Synthesis of amphiphilic peptide-polymer conjugates to investigate the
relationship between polymer structure and self-assembled morphology
By Caleb Cox
Senior Honors Thesis Chemistry
University of North Carolina at Chapel Hill
April 16, 2020
Approved:
1
Abstract
Hybrid peptide-polymer conjugates, peptides coupled with a hydrophobic polymer tail, are of interest due to their potential to combine the precise and diverse functions of peptides with the stability and tunability of polymers. These materials can form hydrophobic-driven aggregates such as micelles, vesicles, or other architectures. Previous studies with peptides coupled with an alkyl chain have shown that the structure and morphology of assembled aggregates strongly depend on the composition of the hydrophobic tail, although, the rational design of peptide conjugates to display specific structures or morphologies is an active area of research. A polymeric hydrophobic tail allows for tuning of hydrophobicity, bulkiness, and length, just by tuning the monomer or synthetic conditions. We expect polymer properties such as hydrophobicity and bulkiness to affect how the conjugates can assemble and pack together, and thus the resulting morphologies. While the landscape of diblock copolymer morphologies has been studied, peptide conjugates are still of interest due to the secondary structures, such as α-helices and β-sheets, that the peptide region of the aggregates can form. Understanding how to control the morphology of the aggregate and the secondary structure of the peptide will provide a new system to study protein binding sites in environments similar to the native state but without having to synthesize the entire protein.
2
Introduction
Peptide-polymer conjugates have garnered attention over the last decade in attempts to combine the beneficial properties of both the peptide and polymer for new functions. Some examples of work have addressed issues in drug delivery; by conjugating a polymer to a drug, properties can be gained such as solubility, increased drug half-life, and increased efficacy by controlling the release of the drug within the body.1–4 In the case of peptide/protein-polymer conjugates, functional proteins can be attached to polymers to increase biocompatibility and reduce biodegradation in the body.1,3,5
Even further, the addition of a hydrophobic alkyl chain to a peptide can lead to the formation of amphiphiles that self-assemble into higher architectures such as micelles or vesicles through hydrophobic-driven aggregation. Previous work has shown that the presence of transition metal ions can also direct this assembly towards separate morphologies.6 Similar hybrid peptide-polymer assemblies have been used as a method to encapsulate drugs that release when triggered by a stimulus such as pH, temperature, or enzymatic cleavage.1,7–9
3 structure not seen in the peptide alone,12 giving evidence that we can modify the surrounding environment of these peptide sequences through self-assembly. Modulating this type of system could allow a different way of studying important binding sites of proteins without having to actually synthesize the entire protein.
To better utilize self-assembly, we want to understand how the properties of the hydrophobic portion of these conjugates, in this case polymers, affect the architecture of the self-assembled structure. To do this, our group has focused on synthesizing peptide-polymer amphiphiles by employing synthetic techniques in polymer chemistry and advances in bio-conjugation chemistry.6,13 We will be able to survey a wide monomer space and examine how properties such as hydrophobicity and bulkiness of pendant groups on the polymer backbone affects the architectures we form in self-assembly. With the knowledge gained from these experiments, we will be able to begin rationally designing compounds to have novel functions that are primarily seen only in large proteins, such as specific metal sequestering.6 Furthermore, by being able to tune the morphology of these self-assembled structures, we can probe questions such as the importance of the peptide matrix for protein binding sites in an environment similar to the native state without having to actually synthesize the entire protein.
4 affects of more hydrophobic and bulky pendant groups, as compared to ethyl acrylate or butyl acrylates. Much of this report will focus on the synthesis and preparation of these oligomers and conjugates since there have not been many reports on similar systems.
Methods
Synthesis of Z33A (sequence: FNMQQQRRFYEALHDKC) was achieved through solid-phase peptide synthesis on a Liberty-Blue peptide synthesizer (CEM corporation). The peptide was synthesized with a cysteine residue at the C-terminus so that the thiol side-chain could be reacted through a Michael addition with our maleimide functionalized polymer.
6 Br O O O R O O N O O O H H O R R O O O O N O O O H H Br + Cu Br PMDETA 50%v Acetone 60 °C, N2,10 min
100:1:1:1 monomer:initiator:Cu-Br:ligand R = Hex, Cy
Br O O O R O O N O O O H H O R SH Et3N
(10 eq.) (11 eq.) DMF, 25 °C, 18hr
S O O O R O O N O O O H H O R S O O O R O O N O O O H H O R
120 °C, 18hr S
O O O R O O N O O O R A) Polymerization B) Substitution C) De-protection S O O O R O O N O O O R FNMQQQRRFYEALHDK N H NH2 O SH + FNMQQQRRFYEALHDK N H NH2 O S S O O O R O O N O O O R
DMF, 85 °C, 1hr TCEP HEPES
1:3:2:20 peptide:polymer:TCEP:HEPES
D) Conjugation
7
Results
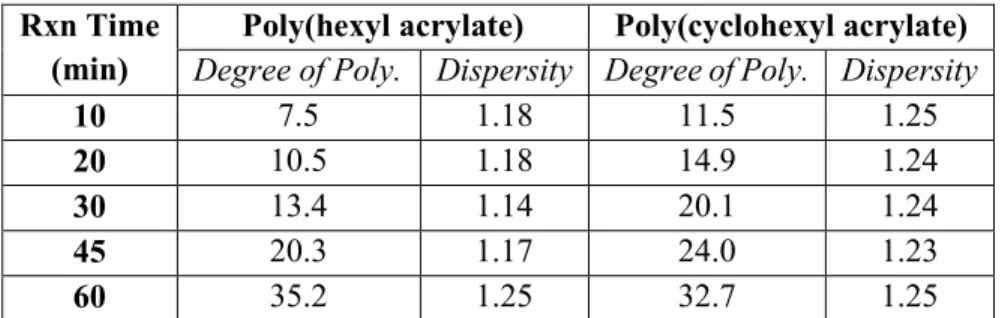
Under the ATRP reaction conditions above, I found that the polymerization proceeded as expected with low dispersities (<1.3) and a nearly linear relationship between reaction time and degree of polymerization, as summarized in Table 1. Moving forward with this study, I replicated the lowest degree of polymerization polymers (10 minute time point) on a larger scale for both the hexyl and cyclohexyl acrylate oligomers. I chose these smaller polymers because they were of similar size to our peptide and we also expected the following substitution and conjugation reactions to have fewer issues, compared to if we began with longer polymers due to intramolecular steric effects.
Rxn Time
(min) Degree of Poly. Dispersity Degree of Poly. Dispersity Poly(hexyl acrylate) Poly(cyclohexyl acrylate)
10 7.5 1.18 11.5 1.25
20 10.5 1.18 14.9 1.24
30 13.4 1.14 20.1 1.24
45 20.3 1.17 24.0 1.23
60 35.2 1.25 32.7 1.25
Below are the 1H-NMR and SEC analysis of the poly(hexyl acrylate) and poly(cyclohexyl acrylate) samples (10 minute reaction times).
8 1H-NMR was used to analyze the reaction products of the polymerization, substitution, and deprotection steps. Figure 2 shows the labeling of important proton peaks on the chain ends and hexyl acrylate pendent group for the 10 minute reaction. The two alkene protons on the protected maleimide chain end were used as reference and the two protons directly neighboring the ester were used to calculate the degree of polymerization. After subtracting the protons from the chain ends and residual monomer, the degree of polymerization was found to be approximately 7. After substitution with propanethiol, 1H-NMR revealed nearly full conversion as indicated by the appearance/shift of the proton near 3.2 ppm. Deprotection by the removal of furan was indicated by the loss of peaks near 5.3 and 6.5 ppm and the shift of the peak from 2.9 to 6.75 ppm. Note that we referenced our ‘deprotected’ spectrum based on the ‘substituted’ spectrum integration of the protons next to the ester (plus 4 Figure 2. 1H-NMR stack of poly(hexyl acrylate) after (A)
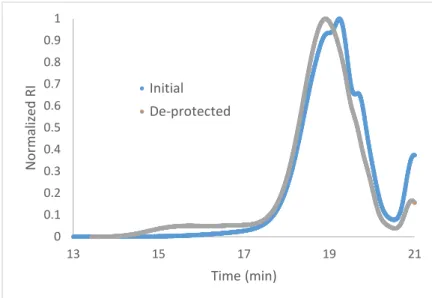
9 extra proton peaks for the chain end) rather than the maleimide proton peaks since there is evidence that some of the maleimide chain ends may have reacted. After deprotection, SEC analysis (Figure 3) showed the formation of a shoulder at the larger molecular weight range, approximately 10 times the molecular weight of our initial sample. This indicates that some polymer reacted with itself and we hypothesized that this deleterious reaction happened through the alkene on the reactive maleimide chain end. Using the integration values of the unreacted polymer and the higher molecular weight shoulder by SEC, approximately 9% of the sample by mass had reacted to form the larger molecular weight substance. If our hypothesis is true that the maleimide chain end reacted to allow polymerization to this higher molecular weight, then we would expect that the maleimide alkene peaks would not integrate to the full two protons when we kept the pendent chain proton integration the same. This was confirmed by integrating the 3.6-4.2 ppm range to the same value after substitution, giving an integration value of ~1.7 rather than 2 for the maleimide alkene protons. This indicates approximately 14% of the chain ends no longer have the reactive alkene group. This is fairly close to the 9% estimate we found using SEC, although we would expect the 1H-NMR to be more accurate since the SEC signal is based on refractive index and the SEC peaks are not well resolved from one another. We did not purify the sample after deprotection since only the unreacted
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
13 15 17 19 21
No
rm
al
ize
d RI
Time (min) Initial
De-protected
10 maleimide would conjugate to our peptide. Moving forward, we did our conjugation based on the effective concentration of the sample since only 86% of the polymer was reactive.
Similar data analysis was used for the polymerization, substitution, and deprotection of the 10 minute poly(cyclohexyl acrylate) sample.
Figure 4 shows the labeling of important proton peaks on the chain ends and cyclohexyl acrylate pendent group. Again, the two alkene protons on the protected maleimide chain end were used as reference and the proton directly neighboring the ester was used to calculate the degree of polymerization. Calculating the degree of polymerization was simpler since there were no overlapping peaks, except for residual monomer. The degree of polymerization was found to be approximately 10 after polymerization, although after substitution this value was recalculated to be approximately 11. Following deprotection, 21% of the maleimide chain ends were estimated to have reacted in a deleterious way, leaving approximately 79% of the sample with reactive maleimide chain ends for conjugation. This was supported by the SEC traces in Figure 5. By Figure 4. 1H-NMR stack of poly(cyclohexyl acrylate) after
11 integration of the shoulder in the ‘deprotected’ trace, approximately 16% of the sample was now in this higher molecular weight form, approximately 20 times the molecular weight of the initial sample. This 16% is in approximate agreement with the 21% loss of maleimide protons found by 1H-NMR. The conjugation reactions of both poly(hexyl acrylate) and poly(cyclohexyl acrylate) with the Z33A peptide were successful, as identified by the masses of peptide-polymer conjugates. This reaction did not go to full completion as indicated by the presence of the peptide mass in the total ion count mass spectra around 4.5 minutes in Figure 6 and Figure 7.
0 0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9 1
13 15 17 19 21
No rm al ize d RI Time (min) Initial De-protected
Figure 5. SEC trace of initial and deprotected poly(cyclohexyl acrylate). Note the formation of the higher molecular weight shoulder near 15 min in the ‘deprotected’ trace.
3 5 7 9 11 13 15 17 19
Tot al Ion C ou nt
Elution Time (min.)
Peptide
Conjugates
Polymers
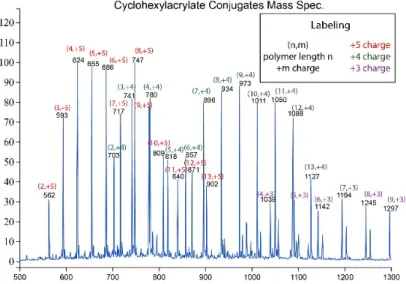
12 The averaged mass spectrum from where the conjugates eluted is included in Figure 8 and Figure 9 with the polymer length and conjugate charge state identified. Figure 8 shows the identified masses of poly(hexyl acrylate)-peptide conjugates with number of monomer units ranging from 2-9. This range is expected with a low degree of polymerization of ~7.
Figure 9 similarly shows the identified masses of poly(hexyl acrylate)-peptide conjugates with number of monomer units ranging from 2-13. This range is expected with the degree of polymerization of ~11, although it is somewhat unexpected that we still see polymers with only 2 Figure 8. Poly(hexyl acrylate)-peptide conjugate mass
spectrum shown from 500 to 1300 Da. Length of polymer in these conjugates ranged 2-9 monomer units over +3 to +5 cahrge states.
3 5 7 9 11 13 15 17 19
Tot
al
Ion
C
ou
nt
Elution time (min.)
Conjugates
Polymers
Peptide
13 monomer units in still significant abundance. This signal could partially be biased towards the shorter conjugates since we would expect the longer polymers to not ionize as well and be detected by the mass spectrum instrument.
Discussion
I have successfully synthesized poly(hexyl acrylate) and poly(cyclohexyl acrylate) as well as conjugates with Z33A peptide, evidenced by 1H-NMR, SEC, and LCMS analysis. While poly(hexyl acrylate) has been synthesized by ATRP in literature15, small oligomers with low dispersities (<1.3) have not. This is the first demonstration of the synthesis of poly(cyclohexyl acrylate) by ATRP as well as the synthesis of small oligomers with low dispersities. While conditions for conjugation of these oligomers to the Z33A peptide remain to be optimized, we have shown that the thiol-maleimide conjugate reaction was successful. Purification by LCMS or other chromatography methods will allow us to isolate the entire conjugate series or a subset (e.g., only oligomers of length 5) for self-assembly analysis. These conjugates will be self-assembled and the morphology will be determined by dynamic light scattering (DLS) and transmission electron microscopy (TEM). Measuring the critical micelle concentration (CMC) will also aid in
14 showing us at what concentration range the conjugates need to self-assemble. Once we make micelles we aim to use circular dichroism (CD) to study the change in secondary structure of the peptide region of the aggregates. These experiments, in context with other peptide-polymer conjugates, will aid in our understanding of how polymer properties affect self-assembly, allowing us to eventually leverage novel ways of studying protein systems.
Conclusion
In this report, I have shown the synthesis of poly(hexyl acrylate) and poly(cyclohexyl acrylate) as well as conjugation with the Z33A peptide. These conjugates will eventually be used to determine property relationships between self-assembled structures and the initial polymer composition. Future work involves expanding this library to include other acrylate polymers (e.g., ethyl acrylate, butyl acrylate) as well as other monomer families. Further, while we know the hydrophobic polymer controls aspects of self-assembly, the peptide region will likely be of similar importance. We plan to use these conjugatable polymers with other peptides to even further expand our library and information space. With these newfound relations, we will continue studying and developing hybrid materials with novel properties.
References
(1) Paik, B. A.; Mane, S. R.; Jia, X.; Kiick, K. L. J. Mater. Chem. B 2017, 5, 8274–8288. (2) Langer, R.; Tirrell, D. Nature 2004, 428, 487–492.
(3) Dong, H.; Dube, N.; Shu, J.; Seo, J.; Mahakian, L.; Ferrara, K.; Xu, T. ACS Nano 2012, 6 (6), 5320–5329.
15 (5) Ruoslahti, E.; Bhatia, S. N.; Sailor, M. J. J. Cell Biol. 2010, 188 (6), 759–768.
(6) Knight, A.; Larsson, J.; Ren, J.; Zerdan, R.; Seguin, S.; Vrahas, R.; Liu, J.; Ren, G.; Hawker, C. J. Am. Chem. Soc. 2018, 140, 1409–1414.
(7) Panda, J. J.; Mishra, A.; Basu, A.; Chauhan, V. S. Biomacromolecules 2008, 9, 2244– 2250.
(8) Bacinello, D.; Garanger, E.; Taton, D.; Chui, K.; Lecommandoux, S. Eur. Polym. J. 2015, 62, 363–373.
(9) Kalafatovic, D.; Nobis, M.; Javid, N.; Frederix, P. W. J. M.; Anderson, K. I.; Saunders, R.; Ulijn, R. V. Biomater. Sci. 2015, 3, 246–249.
(10) Ku, T.; Chien, M.; Thompson, M. P.; Sinkovits, R. S.; Norman, H.; Baker, T. S.; Gianneschi, N. C. J. Am. Chem. Soc. 2013, 133 (22), 8392–8395.
(11) Wang, C.; Chen, Q.; Wang, Z.; Zhang, X. Angew. Chemie - Int. Ed. 2010, 49, 8612–8615. (12) Nieuwland, M.; Ruizendaal, L.; Brinkmann, A.; Kroon-Batenburg, L.; Van Hest, J. C. M.;
Löwik, D. W. P. M. Faraday Discuss. 2013, 166, 361–379. (13) Kalia, J.; Raines, R. Curr. Org. Chem. 2010, 14 (2), 138–147.
(14) Li, Y.; Wang, Y.; Ou, S.; Lock, L. L.; Xu, X.; Ghose, S.; Li, Z. J.; Cui, H. Biomacromolecules 2017, 18, 3611–3620.
(15) Datta, H.; Singha, N. K. J. Polym. Sci. Part A Polym. Chem. 2008, 3499–3511.
(16) Mantovani, G.; Tao, L.; Haddleton, D. M. J. Am. Chem. Soc. 2005, 127 (7), 2966–2973. (17) Anastasaki, A.; Willenbacher, J.; Fleischmann, C.; Gutekunst, W. R.; Hawker, C. J.