches across the entire length the strings, a local alignment can begin and end anywhere within the two sequences.
The Smith-Waterman [6] and Gotoh [7] algorithm is a dynamic programming algorithm that performs local sequence alignment on two strings of data, and . The size of these strings is and , respectively, as stated previously. The dynamic programming approach uses a table or matrix to preserve values and avoid re-computation. This method creates data dependencies among the different values.
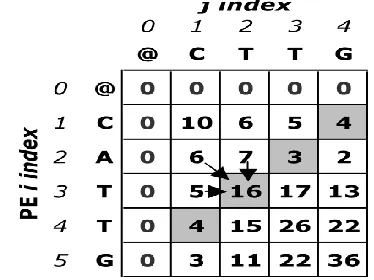
[image:3.612.343.530.187.327.2]A matrix entry cannot be computed without prior computation of its north, west and northwestern neighbors as seen in Figure 1 Equations 1-4 describe the recursive relationships between the computations.
Figure 1: An example of the sequential Smith-Waterman matrix.
The dependencies of cell (3, 2) are shown with arrows. While the calculated values for the entire matrix are
given, the shaded anti-diagonal (where all values are
equal) shows one wave front or logical parallel step since they can be computed concurrently. Fine gap penalties are used in this example as well as in the parallel code that produces the top alignment and other top scoring alignments.
The Smith-Waterman algorithm, allows for insertion and deletions of base pairs, referred to as indels. To find the best scoring alignment with all possible indels and alignments is computationally and memory intensive, therefore a good candidate for parallelization.
In the following section a generic functional approach of Smith Waterman technique has been elaborated. Further discussion with numerous significances has been discussed in Chapter-3.
As outlined in [7], several values are required to be computed for every possible combination of deletions insertions and matches . For a deletion with affine gap penalties, Equation 1.1 can be employed for computing the current cell's value using the north neighbors’ values
for a match minus the cost to open up a new gap .
The other value used from the north neighbor is
the cost of an already opened gap from the north. From those, the gap extension penalty is subtracted.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
900 An insertion is similar in Equation 1.2, using the western
neighbors match and an existing open gap values,
subtracting the cost to extend a gap.
1.2
To compute a match where a character from both sequences is aligned, the values for is required to be estimated, where the actual base pairs, can be obtained by Equation 1.3.
1.3
This value is then combined with the overall score of the northwest neighbor, and the maximum value from and zero becomes the new final score for that
cell (Equation 1.4).
1.4
Once the matrix has been fully computed the second, distinct part of the SW algorithm performs a traceback. Starting with the maximum value in the matrix, the algorithm will backtrack based on which of the three values
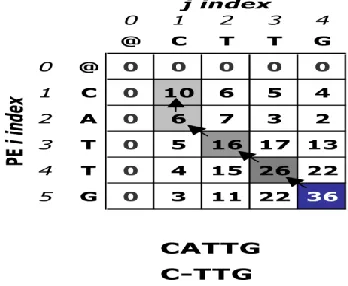
[image:4.612.73.247.515.658.2]was used to compute the maximum final value. The backtracking stops when a zero is reached. Below is an example of a completed matrix in Figure 2, showing the traceback and the corresponding local alignment.
Figure 2: Smith-Waterman matrix with traceback and resulting alignment.
IV. PARALLEL COMPUTING MODELS
The main parallel model used to develop and extend Smith-Waterman sequence alignment is the Associative Computing (ASC) [11]. The goal of this research was to develop and extend efficient parallel versions of the Smith-Waterman algorithm. These models as well as another that were used for this research are described in detail here in this chapter. In order to understand a better system model to be developed for parallel programming and computing
paradigm, the associated system frameworks or
computational architecture is required to be understood. The following section of the presented manuscript, discusses the models of parallel computing in real time computational problems.
A. Models of Parallel Computation
In this section of the presented manuscript few of the dominant technical factors have been defined. Two terms of interest from Flynn's Taxonomy of computer architectures are MIMD and SIMD, the two different models of parallel computing utilized in this research. A cluster of computers, classified as a multiple-instruction, multiple-data (MIMD) model is used as a proof-of-concept to overcome memory limitations in extremely large-scale alignments. As considering the presented research or thesis work, the author has taken into consideration of the Smith Waterman algorithm, which is efficient for being functional with multiple instructions, multiple data case or MIMD scenario. A brief of MIMD has been given as follows. In this research work, author emphasizes to enhance existing MIMD systems with introducing certain optimization paradigm in terms of associated functional components and performance parameters.
B. Multiple Instruction, Multiple Data (MIMD)
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
901 The data is moved along the network asynchronously by individual processors under the control of their individual program they are executing. Typically, communication is handled by one of several different parallel languages that support message-passing. A very common library for this is known as the Message Passing Interface (MPI). Communication in a \SIMD-like" fashion is possible, but the data movements will be asynchronous. Parallel computations by MIMDs usually require extensive communication and frequent synchronizations unless the various tasks being executed by the processors are highly independent.
Data-parallel programming is also an important technique for MIMD programming, but here all the tasks perform the same operation on different data and are only synchronized at various critical points. The majority of algorithms for MIMD systems are written in the Single-Program, Multiple-Data (SPMD) programming paradigm. Each processor has its own copy of the same program, executing the sections of the code specific to that processor or core on its local data. The popularity of the SPMD paradigm stems from the fact that it is quite difficult to write a large number of different programs that will be executed concurrently across different processors and still be able to cooperate on solving a single problem.
C. Single Instruction, Multiple Data (SIMD)
The SIMD model consists of multiple, simple arithmetic processing elements called PEs. Each PE has its own local memory that it can fetch and store from, but it does not have the ability to compile or execute a program. The compilation and execution of programs are handled by a processor called a control unit [12]. The control unit is connected to all PEs, usually by a bus. All active PEs execute the program instructions received from the control unit synchronously in lock-step. "In any time unit, a single operation is in the same state of execution on multiple processing units, each manipulating different data" [12]. While the same instruction is executed at the same time in parallel by all active PEs, some PEs may be allowed to skip
any particular instruction [13]. This is usually
accomplished using an if-else" branch structure where some of the PEs execute the if instructions and the remaining PEs execute the else part. This model is ideal for problems that are parallel in nature that have at most a small number of if-else branching structures that can occur simultaneously, such as image processing and matrix operations.
Data can be broadcast to all active PEs by the control unit and the control unit can also obtain data values from a particular PE using the connection (usually a bus) between the control unit and the PEs. Additionally, the set of PE are connected by an interconnection network, such as a linear array, 2-D mesh, or hypercube that provides parallel data movement between the PEs. Data is moved through this network in synchronous parallel fashion by the PEs, which executes the instructions including data movement, in lock-step. It is the control unit that broadcasts the instructions to the PEs. In particular, the SIMD network does not use the message-passing paradigm used by most parallel computers today. An important advantage of this is that SIMD network communication is extremely efficient and the maximum time required for the communication can be determined by the worst-case time of the algorithm controlling that particular communication.
This is the matter of fact that there have been great evolution in parallel computing and its optimization, still it possess a huge potential to be explored for making it optimized. A Proper tracking approach might make the smith Waterman algorithm to function with different dimensional computation. One of the predominant problems with smith waterman approach is its tracking facility and optimal alignment sequencing. Being a local sequential alignment approach this algorithm suffers a lot when it has to be employed with huge gene data backs. While considering its applications for huge data processing and computation, still it cannot be stated as the optimal approach. Therefore there is the need to develop such a system model, which can not only optimize the computational efficiency but also the time factor, so as to grab optimum performance in case of biological sequencing applications and parallel computing facilities. On the other hand, it can be realized that an optimized approach of trace back and tracking with optimum mid points and alignment sequencing can provide a better solution for Smith Waterman Algorithm and hence its applicability for gene sequential alignments or parallel computing applications.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
902 In this research work, the author has developed an optimum biological sequential approach based on optimized Smith waterman algorithm. The systemic development and its realization have been discussed in Chapter-4.
D. Motivation
As per the increase in the computational complexity and more speedy increase in highly efficient computing systems, certain optimized parallel computing approaches are required. A number of approaches have been developed for parallel computing and sequential alignments problem. Few of the predominant systems were based on local alignment schemes. One motivation for local alignment is the difficulty of obtaining correct alignments in regions of low similarity between distantly related biological sequences, because mutations have added too much 'noise' over evolutionary time to allow for a meaningful comparison of those regions. Local alignment avoids such regions altogether and focuses on those with a positive score, i.e. those with an evolutionary conserved signal of similarity. A prerequisite for local alignment is a negative expectation score. The expectation score is defined as the average score that the scoring system (substitution matrix and gap penalties) would yield for a random sequence.
Another motivation for using local alignments approach for parallel sequencing and alignments is that there is a reliable statistical model for optimal local alignments. The alignment of unrelated sequences tends to produce optimal local alignment scores which follow an extreme value distribution. This property allows programs to produce an expectation value for the optimal local alignment of two sequences, which is a measure of how often two unrelated sequences would produce an optimal local alignment whose score is greater than or equal to the observed score. Very low expectation values indicate that the two sequences in question might be homologous, meaning they might share a common ancestor.
The Smith–Waterman algorithm is fairly demanding of
time: To align two sequences of
lengths m and n, 0(mn) time is required. Smith–Waterman local similarity scores can be calculated in 0(m) (linear) space if only the optimal alignment needs to be found, but naive algorithms to produce the alignment require 0(mn) space. A linear space strategy to find the best local alignment has been described. The approach of BLAST (Basic Local Alignment Search Tool) and FASTA reduce the amount of time required by identifying conserved regions using rapid lookup strategies, at the cost of exactness.
Intra-task parallelization approach makes the system functional not only by means of higher computational efficacy but also reduced memory occupancy. On the other hand DP lookup table generation makes the system operational while optimizing resource utilization and table consideration for sequencing problems. These are the factors which can enhance the conventional approach of gene sequencing and therefore the optimization of these approaches must be considered to come up with better results for aforementioned requirements.
This is the matter of fact that the existing approaches of the Smith Waterman with its two approaches, serial and parallel sequencing has performed well, but considering present needs and data computation scenario, that can’t be assured for delivering optimum. The parallel sequencing approach is better but while considering other strategic implementations such as Millers and Mayors scheme, it can be expected that the resulting computational efficiency would be far better. And if this scheme is implemented with Backtracking scheme with diagonal architecture, that it can be a revolution in the field of parallel programming and computation for gene sequencing. Considering certain mathematical alignment issues and parallel computing aspects it can be realized that the diagonal computation with very precise tracking or backtracking facility might come up with an optimum solution for sequencing and parallel computing and sequential alignment with huge data sets.
Thus, this consideration and motivations have energized the author to come up with a hybrid system model that could deliver not only higher speed and optimized computation efficacy but also a system with minimum memory occupancy.
E. Research Objective
Considering the requirement of a highly robust and efficient system model and approach for parallel computing applications, here in this research work, the author proposes to develop a highly optimized parallel programming approach that come up with the optimum sequencing scheme in the form of Diagonal alignment of genes. The overall objectives of this research work have been classified into two categories. The first states for General Objectives while another represents Specific Objectives.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
903 While Specific objectives are those objectives which do specifies the exact and precise presentation of approaches and techniques to be used to accomplish the general objectives. These research objectives have been given as follows:
F. General Objectives
To develop a novel scheme for Biological gene
sequencing application.
To develop a highly robust and efficient approach to parallel computing that could deliver higher computational efficiency even with minimum time and space requirements for varying sequential length.
To develop Diagonal Parallel Sequencing and
Alignment Approach (DPSAA) for parallel
computing applications.
To develop an approach for enhancing conventional Smith Waterman (SW) algorithm for gene sequencing and alignment applications.
To compare the proposed Diagonal sequencing or
alignment approach with other existing parallel and serial, sequential alignment techniques and justify that diagonal sequencing can be of great significance.
G. Specific Objectives
To develop a highly robust and optimized Smith Waterman (SW) algorithm based system model for optimal and efficient local sequence alignment applications.
To develop an Intra-task kernel based parallelization
technique for Diagonal Parallel Gene Sequence
Alignment Approach that could bring minimum time complexity with higher computation rate with minimum instruction sets required to execute sequential queries as compared to other parallel as well as serial gene sequencing techniques.
To implement the optimized Myers-Miller algorithm approach with a goal to minimize the memory occupancy and speed up the processing rate even with enhanced computation.
To develop an optimization approach for
accomplishing enhanced Backtracking facility,
optimum sequential distance computation, pairwise
estimation and parallelization of progressive
sequential alignment facility for Diagonal sequencing alignment applications such as biological gene sequencing.
V. PROPOSED SYSTEM
Considering the requirement of a highly optimized and efficient system for gene alignment and biological sequencing with the parallel computing process, the author of this research has proposed Smith Waterman based
system developed that could accomplish higher
computation rate and gene sequencing.
In this research work, the author has proposed a Smith Waterman algorithm based diagonal sequencing approach that could accomplish the higher rate sequencing with diagonal scheme. Here in this research work, the author proposes to implement Myers and Millers algorithm enriched sequencing system that can accomplish optimum performance even with minimum space consumption and complexity. The author proposes to implement Intra-task parallelization based backtracking and sequencing while
optimizing trace-backing and optimum midpoint
estimation. In this work, in order to accomplish a fast sequencing and processing facility the author advocate for a sequential distance computation algorithm that estimates the distance between metrics and nodes so as to speed up the overall processing of alignments. For developing enhanced sequential distance estimation algorithm, here in this work the author proposes to develop a novel pairwise alignment scheme that achieves the goal of optimal local alignment by means of a forward pass and a reverse pass on the alignment matrix of two different sequences using the Smith-Waterman (SW) algorithm. Here in this work, the author takes into consideration of intra-task parallelogram kernels so as to reduce memory as well as computational counts. On the other hand the consideration of DP lookup table has been proposed only for reducing computational cost and speedup ratio while performing on gene sequence with longer query length. The emphasis on trace backing has been advocated so as to enrich forward and reverse sequencing paradigm and with reduced exploration overheads. The author proposes to develop an enhanced algorithm for optimum trace backing, mean estimation and sequence distance computation. Even here in this thesis, for precise and efficient alignment purpose the author has proposed for optimized pairwise alignment issue that takes care of every associated parametric optimization on Smith Waterman algorithm.
VI. PROBLEM DEFINITION
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
904 This task cannot be adequately solved by traditional string matching methods because genomic sequences that share the same biological purpose mutate over time when exposed to evolutionary events, and may no longer match identically. Genomic sequence comparison tools such as BLAST (Basic Local Alignment Search Tool) and Hidden Markov Models are based upon approximate string matching principles that measure the overall similarity between strings and are thus more tolerant of mismatches. This fuzzy string matching problem can be formulated in two different ways. The similarity between two strings can be assessed explicitly, by minimizing an ad hoc cost function (e.g. edit distance) over all possible alignments between the strings. Alternatively, a similarity score can also be computed stochastically, by finding the maximum likelihood path through a hidden Markov model (HMM) trained from an input string. Both of these approaches are optimization problems that require dynamic programming (DP) to solve. The DP step is often very computationally expensive, especially when comparing large strings. Fortunately, the data dependencies in the recurrence relations allow some degree of parallelism and the computation for some cell entries of the DP table can be distributed across a set of processors.
The Smith-Waterman (SW) algorithm searches for a sequence database to identify the similarities between a query sequence and subject sequences. However, this algorithm is prohibitively high in terms of time and space complexity; the exponential growth of sequence databases also poses computational challenges. The Smith-Waterman (SW) algorithm [14][15] is a dynamic programming-based approach to identify optimal local alignments of biological sequence pairs. Due to its maximal sensitivity for local alignments, this algorithm is a fundamental operation in bioinformatics, including biological sequence database search, multiple sequence alignment [16][17] and next-generation sequencing read alignment [18][19].
In biological sequence database search, the similarities between sequences can be inferred from optimal local alignment scores calculated by the SW algorithm. To calculate optimal local alignment scores, the SW algorithm has a linear space complexity and a quadratic time complexity. However, this quadratic time complexity makes the SW algorithm computationally demanding for large-scale sequence database search. This is further compounded by the rapid growth of sequence databases. Therefore, several heuristics such as FASTA [20] and BLAST have been proposed to accelerate the sequence database search, but not guaranteeing to discover optimal local alignments.
These heuristics usually produce considerably good results, but might fail to detect some distantly related sequences due to the loss of sensitivity. Hence, it has great significance to accelerate the SW algorithm so as to maintain optimal results. Consequently, a lot of efforts have been made to parallelize this computation on high-performance computing architectures ranging from loosely-coupled to tightly-loosely-coupled ones.
One of the most restrictive characteristics of SW and its variants is the quadratic space needed to store the Dynamic Programming matrices. For instance, in order to compare two 33 MBP (Million Base Pairs) sequences, there is the need of minimum 4.3 PB of memory. Another restrictive characteristic of the SW algorithm is that it is usually slow due to its quadratic time complexity. In order to accelerate the comparison between long sequences, heuristic tools such as LASTZ and MUMMER were created. They use seeds (LASTZ) and suffix trees (MUMMER) to scan the sequences, providing a big picture of the main differences/similarities between them. On the other hand, Smith-Waterman provides the optimal local alignment, where the regions of differences/similarities are much more accurate, as well as the gapped regions that represent inclusion/deletion of bases. Therefore, both kinds of tools can be considered for implementing in a complementary way: first, MUMMER or LASTZ might be executed which would in later stage be processed for SW that would be run to obtain the optimal local alignment for the cases where a more detailed analysis should be made.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
905 Here, in this research work, the author has defined certain optimization and formulated an approach based on Mayors and Millers algorithm that estimates highly optimum trace path and midpoint estimation facility for sequential alignment. The author has formulated the scheme for diagonal reverse estimation and optimal intra-task parallelization scheme for sequential alignment. The intra-task parallelization would cause the reduction in memory occupancy and thus the enhancement with DP lookup table would cause the reduction in total computation cost. Considering the mathematical aspects of alignment and sequencing the diagonal parallel block based sequencing could not only reduce the overall computational cost but also the complexity incurred in exploring entire sequential query length. Here in this approach the author has also introduced a scheme armored with two pair wise alignment implementations, one forward and one reverse and even the adhoc cost gets reduced. The reverse alignment has been optimized with backtracking algorithms and even in diagonal approach that comes up to be least complicated and minimum space consuming. In fact Smith Waterman algorithm would the potential candidate that
could accomplish such modifications without
compromising with its robustness as per increase in sequence length or query length. Therefore, the author has advocated the application of SW approach.
The overall problem formulation in this research work establishes itself for optimal functional for accomplishing ultimate goal of gene sequencing and parallel computing.
REFRENCES
[1] F. Guinand, “Parallelism for computational molecular biology," in ISThmus 2000 Conference on Research and Development for the Information Society, Poznan, Poland, 2000.
[2] L. D'Antonio, “Incorporating bioinformatics in an algorithms course," in Proceedings of the 8th annual conference on Innovation and Technology in Computer Science Education, vol. 35 (3), 2003, pp. 211{214.
[3] H. B. J. Nicholas, D. W. D. II, and A. J. Ropelewski. (Revised 1998) Sequence analysis tutorials: A tutorial on search sequence databases and sequence scoring methods. [Online]. Available: http://www.nrbsc.org/old/education/tutorials/
sequence/db/index.html
[4] X. Huang, Chapter 3: Bio-Sequence Comparison and Alignment, ser. Current Topics in Computational Molecular Biology. Cambridge, MA: The MIT Press, 2002.
[5] S. Needleman and C. Wunch, “A general method applicable to the search for similarities in the amino acid sequences of two proteins," Journal of Molecular Biology, vol. 48, no. 3, pp. 443{453, 1970. [6] T. F. Smith and M. S. Waterman, “Identification of common
molecular subsequences," Journal of Molecular Biology, vol. 147, no. 1, pp. 195{197, 1981.
[7] O. Gotoh, “An improved algorithm for matching biological sequences," Journal of Molecular Biology, vol. 162, no. 3, pp. 705-708, 1982.
[8] X. Huang and W. Miller, “A time-efficient linear-space local similarity algorithm," Adv.Appl.Math., vol. 12, no. 3, pp. 337{357, 1991.
[9] M. Camerson and H. Williams., “Comparing compressed sequences for faster nucleotide blast searches," IEEE/ACM Transactions on Computational Biology and Bioinformatics, vol. 4, no. 3, pp. 349{364, 2007.
[10] J. D. Frey, “The use of the smith-waterman algorithm in melodic song identification." Master's Thesis, Kent State University, 2008. [11] . Potter, J. W. Baker, S. Scott, A. Bansal, C. Leangsuksun, and C.
Asthagiri, “Asc: an associative-computing paradigm," Computer, vol. 27, no. 11, pp. 19{25, 1994.
[12] M. J. Quinn, Parallel Computing: Theory and Practice, 2nd ed. New York: McGraw-Hill, 1994.
[13] J. Baker. (2004) Simd and masc: Course notes from cs 6/73301: Parallel and distributed computing - power point slides.
[14] Smith T, Waterman M: Identification of common molecular subsequences. J Mol Biol 1981, 147:195–197.
[15] Gotoh O: An improved algorithm for matching biological sequences. J Mol Biol 1982, 162:707–708.
[16] Thompson JD, Higgins DG, Gibson TJ: CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22:4673–4680.
[17] Liu Y, Schmidt B, Maskell DL: MSA-CUDA: Multiple Sequence Alignment on Graphics Processing Units with CUDA. 20th IEEE International Conference on Application-specific Systems, Architectures and Processors; 2009:121–128.
[18] Li H, Durbin R: Fast and accurate short read alignment with Burrows Wheeler transform. Bioinformatics 2009, 25(14):1755– 1760.
[19] Liu Y, Schmidt B, Maskell DL: CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows-Wheeler transform. Bioinformatics 2012, 28(14):1830–1837.
International Journal of Emerging Technology and Advanced Engineering
Website: www.ijetae.com (ISSN 2250-2459, ISO 9001:2008 Certified Journal, Volume 4, Issue 3, March 2014)
906 BIBLIOGRAPHY
Ananth Prabhu G received the BE degree in computer science and Engineering in 2007 from VTU, Belgaum and the MTech degree in computer science and engineering in 2010, from the Manipal University, Manipal. Currently, he is working towards the PhD degree in the department of computer science & engineering from VTU, Belgaum. He works as an assistant professor in the department of computer science and engineering in Sahyadri College of Engineering & Management, Mangalore. His research interests include high performance computing, parallel programming, GPGPUs, and load balancing.