0095-1137/95/$04.0010
Copyrightq1995, American Society for Microbiology
PCR Assay Based on DNA Coding for 16S rRNA for Detection
and Identification of Mycobacteria in Clinical Samples
L. F. F. KOX,
1,2* J.
VAN
LEEUWEN,
1S. KNIJPER,
1H. M. JANSEN,
2ANDA. H. J. KOLK
1Department of Biomedical Research, Royal Tropical Institute,
1and Division of
Pulmonary Diseases, Academic Medical Centre,
2Amsterdam, The Netherlands
Received 12 May 1995/Returned for modification 12 July 1995/Accepted 19 September 1995
A PCR and a reverse cross blot hybridization assay were developed for the detection and identification of
mycobacteria in clinical samples. The PCR amplifies a part of the DNA coding for 16S rRNA with a set of
primers that is specific for the genus Mycobacterium and that flanks species-specific sequences within the genes
coding for 16S rRNA. The PCR product is analyzed in a reverse cross blot hybridization assay with probes
specific for M. tuberculosis complex (pTub1), M. avium (pAvi3), M. intracellulare (pInt5 and pInt7), M. kansasii
complex-M. scrofulaceum complex (pKan1), M. xenopi (pXen1), M. fortuitum (pFor1), M. smegmatis (pSme1),
and Mycobacterium spp. (pMyc5a). The PCR assay can detect 10 fg of DNA, the equivalent of two mycobacteria.
The specificities of the probes were tested with 108 mycobacterial strains (33 species) and 31 nonmycobacterial
strains (of 17 genera). The probes pAvi3, pInt5, pInt7, pKan1, pXen1, and pMyc5a were specific. With probes
pTub1, pFor1, and pSme1, slight cross hybridization occurred. However, the mycobacterial strains from which
the cross-hybridizing PCR products were derived belonged to nonpathogenic or nonopportunistic species which
do not occur in clinical samples. The test was used on 31 different clinical specimens obtained from patients
suspected of having mycobacterial disease, including a patient with a double mycobacterial infection. The
samples included sputum, bronchoalveolar lavage, tissue biopsy samples, cerebrospinal fluid, pus, peritoneal
fluid, pleural fluid, and blood. The results of the PCR assay agreed with those of conventional identification
methods or with clinical data, showing that the test can be used for the direct and rapid detection and
identification of mycobacteria in clinical samples.
The human immunodeficiency virus (HIV) epidemic is
hav-ing a profound impact on the tuberculosis problem (2, 5, 10).
Serious diseases caused by mycobacteria other than
Mycobac-terium tuberculosis, mostly those belonging to the M. avium-M.
intracellulare (MAI) complex, are now commonly associated
with severe immunosuppression (21, 35, 58, 59). Opportunistic
mycobacteria commonly associated with HIV are M. kansasii,
M. xenopi, M. fortuitum, and M. scrofulaceum (46, 58).
Addi-tional risk factors for mycobacterial disease other than
tuber-culosis are iatrogenic immunosuppression, preexisting lung
disease, e.g., chronic obstructive pulmonary disease, and a
pre-vious tuberculosis (37).
A correct diagnosis of the mycobacterial disease is important
because the treatment of tuberculosis differs from that for
diseases caused by other mycobacteria, which are often
resis-tant to the drugs used for treating tuberculosis (19, 27).
The methods of identification in current use require
cul-tured mycobacteria. Culture from clinical samples is hampered
by the slow growth of mycobacteria. Inoculation on solid media
requires a mean incubation time of 4 weeks before sufficient
growth is obtained to enable identification to begin. Two
speedier methods for culture are the radiometric method (1,
41, 49) and a biphasic (broth-agar) system (20, 45). By
con-ventional biochemical methods (52, 57), the identification of
the species to which the cultured mycobacterial strain belongs
may require an additional 2 to 4 weeks. More rapid methods
for the identification of cultured mycobacteria are the analysis
of lipid composition (7, 11, 33), the use of species-specific
antibodies (44, 56), and species-specific DNA or RNA probes
(14, 16, 32).
Nucleic acid amplification techniques provide new
possibil-ities for the rapid diagnosis of mycobacterial diseases. Most of
the tests are based on PCR amplification of the M.
tuberculosis-specific repetitive sequence IS6110 (6, 8, 9, 18, 26, 28, 29, 38).
RNA amplification tests with 16S rRNA sequences have also
been used (23, 36, 39, 53). PCR assays based on DNA coding
for 16S rRNA (16S rDNA) sequences have been developed for
the detection and identification of mycobacteria other than M.
tuberculosis (3, 22, 24, 40, 54). These tests, however, have not
been used for the direct detection and identification of
myco-bacteria in clinical samples. Our study is the first in which a 16S
rDNA-based PCR assay was used directly on clinical samples.
We report the development of a 16S rDNA-based PCR and
a reverse cross blot hybridization assay to detect and identify
mycobacteria directly in clinical samples. We present the
re-sults with 31 different clinical specimens obtained from
pa-tients suspected of having mycobacterial disease.
MATERIALS AND METHODS
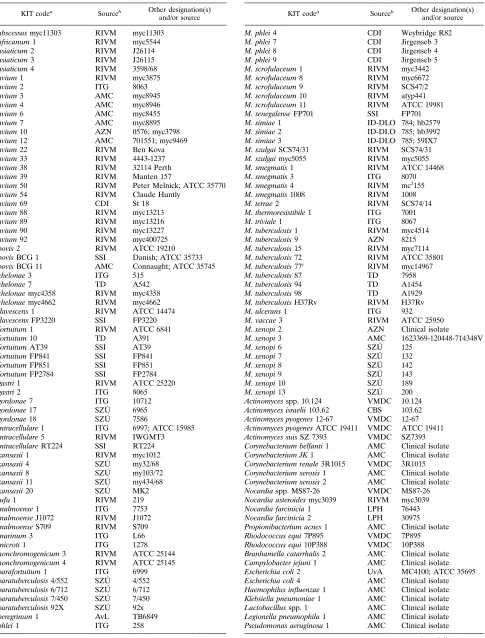
Bacterial strains.The bacterial strains that are used in this study are listed in
Table 1.
DNA isolation from bacteria.DNA was isolated as previously described (17)
and dissolved in Tris-EDTA (TE) buffer (10 mM Tris-HCl [pH 8.3], 1 mM EDTA) containing ribonuclease at a concentration of 5mg/ml (RNase from bovine pancreas, DNase free; Boehringer, Mannheim, Germany). The DNA concentration was determined by measuring the A260by using the following
conversion factor: an A260of 1 corresponds to a DNA concentration of 50mg/ml.
A part of the DNA solution was diluted to a concentration of 10 ng/ml and then further diluted in TE buffer to provide five samples with a concentration ranging from 1 ng to 100 fg of DNA per ml.
Bacterial lysates.With a loop, a small amount of bacteria (approximately 1
mg; about 108
bacteria) was suspended in a vial containing 0.5 ml of TE buffer and 0.5 ml of glass beads with a diameter of 0.10 to 0.15 mm (Biospec Products,
* Corresponding author. Mailing address: Department of Biomedi-cal Research, Royal TropiBiomedi-cal Institute, Meibergdreef 39, 1105 AZ Amsterdam, The Netherlands. Phone: 5665441. Fax: 31-20-6971841.
3225
on May 15, 2020 by guest
http://jcm.asm.org/
TABLE 1. Bacterial strains used in this study
KIT codea
Sourceb Other designation(s)
and/or source
M. abscessus myc11303 RIVM myc11303 M. africanum 1 RIVM myc5544 M. asiaticum 2 RIVM J26114 M. asiaticum 3 RIVM J26115 M. asiaticum 4 RIVM 3598/68 M. avium 1 RIVM myc3875
M. avium 2 ITG 8063
M. avium 3 AMC myc8945
M. avium 4 AMC myc8946
M. avium 6 AMC myc8455
M. avium 7 AMC myc8895
M. avium 10 AZN 0576; myc3798 M. avium 12 AMC 701551; myc9469 M. avium 22 RIVM Ben Kova M. avium 33 RIVM 4443-1237 M. avium 38 RIVM 32114 Perth M. avium 39 RIVM Manten 157
M. avium 50 RIVM Peter Melnick; ATCC 35770 M. avium 54 RIVM Claude Huntly
M. avium 69 CDI St 18 M. avium 88 RIVM myc13213 M. avium 89 RIVM myc13216 M. avium 90 RIVM myc13227 M. avium 92 RIVM myc400725 M. bovis 2 RIVM ATCC 19210 M. bovis BCG 1 SSI Danish; ATCC 35733 M. bovis BCG 11 AMC Connaught; ATCC 35745 M. chelonae 3 ITG 515
M. chelonae 7 TD A542 M. chelonae myc4358 RIVM myc4358 M. chelonae myc4662 RIVM myc4662 M. flavescens 1 RIVM ATCC 14474 M. flavescens FP3220 SSI FP3220 M. fortuitum 1 RIVM ATCC 6841 M. fortuitum 10 TD A391 M. fortuitum AT39 SSI AT39 M. fortuitum FP841 SSI FP841 M. fortuitum FP851 SSI FP851 M. fortuitum FP2784 SSI FP2784 M. gastri 1 RIVM ATCC 25220
M. gastri 2 ITG 8065
M. gordonae 7 ITG 10712 M. gordonae 17 SZU´ 6965 M. gordonae 18 SZU´ 7586
M. intracellulare 1 ITG 6997; ATCC 15985 M. intracellulare 5 RIVM IWGMT3 M. intracellulare RT224 SSI RT224 M. kansasii 1 RIVM myc1012 M. kansasii 4 SZU´ my32/68 M. kansasii 8 SZU´ my103/72 M. kansasii 11 SZU´ my434/68 M. kansasii 20 SZU´ MK2
M. lufu 1 RIVM 219
M. malmoense 1 ITG 7753 M. malmoense J1072 RIVM J1072 M. malmoense S709 RIVM S709
M. marinum 3 ITG L66
M. microti 1 ITG 1278 M. nonchromogenicum 3 RIVM ATCC 25144 M. nonchromogenicum 4 RIVM ATCC 25145 M. parafortuitum 1 ITG 6999 M. paratuberculosis 4/552 SZU´ 4/552 M. paratuberculosis 6/712 SZU´ 6/712 M. paratuberculosis 7/450 SZU´ 7/450 M. paratuberculosis 92X SZU´ 92x M. peregrinum 1 AvL TB6849
M. phlei 1 ITG 258
Continued
TABLE 1—Continued
KIT codea
Sourceb Other designation(s)
and/or source
M. phlei 4 CDI Weybridge R82 M. phlei 7 CDI Jirgenseb 3 M. phlei 8 CDI Jirgenseb 4 M. phlei 9 CDI Jirgenseb 5 M. scrofulaceum 1 RIVM myc3442 M. scrofulaceum 8 RIVM myc6672 M. scrofulaceum 9 RIVM SCS47/2 M. scrofulaceum 10 RIVM atyp441 M. scrofulaceum 11 RIVM ATCC 19981 M. senegalense FP701 SSI FP701 M. simiae 1 ID-DLO 784; hb2579 M. simiae 2 ID-DLO 785; hb3992 M. simiae 3 ID-DLO 785; 59IX7 M. szulgai SCS74/31 RIVM SCS74/31 M. szulgai myc5055 RIVM myc5055 M. smegmatis 1 RIVM ATCC 14468 M. smegmatis 3 ITG 8070 M. smegmatis 4 RIVM mc2155
M. smegmatis 1008 RIVM 1008 M. terrae 2 RIVM SCS74/14 M. thermoresistibile 1 ITG 7001
M. triviale 1 ITG 8067
M. tuberculosis 1 RIVM myc4514 M. tuberculosis 9 AZN 8215 M. tuberculosis 15 RIVM myc7114 M. tuberculosis 72 RIVM ATCC 35801 M. tuberculosis 77c
RIVM myc14967
M. tuberculosis 87 TD 7958 M. tuberculosis 94 TD A1454 M. tuberculosis 98 TD A1929 M. tuberculosis H37Rv RIVM H37Rv
M. ulcerans 1 ITG 932
M. vaccae 3 RIVM ATCC 25950 M. xenopi 2 AZN Clinical isolate
M. xenopi 3 AMC 1623369-120448-714348V
M. xenopi 6 SZU´ 125
M. xenopi 7 SZU´ 132
M. xenopi 8 SZU´ 142
M. xenopi 9 SZU´ 143
M. xenopi 10 SZU´ 189 M. xenopi 13 SZU´ 200 Actinomyces spp. 10.124 VMDC 10.124 Actinomyces israelii 103.62 CBS 103.62 Actinomyces pyogenes 12-67 VMDC 12-67 Actinomyces pyogenes ATCC 19411 VMDC ATCC 19411 Actinomyces suis SZ 7393 VMDC SZ7393 Corynebacterium belfanti 1 AMC Clinical isolate Corynebacterium JK 1 AMC Clinical isolate Corynebacterium renale 3R1015 VMDC 3R1015 Corynebacterium xerosis 1 AMC Clinical isolate Corynebacterium xerosis 2 AMC Clinical isolate Nocardia spp. MS87-26 VMDC MS87-26 Nocardia asteroides myc3039 RIVM myc3039 Nocardia farcinicia 1 LPH 76443 Nocardia farcinicia 2 LPH 30975 Propionibacterium acnes 1 AMC Clinical isolate Rhodococcus equi 7P895 VMDC 7P895 Rhodococcus equi 10P388 VMDC 10P388 Branhamella catarrhalis 2 AMC Clinical isolate Campylobacter jejuni 1 AMC Clinical isolate Escherichia coli 2 UvA MC4100; ATCC 35695 Escherichia coli 4 AMC Clinical isolate Haemophilus influenzae 1 AMC Clinical isolate Klebsiella pneumoniae 1 AMC Clinical isolate Lactobacillus spp. 1 AMC Clinical isolate Legionella pneumophila 1 AMC Clinical isolate Pseudomonas aeruginosa 1 AMC Clinical isolate
Continued on following page
on May 15, 2020 by guest
http://jcm.asm.org/
Bartlesville, Okla.). The suspension was incubated for 15 min at 1008C to kill the bacteria. The cells were lysed by shaking the suspensions in a mechanical dis-ruptor (MiniBeadBeater model 3110; Biospec Products) for 1 min at maximum speed. After the glass beads had settled, a part of the bacterial lysate was diluted 100 times in TE buffer. This dilution contained about 10 ng of DNA per ml. Ten microliters of this solution was used for the PCR.
Clinical samples.Clinical samples were obtained from various hospitals in The
Netherlands. Microscopic examinations and cultures of the clinical samples were performed at the microbiology laboratories of the hospitals where the patients were diagnosed. The PCR was performed at the Royal Tropical Institute.
Microscopy.All clinical samples were concentrated and examined under the
microscope after Ziehl-Neelsen staining (48).
Decontamination of sputum for culture.Sputum samples were
decontami-nated, depending on the preference of the laboratory, by the NaOH Petroff method (15), by the sodium laurylsulfate method (15), or with NaOH-sodium citrate-N-acetyl-L-cysteine (30). In all these cases, samples were concentrated by centrifugation after decontamination.
Routine culture.Samples were cultured on two solid media, Lo
¨wenstein-Jensen and Coletsos or Ogawa. The samples from extrapulmonary sites were cultured in addition on Middlebrook 7H9 broth (Difco Laboratories, Detroit, Mich.) and/or Septi-Chek AFB (Becton Dickinson Microbiology Systems, Cock-eysville, Md.). Cultured mycobacteria were identified by standard biochemical methods (52, 57).
DNA isolation from clinical samples.The clinical samples were sent at
ambi-ent temperature to the Royal Tropical Institute, where the DNA isolation pro-cedures were performed as previously described (29). Samples that could not be tested on the same day that they arrived were treated up to and including the lysis buffer step or the proteinase K step.
Oligonucleotides.The oligonucleotides (Isogen Biosciences, Amsterdam, The
Netherlands) that were used as primers in the PCR and as probes for hybrid-ization with 16S rDNA sequences are listed in Table 2. The forward primer pMyc14 was biotin labeled at the 59end of the DNA via a six-carbon-spacer arm (pMyc14bio).
PCR.The composition of the PCR mixture for the amplification of 16S rDNA sequences was 10 mM Tris-HCl (pH 8.3), 50 mM NaCl, 3.0 mM MgCl2, 0.01%
(wt/vol) gelatin, 0.2 mM (each) deoxynucleoside triphosphates (dATP, dGTP, dCTP, and dUTP), 0.2mM (each) primers pMyc14bio and pMyc7, 1 U of Taq DNA polymerase (Perkin-Elmer Cetus, Norwalk, Conn.), and 0.2 U of
uracil-N-glycosylase (UNG; GIBCO BRL, Gaithersburg, Md.) per 50-ml reaction mix-ture volume. For the amplification of IS6110 sequences with the primers INS1 and INS2 (26) we used a PCR mixture similar to that described previously (29). The PCR mixture was aliquoted to a final volume of 35ml per reaction vial and stored at2208C. When a test was performed, the reaction vials were removed from the freezer and 50ml of mineral oil (Sigma, St. Louis, Mo.) was added before the 10-ml sample was placed under the oil. Each clinical sample was tested in duplicate in the PCR. To test for inhibitors, one part of the sample was spiked with DNA (50 fg in the IS6110-based PCR and 200 fg in the 16S rDNA-based PCR) of a M. smegmatis strain containing a modified IS6110 sequence (25); the other part was not spiked. PCR was performed in a water bath thermocycler (Bio-med 60 processor; Bio-med, Theres, Germany) or a heating block thermo-cycler (Gene ATAQ controller; Pharmacia LKB, Uppsala, Sweden). Prior to amplification the PCR mixtures were incubated for 10 min at 408C to allow UNG to excise uracil from any contaminating dU-containing PCR product. A PCR cycle included 1.5 min of denaturation at 948C, 2 min of annealing at 658C, and 3 min of extension at 728C. At these high cycling temperatures, the UNG is inactivated and any possible contaminants are broken into small fragments. A total of 40 PCR cycles were performed. After the last PCR cycle, the vials were maintained at 728C until they were removed from the PCR machine to avoid the degradation of newly synthesized dU-containing PCR products by residual or regained activity of UNG. Reaction mixtures were stored at2208C.
Agarose gel electrophoresis.Electrophoresis of PCR products (16ml) was
performed on 2% agarose gels stained with ethidium bromide. PCR products were visualized by UV fluorescence. The results were recorded by Bio-print photodocumentation (Vilber-Lourmat, Marne-La-Valle´e, France).
Tailing of oligonucleotide probes with dTTP.The tailing reactions were
per-formed with 200 pmol of the oligonucleotide probe in a solution containing 25 mM Tris-HCl (pH 6.6), 200 mM potassium cacodylate, 0.025% (wt/vol) bovine serum albumin, 5 mM CoCl2, 0.5 mM dTTP, and 25 U of terminal transferase
(Boehringer) per 40-ml reaction mixture volume. The reaction mixtures were incubated at 378C. After 2 h of incubation the reactions were stopped by adding 4ml of a 0.2-mM EDTA solution. Tailing the oligonucleotide probes is necessary for the efficient capture of the PCR products in the reverse cross blot hybrid-ization assay.
Reverse cross blot hybridization assay.The reverse cross blot hybridization
assays were performed in a cross blotter (Accutran-Cross ACC 100/0; Schleicher & Schuell, Dassel, Germany) in which different molds can be used. The cross blot
TABLE 2. Nucleotide sequences of oligonucleotides (primers and probes) for amplification and hybridization of 16S rDNA sequences
Oligonucleotide Relevant characteristic(s) Strand type
and positiona Nucleotide sequence (59to 39)
pMyc14bio Forward primer, 59biotinylated .49–68 GRGRTACTCGAGTGGCGAACb
pMyc7 Reverse primer ,261–244 GGCCGGCTACCCGTCGTC
pTub1 M. tuberculosis complexcprobe ,168–148 ACCACAAGACATGCATCCCG
pAvi3 M. avium-M. paratuberculosisdprobe ,168–148 ACCAGAAGACATGCGTCTTG
pInt5 M. intracellulare probe Ie ,169–147 CACCTAAAGACATGCGCCTAA
pInt7 M. intracellulare probe IIe ,169–147 CACCAAAAGACATGCGTCTAA
pKan1 M. kansasii complexf-M. scrofulaceum complexgprobe ,168–148 ACCACAAGGCATGCGCCAAG
pXen1 M. xenopi probe ,168–148 ACCACCCCACATGCGCAGAA
pFor M. fortuitum-M. senegalensehprobe ,168–148 ACCACACACCATGAAGCGCG
pSme1 M. smegmatis probe ,158–140 CATGCGACCAGCAGGGTGT
pMyc5a Mycobacterium probe ,201–184 GGGCCCATCCCACACCGC
a.
, coding strand;,, noncoding strand. Numbering is according to the alignment of Rogall et al. (42).
b
R, A or G.
c
The M. tuberculosis complex consists of M. tuberculosis, M. africanum, M. bovis, M. bovis BCG, and M. microti (13).
d
M. paratuberculosis is considered to be a subspecies of M. avium (51). e
M. intracellulare is characterized by different 16S rDNA sequences (4), most of which hybridize with probe pInt5. f
The M. kansasii complex consists of M. kansasii plus M. gastri (13).
g
The M. scrofulaceum complex consists of M. scrofulaceum and M. simiae (13).
h
[image:3.612.59.555.533.661.2]M. senegalense is pathogenic for animals and is related to M. fortuitum (47). TABLE 1—Continued
KIT codea
Sourceb Other designation(s)
and/or source
Salmonella typhimurium 1 AMC Clinical isolate Shigella flexneri 1 AMC Clinical isolate Staphylococcus aureus 1 AMC Clinical isolate Streptococcus pneumoniae 1 AMC Clinical isolate Streptococcus pyogenes 1 AMC Clinical isolate
aSpecies were determined by biochemical means by the supplier of the strain.
KIT, Royal Tropical Institute, Amsterdam.
bAMC, Academic Medical Centre, Amsterdam, The Netherlands; AvL,
toni van Leeuwenhoek Hospital, Amsterdam, The Netherlands; AZN, St. An-tonius Hospital Nieuwegein, Utrecht, The Netherlands; CBS, Central Bureau for Fungal Cultures, Baarn, The Netherlands; CDI, Central Veterinary Institute, Lelystad, The Netherlands; ITG, Institute for Tropical Medicine, Antwerp, Bel-gium; KIT, Royal Tropical Institute, Amsterdam, The Netherlands; LVF, Lab-oratory for Public Health, Leeuwarden, The Netherlands; RIVM, National In-stitute of Public Health and Environmental Protection, Bilthoven, The Netherlands; SSI, Statens Serum Institute, Copenhagen, Denmark; SZU´ , Na-tional Institute for Public Health, Prague, Czech Republic; TD, Tuberculosis Division, Ministry of Public Health, Bangkok, Thailand; UvA, University of Amsterdam, Amsterdam, The Netherlands; VMDC, Veterinary Microbiological Diagnostic Centre, Utrecht, The Netherlands.
cM. tuberculosis 77 is a strain lacking IS6110.
on May 15, 2020 by guest
http://jcm.asm.org/
system is illustrated in Fig. 1. The reverse cross blot hybridization assay was performed as follows.
(i) Preparation of membranes.The rubber mat of the blot assembly was
covered with Parafilm (Parafilm ‘‘M’’ laboratory film; American National Can, Greenwich, Conn.), two layers of blotting paper (GB 002; Schleicher & Schuell), a piece (6 by 13 cm) of reinforced nitrocellulose membrane (Optitran BA-S 83; Schleicher & Schuell), and a mold (7 by 14 cm) with 34 horizontal slots num-bered 0 to 33 (50 by 2 mm) on top of the membrane. To each slot of the mold, 25 pmol of a dTTP-tailed oligonucleotide probe in 500ml of 103SSC (13SSC is 0.15 M NaCl and 0.015 M sodium citrate [pH 7.0]) was added. The cross blotter was placed on a rotary shaker (TPM-2; Sarstedt, Nu¨mbrecht, Germany) at 100 rpm for 16 h. The slots of the mold in the cross blotter were washed with 500ml of 103SSC, and then the membrane was washed separately with 103SSC and incubated on the rotary shaker. The probes were fixed to the membrane by UV light of 312 nm until 1 J/cm2
was reached (Fluo-link TFL20M; Vilber-Lourmat). Baking in an oven at 1208C for 15 min gives the same result.
(ii) Hybridization assay.The membrane was put in the cross blotter on a layer
of Parafilm on the rubber mat, and a different mold with four blocks of 14 vertical slots marked A to N (2 by 30 mm) was placed on top of it. The slots of the membrane were prehybridized in 200ml of hybridization solution containing 53
SSC, 1% (wt/vol) blocking agent (Boehringer), 0.1% (wt/vol) N-laurylsarcosine, and 0.02% (wt/vol) sodium dodecyl sulfate (SDS) at room temperature on the rotary shaker. After at least 5 min of prehybridization, the hybridization solution was discarded. Samples of 15ml of PCR product were denatured by boiling for 5 min and subsequent cooling on ice. After the addition of 200ml of ice-cold hybridization solution, the samples were added to the slots of the cross blotter and incubated at 508C for 1 h on the rotary shaker. The cross blotter was emptied by flicking, and the slots were incubated with 200ml of 0.1% (wt/vol) SDS in 23
SSC at 508C for 5 min on the rotary shaker. The hybridized PCR products on the membrane were detected by incubation with streptavidin-alkaline phosphatase and a color substrate (4-nitroblue tetrazolium chloride and 5-bromo-4-chloro-3-indolylphosphate) according to the instructions of the manufacturer (Boehr-inger).
RESULTS
Sensitivity and specificity of the PCR after agarose gel
elec-trophoresis.
The sensitivity of the PCR with primers pMyc14
bio and pMyc7 (Table 2) was tested in duplicate with 100, 10,
and 1 pg and 100, 10, and 1 fg of DNA from M. tuberculosis 1,
M. avium 2, M. intracellulare 1, M. kansasii 1, M. xenopi 2, M.
fortuitum 1, and M. smegmatis 1008 (Table 1). The PCR
prod-ucts were electrophoresed on 2% agarose gels stained with
ethidium bromide. Figure 2 shows that with M. tuberculosis
DNA a 208-bp fragment is amplified and that the detection
limit is 100 fg of DNA, which amounts to 20 mycobacteria. The
detection limits were the same for the other species of
myco-bacteria tested (results not shown).
DNA from lysates of the other 101 mycobacterial strains (33
species) listed in Table 1 could be amplified in the PCR. Of the
31 nonmycobacterial strains (of 17 genera) tested, only DNA
from lysates from some species belonging to genera closely
related to the genus Mycobacterium, namely, Corynebacterium
renale, Corynebacterium xerosis, Corynebacterium JK, Nocardia
asteroides, Nocardia farcinicia, and Rhodococcus equi, could be
amplified as tested by agarose gel electrophoresis (results not
shown). These products are formed because of the low number
of mismatches between the primers pMyc7 and pMyc14bio and
the 16S rDNA sequences of these bacteria (up to two
mis-matches per primer).
Sensitivity and specificity of the PCR after the reverse cross
blot hybridization assay.
Membranes were spotted with the
dTTP-tailed probes: pTub1, pAvi3, pInt5, pKan1, pXen1,
pFor1, pSme1, and the Mycobacterium-specific probe pMyc5a
(Table 2). The sensitivity of the reverse cross blot hybridization
assay with biotinylated 16S rDNA PCR fragments was tested
with 15
m
l of the same PCR products used for agarose gel
electrophoresis. Figure 3 shows that for M. tuberculosis DNA
with the probe pTub1 and the Mycobacterium-specific probe
pMyc5a, 10 fg of DNA, the DNA equivalent of two
mycobac-teria, can be detected. The negative result with 1 fg of DNA is
not shown. For the other six mycobacterial species tested, the
detection limits of the hybridization assays with the
corre-sponding species-specific probe and the genus-specific probe
pMyc5a were the same. When we used the PCR product of 100
pg of mycobacterial DNA, no cross hybridization was found.
The PCR products from lysates of N. asteroides, C. xerosis, and
C. JK (each containing about 100 pg of DNA), although giving
a positive result after agarose gel electrophoresis, showed no
hybridization with the species- and genus-specific probes
men-tioned above.
[image:4.612.71.285.112.360.2]Tables 3 and 4 show the overall results of the hybridization
of the probes with PCR products from all the strains tested.
Mismatches between the species-specific probes and the
cor-responding regions of other mycobacterial species are detailed
in Table 5. All PCR products from the 108 mycobacterial
FIG. 1. Diagram of cross blot hybridization apparatus.
FIG. 2. Determination of the sensitivity of PCR after agarose gel electro-phoresis. Lane 1, 800 ng of HaeIII-digested PhiX174 DNA (in replicative form) as a molecular size marker; lanes 2 to 6, PCR products from 100, 10, and 1 pg and 100 and 10 fg of M. tuberculosis DNA, respectively.
on May 15, 2020 by guest
http://jcm.asm.org/
strains (33 species) hybridized with the Mycobacterium-specific
probe pMyc5a, while none of the PCR products from the 31
nonmycobacterial strains (17 genera) hybridized with either
the Mycobacterium-specific or the species-specific probes. Even
the PCR products from reactions with C. renale, N. farcinicia,
and R. equi, which, like those of N. asteroides, C. xerosis, and C.
JK, give a clearly visible band after gel electrophoresis, did not
hybridize with the genus-specific probe pMyc5a. The PCR
products of all 13 M. tuberculosis complex strains hybridized
with the species-specific probe pTub1, including M. tuberculosis
77, which lacks the IS6110 sequence. Cross hybridization was
observed with the PCR product from the M. asiaticum strain
(one mismatch), and a weak signal was observed with the PCR
product from M. terrae (three mismatches). The PCR products
from all 22 MAI complex and 4 M. paratuberculosis strains
hybridized with one of the three MAI complex probes (pAvi3,
pInt5, and pInt7). Table 4 shows the results with the MAI
probes in more detail. The PCR products from the three
strains that were identified as M. intracellulare by biochemical
methods hybridized with probe pInt5. All PCR products from
the M. paratuberculosis strains hybridized with probe pAvi3,
which is consistent with the fact that M. paratuberculosis is now
considered to be a subspecies of M. avium (51). Of the PCR
products from the 19 strains that were identified as M. avium
by biochemical means, 13 hybridized with probe pAvi3, 5
hy-bridized with probe pInt5, and 1 hyhy-bridized with probe pInt7
(and weakly hybridized with probes pAvi3 and pInt5). Both
serotyping and hybridization suggest that these last six strains
are M. intracellulare. The PCR products from M. kansasii, M.
gastri, M. scrofulaceum, and M. simiae were the only ones to
hybridize with probe pKan1. The PCR products of all M.
for-tuitum strains and the M. senegalense strain hybridized with
probe pFor1. There was cross hybridization with the PCR
products from M. peregrinum (two mismatches), formerly
named M. fortuitum subsp. peregrinum (31), with probe pFor1.
Note that the PCR products of the M. chelonae strains and the
M. abscessus strain, belonging to the M. fortuitum complex (13),
did not hybridize with probe pFor1. In addition to this, the
PCR products from M. nonchromogenicum strains hybridized
with pFor1 (three mismatches). No other PCR products
bridized with probe pFor1. The only PCR products that
hy-bridized with probe pXen1 were those of the M. xenopi strains.
The hybridization of PCR products with probe pSme1 was
positive for the PCR products of the M. smegmatis strains but
also for the PCR products of M. thermoresistibile (one
mis-match) and M. flavescens (four mismatches). No other cross
hybridization was observed.
[image:5.612.62.296.70.191.2]Results of the 16S rDNA-based PCR assay compared with
those of the IS6110-based PCR assay and conventional
meth-ods.
We tested in our 16S rDNA-based PCR assay 18 clinical
samples which were positive in the IS6110-based PCR assay for
the detection of M. tuberculosis (26, 29). Table 6 shows that 17
of the 18 clinical samples had a positive result with the M.
tuberculosis probe pTub1 in the 16S rDNA-based PCR assay.
Biopsy sample 6 was positive in the IS6110-based PCR assay,
indicating that M. tuberculosis was present in the clinical
sam-ple. However, only M. avium was cultured. In the 16S
rDNA-based PCR assay, the PCR product of biopsy sample 6
hybrid-ized with both probes pTub1 and pAvi3. This indicated that
both M. tuberculosis and M. avium were present. The patient
had a double infection. Sample 7, a bronchoalveolar lavage,
was positive by microscopy but negative in culture. The fact
that these mycobacteria could not be cultured indicates that
FIG. 3. Determination of the sensitivity and specificity of PCR by the reverse cross blot hybridization assay. Lanes 1 to 4, PCR product from 100 pg, 1 pg, 100 fg, and 10 fg of M. tuberculosis DNA, respectively; lane 5, PCR product from 100 pg of M. avium DNA; lane 6, 100 pg of M. intracellulare DNA; lane 7, PCR product from 100 pg of M. kansasii DNA; lane 8, PCR product from 100 pg of
M. xenopi DNA; lane 9, PCR product from 100 pg of M. fortuitum DNA; lane 10,
PCR product from 100 pg of M. smegmatis DNA; lane 11, PCR product from N.
asteroides lysate; lane 12, PCR product from C. xerosis lysate; lane 13, PCR
product from C. JK lysate; lane 14, PCR product from water. Details of the probes are given in Table 2.
TABLE 3. Results of hybridization of mycobacterial and nonmycobacterial 16S rDNA PCR products with
specific oligonucleotide probes
Species (n)a Result of hybridization with probe: b
pTub1 pMAIcpKan1 pXen1 pFor1 pSme1 pMyc5a
M. tuberculosis (8) 1 2 2 2 2 2 1
M. africanum (1) 1 2 2 2 2 2 1
M. bovis (3) 1 2 2 2 2 2 1
M. microti (1) 1 2 2 2 2 2 1
M. asiaticum (3) 1/2 2 2 2 2 2 1
M. terrae (1) 1/2 2 2 2 2 2 1
MAI complexd
(22) 2 1 2 2 2 2 1
M. paratuberculosis (4) 2 1 2 2 2 2 1
M. kansasii (5) 2 2 1 2 2 2 1
M. gastri (2) 2 2 1 2 2 2 1
M. scrofulaceum (5) 2 2 1 2 2 2 1
M. simiae (3) 2 2 1 2 2 2 1
M. xenopi (8) 2 2 2 1 2 2 1
M. fortuitum (6) 2 2 2 2 1 2 1
M. senegalense (1) 2 2 2 2 1 2 1
M. peregrinum (1) 2 2 2 2 1/2 2 1
M. nonchromogenicum (2) 2 2 2 2 1/2 2 1
M. smegmatis (4) 2 2 2 2 2 1 1
M. thermoresistibile (1) 2 2 2 2 2 1 1
M. flavescens (2) 2 2 2 2 2 1/2 1
M. chelonae (4) 2 2 2 2 2 2 1
M. abscessus (1) 2 2 2 2 2 2 1
M. gordonae (3) 2 2 2 2 2 2 1
M. lufu (1) 2 2 2 2 2 2 1
M. malmoense (3) 2 2 2 2 2 2 1
M. marinum (1) 2 2 2 2 2 2 1
M. parafortuitum (1) 2 2 2 2 2 2 1
M. phlei (5) 2 2 2 2 2 2 1
M. szulgai (2) 2 2 2 2 2 2 1
M. triviale (1) 2 2 2 2 2 2 1
M. ulcerans (1) 2 2 2 2 2 2 1
M. vaccae (1) 2 2 2 2 2 2 1
Non-Mycobacteriume(31) 2 2 2 2 2 2 2 a
n, number of strains. b
Details of the probes are given in Table 2.1, positive;1/2, weakly positive;
2, negative.
c
pMAI probes used were pAvi3, pInt5, or pInt7.
d
Results with MAI probes (pAvi3, pInt5, and pInt7) are given in more detail in Table 4.
e
The non-Mycobacterium species are represented by 17 genera.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:5.612.315.553.335.670.2]they were not viable. All seven microscopy-positive samples
and 10 of 11 microscopy-negative samples were positive with
probe pTub1 in the 16S rDNA-based PCR assay.
Results of the 16S rDNA-based PCR assay compared with
results of conventional methods.
The 16S rDNA PCR assay
[image:6.612.60.298.103.377.2]was performed on 13 clinical samples from 8 patients
sus-pected of having mycobacterial infections other than
tubercu-losis (Table 7). At the time that the samples were tested, the
results of cultures were not known. Samples 19 and 20 were
sputum samples from a patient with chronic obstructive
pul-monary disease. Both samples had a positive result with probe
pAvi3, and M. avium was cultured from both. Samples 21 and
22 (bronchoalveolar lavages) and 3 (sputum) were from
an-other patient with chronic obstructive pulmonary disease who
had had an M. avium infection one year previously, proven by
culture and biochemical identification. The 16S rDNA-based
PCR assay gave a positive result with probe pAvi3. M. avium
was cultured from all three samples. Sample 24 was a blood
sample giving a positive result with probe pAvi3. The patient
was an 83-year-old woman with pulmonary infiltrates who was
diagnosed with Wegener’s disease. She was treated with
ste-roids and azathioprine. The blood was not cultured for
myco-bacteria. The death of the patient precluded confirmation of
the PCR result by testing another blood sample. Sample 25
(sputum) from another patient was positive with probe pInt5.
The cultured mycobacterium was identified as belonging to the
MAI complex. Samples 26 (sputum), 27 (bronchoalveolar
la-vage), and 28 (sputum), from three separate patients, were
positive with probe pKan1. These results were confirmed
be-cause M. kansasii was cultured. Samples 29, 30 (peritoneal
fluids), and 31 (endometrium biopsy), which were obtained
from one patient, were all positive with probe pFor1,
suggest-ing an infection with M. fortuitum. No mycobacteria were
cul-tured from these samples. The patient was first treated with
antituberculous drugs (rifampin, isoniazid, and pyrazinamide)
but without effect. She improved when other antimycobacterial
TABLE 4. Results of hybridization of the MAI complex and M. paratuberculosis 16S rDNA PCR products with MAI
complex-specific oligonucleotide probes
Serotypea KIT codeb
Result of hybridization with probe:c
pAvi3 pInt5 pInt7
2 M. avium 1 1 2 2
2 M. avium 2 1 2 2
2 M. avium 69 1 2 2
2/3 M. avium 3 1 2 2
3 M. avium 22 1 2 2
4 M. avium 4 1 2 2
4 M. avium 6 1 2 2
4 M. avium 7 1 2 2
5 M. avium 33 1 2 2
7 M. avium 38 2 1 2
7 M. avium 39 2 1 2
8 M. avium 12 1 2 2
16 M. avium 10 2 1 2
16 M. intracellulare 1 2 1 2
18 M. avium 50 1/2 1/2 1
19 M. avium 54 2 1 2
ND M. avium 88 1 2 2
ND M. avium 89 1 2 2
ND M. avium 90 1 2 2
ND M. avium 92 2 1 2
ND M. intracellulare 5 2 1 2
ND M. intracellulare RT244 2 1 2 M. paratuberculosis 4/552 1 2 2 M. paratuberculosis 6/712 1 2 2 M. paratuberculosis 7/450 1 2 2 M. paratuberculosis 92X 1 2 2 aSerotypes 1 to 6, 8 to 11, and 21 are M. avium. Serotypes 7, 12 to 20, and 25
are M. intracellulare. It is unclear to what species serotypes 22 to 24 and 26 to 28 belong (43). ND, not determined.
bSpecies were determined by biochemical means by the supplier of the strains.
KIT, Royal Tropical Institute, Amsterdam.
cDetails of the probes are given in Table 2.1, positive;1/2, weakly positive;
2, negative.
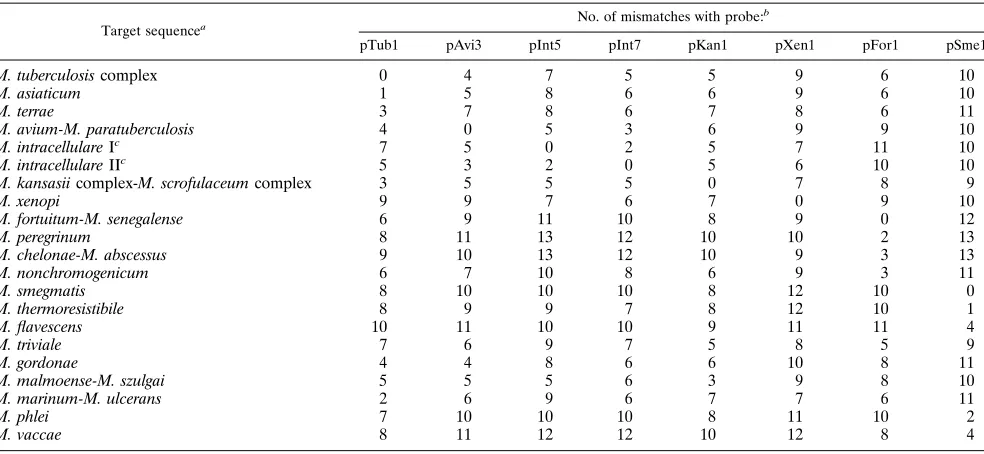
TABLE 5. Mismatches between species-specific probes and the corresponding regions in the 16S rDNA of other mycobacterial species
Target sequencea No. of mismatches with probe: b
pTub1 pAvi3 pInt5 pInt7 pKan1 pXen1 pFor1 pSme1
M. tuberculosis complex 0 4 7 5 5 9 6 10
M. asiaticum 1 5 8 6 6 9 6 10
M. terrae 3 7 8 6 7 8 6 11
M. avium-M. paratuberculosis 4 0 5 3 6 9 9 10
M. intracellulare Ic 7 5 0 2 5 7 11 10
M. intracellulare IIc 5 3 2 0 5 6 10 10
M. kansasii complex-M. scrofulaceum complex 3 5 5 5 0 7 8 9
M. xenopi 9 9 7 6 7 0 9 10
M. fortuitum-M. senegalense 6 9 11 10 8 9 0 12
M. peregrinum 8 11 13 12 10 10 2 13
M. chelonae-M. abscessus 9 10 13 12 10 9 3 13
M. nonchromogenicum 6 7 10 8 6 9 3 11
M. smegmatis 8 10 10 10 8 12 10 0
M. thermoresistibile 8 9 9 7 8 12 10 1
M. flavescens 10 11 10 10 9 11 11 4
M. triviale 7 6 9 7 5 8 5 9
M. gordonae 4 4 8 6 6 10 8 11
M. malmoense-M. szulgai 5 5 5 6 3 9 8 10
M. marinum-M. ulcerans 2 6 9 6 7 7 6 11
M. phlei 7 10 10 10 8 11 10 2
M. vaccae 8 11 12 12 10 12 8 4
a
Sequence data were retrieved from GenBank.
b
Details of the probes are given in Table 2.
c
M. intracellulare is characterized by different 16S rDNA sequences (4).
on May 15, 2020 by guest
http://jcm.asm.org/
[image:6.612.61.553.476.703.2]drugs (pyrazinamide, ethambutol, ciprofloxacin, and
clarithro-mycin) were used.
DISCUSSION
We developed a PCR and a hybridization assay for the rapid
detection and identification of mycobacteria in clinical
sam-ples. Two oligonucleotides based on mycobacterial 16S rDNA
sequences were used as primers for the PCR. The primers span
a region of the mycobacterial 16S rDNA that is specific at the
species level. The species to which the mycobacterium belongs
is identified by hybridization of the PCR product with
species-specific probes and a Mycobacterium-species-specific probe in a reverse
cross blot hybridization assay. After hybridization of the PCR
product with the Mycobacterium-specific probe pMyc5a, the
DNA equivalent of two mycobacteria could be detected and
the organism was confirmed to belong to the genus
Mycobac-terium.
We chose hybridization analysis of the 16S rDNA PCR
products because it is technically simple. When a cross blot
system is used, one PCR product can be hybridized with
dif-ferent probes simultaneously. Our reverse cross blot
hybrid-ization assay is as simple as our previously described dot blot
hybridization assay (26, 29). The PCR products can also be
analyzed by sequencing (22, 24), but this is not a technique that
is suitable for a routine laboratory. Analysis of the PCR
prod-ucts by restriction enzyme analysis (22, 40, 54) is technically
simpler than sequencing, but analysis of the data is laborious.
Furthermore, when sequencing or restriction enzyme analysis
is performed on 16S rDNA PCR products from a culture or a
clinical sample containing more than one species of
mycobac-teria, it may be difficult, if not impossible, to identify them.
[image:7.612.59.298.101.309.2]We selected eight species-specific probes for the
identifica-tion of the most important pathogenic and opportunistic
my-cobacteria (Table 2). The specificities of the probes were
tested with 108 mycobacterial strains (33 species) and 31
non-mycobacterial strains (17 genera). Our testing showed no cross
hybridization with the probes pAvi3, pInt5, pInt7, pKan1, and
pXen1. There was some slight cross hybridization of PCR
products from a number of mycobacterial strains with the
probes pTub1, pFor1, and pSme1. This slight cross
hybridi-zation plays no role when testing clinical samples, since the
mycobacterial strains from which the cross-hybridizing PCR
products were derived never belonged to pathogenic or
oppor-tunistic species. For example, the PCR products from M. terrae
and M. asiaticum cross hybridized slightly with probe pTub1. It
has been reported that there are some M. terrae strains that
give positive reactions with a commercially available M.
tuber-culosis complex probe (12). Cross hybridization of M. asiaticum
16S rDNA PCR products with a M. tuberculosis
complex-spe-cific probe has also been reported by Mabilat and coworkers,
who developed an automated hybridization assay for the
iden-tification of M. tuberculosis complex isolates (34). Cross
hybrid-ization can be explained by a small number of mismatches
between species-specific probes and the corresponding regions
in the 16S rRNA of other mycobacterial species (Table 5). In
addition, the position of the mismatches may also influence the
result. For example, M. terrae PCR products that have three
mismatches with probe pTub1 (at positions 7, 8, and 20 from
the 5
9
site of the probe) give slight cross hybridization with
TABLE 6. Results of samples positive by the IS6110-based PCR assay compared with those tested by the 16S rDNA-based
PCR assay, microscopy, and culture
Code Sample typea
Patient
16S rDNA PCR assay probeb
Micros-copy Culture
1 BAL A pTub1 Positive M. tuberculosis
2 Sputum B pTub1 Positive M. tuberculosis
3 Peritoneal fluid C pTub1 Positive M. tuberculosis
4 Sputum D pTub1 Positive M. tuberculosis
5 Sputum E pTub1 Positive M. tuberculosis
6 Lung biopsy F pTub11
pAvi3
Positive M. avium
7 BAL G pTub1 Positive Negative
8 CSF H pTub1 Negative M. tuberculosis
9 Pus I pTub1 Negative M. tuberculosis
10 Pleural fluid J pTub1 Negative M. tuberculosis
11 Sputum K pTub1 Negative Negative
12 Lung biopsy L pTub1 Negative Negative
13 CSF M pTub1 Negative Negative
14 Pleura biopsy N pTub1 Negative Negative
15 CSF O pTub1 Negative Negative
16 CSF O pTub1 Negative Negative
17 Lung biopsy P pTub1 Negative Not done
18 Pus Q Negative Negative Negative
a
BAL, bronchoalveolar lavage; CSF, cerebrospinal fluid.
b
The probes pTub1 and pAvi3 are described in Table 2.
TABLE 7. Results of the 16S rDNA-based PCR assay compared with those of microscopy and culture of samples from patients suspected of having mycobacterial disease other than tuberculosis
Code Sample typea Patient 16S rDNA PCR
assay probeb Microscopy Culture Other diseasec
19 Sputum R pAvi3 Positive M. avium COPD
20 Sputum R pAvi3 Positive M. avium COPD
21 BAL S pAvi3 Positive M. avium COPD
22 BAL S pAvi3 Positive M. avium COPD
23 Sputum S pAvi3 Positive M. avium COPD
24 Blood T pAvi3 Not done Not done Wegener’s disease
25 Sputum U pInt5 Positive MAI complex None
26 Sputum V pKan1 Positive M. kansasii None
27 BAL W pKan1 Positive M. kansasii AIDS
28 Sputum X pKan1 Positive M. kansasii None
29 Peritoneal fluid Y pFor1 Negative Negative None
30 Peritoneal fluid Y pFor1 Negative Negative None
31 Endometrium biopsy Y pFor1 Negative Negative None
a
BAL, bronchoalveolar lavage.
b
Details of the probes are given in Table 2.
c
COPD, chronic obstructive pulmonary disease.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:7.612.60.556.555.702.2]probe pTub1, while M. marinum PCR products that have only
two mismatches with probe pTub1 (at positions 7 and 14 from
the 5
9
site of the probe) do not cross hybridize with this probe.
This suggests that the maximum length of matching
nucleo-tides is related to the extent of cross hybridization. For M.
marinum PCR products and probe pTub1 this is only 7
nucle-otides, and for M. terrae PCR products and probe pTub1 this is
11 nucleotides. All cross-hybridizing PCR products from other
species have maximum lengths of at least 10 matching
nucle-otides (results not shown).
It may happen that a PCR product hybridizes with the
Mycobacterium-specific probe but not with one of the
species-specific probes. In such a case, the nucleotide sequence of the
PCR product can be determined for further characterization of
the mycobacterium. Alternatively, we suggest performing a
second reverse hybridization assay with another panel of
spe-cific probes. For this purpose, we are extending the test with
probes for M. chelonae, M. abscessus, M. gordonae, M.
gena-vense, and M. leprae.
PCR assays for the identification of mycobacteria based on
16S rDNA sequences have also been described by others (3, 22,
24, 40, 54). Our study is the first in which a 16S rDNA-based
PCR assay was used directly on a variety of clinical samples.
When we tested 18 clinical samples that were positive by the
IS6110-based PCR, 17 samples were positive with probe pTub1.
In one patient we detected a double infection with M.
tuber-culosis complex and M. avium. The one sample that was
neg-ative in the 16S rDNA-based PCR assay gave a weak positive
result in the IS6110-based PCR assay. There are multiple
cop-ies of the target for this assay in the M. tuberculosis genome and
only one target for the 16S rDNA, explaining the discrepancy
in the results with this sample. An advantage of the 16S
rDNA-based PCR assay over the IS6110-rDNA-based PCR is that the
ex-ceptional strains without IS6110 (50, 55) can be easily detected
as a strain of the M. tuberculosis complex. For optimal
sensi-tivity for the detection and identification of M. tuberculosis
complex and other mycobacteria, a combination of both PCRs
(a multiplex PCR) is favored.
When the test was applied to 13 clinical samples from eight
patients suspected of having mycobacterial disease other than
tuberculosis, results with the 16S rDNA-based PCR assay
cor-responded to results obtained by the biochemical identification
of mycobacteria in all six patients from whom samples could be
cultured. For the other two patients, the results of the 16S
rDNA-based PCR were in agreement with clinical findings.
A great advantage of the PCR-based method is that it can be
performed directly with clinical samples, unlike traditional
typ-ing, for which a cultured mycobacterium strain is needed.
Re-sults with the 16S rDNA-based PCR assay can be obtained
within 72 h of receipt of the clinical sample. Even when
per-formed on cultured mycobacteria, the 16S rDNA-based PCR
assay allows a more rapid and more accurate identification of
mycobacteria than biochemical identification methods, which
can take 2 to 4 weeks. In this study, we demonstrated that our
16S rDNA PCR assay is a very sensitive tool for the direct and
rapid detection and identification of mycobacteria in clinical
samples.
ACKNOWLEDGMENTS
L.F.F.K. was supported by a grant from the Amsterdam Society and Research Fund for Prevention and Cure of Tuberculosis. This study was supported by the commission of the European Communities Di-rectorate for Science Technology and Development (project number TS3-CT91-0036).
We thank E. M. Rankin for a critical review of the manuscript.
REFERENCES
1. Anargyros, P., D. S. J. Astill, and I. S. L. Lim. 1990. Comparison of improved BACTEC and Lo¨wenstein-Jensen media for culture of mycobacteria from clinical specimens. J. Clin. Microbiol. 28:1288–1291.
2. Bloom, B. R., and C. J. L. Murray. 1992. Tuberculosis: commentary on a re-emergent killer. Science 257:1055–1064.
3. Bo¨ddinghaus, B., T. Rogall, T. Flohr, H. Blo¨cker, and E. C. Bo¨ttger.1990. Detection and identification of mycobacteria by amplification of rRNA. J. Clin. Microbiol. 28:1751–1759.
4. Bo¨ddinghaus, B., J. Wolters, W. Heikens, and E. C. Bo¨ttger.1990. Phyloge-netic analysis and identification of different serovars of Mycobacterium
in-tracellulare at the molecular level. FEMS Microbiol. Lett. 70:197–204.
5. Braun, M. M., R. H. Byers, W. L. Heyward, C. A. Ciesielski, A. B. Bloch, R. L.
Berkelman, and D. E. Snider.1990. Acquired immunodeficiency syndrome
and extrapulmonary tuberculosis in the United States. Arch. Intern. Med.
150:1913–1916.
6. Brisson-Noe¨l, A., C. Aznar, C. Chureau, S. Nguyen, C. Pierre, M. Bartoli, R.
Bonete, G. Pialoux, B. Gicquel, and G. Garrigue.1991. Diagnosis of
tuber-culosis by DNA amplification in clinical practice evaluation. Lancet 338:364– 366.
7. Butler, W. R., K. C. Jost, Jr., and J. O. Kilburn. 1991. Identification of mycobacteria by high-performance liquid chromatography. J. Clin. Micro-biol. 29:2468–2472.
8. Clarridge, J. E., III, R. M. Shawar, T. M. Shinnick, and B. B. Plikaytis. 1993. Large-scale use of polymerase chain reaction for detection of Mycobacterium
tuberculosis in a routine mycobacteriology laboratory. J. Clin. Microbiol. 31:
2049–2056.
9. Eisenach, K. D., M. D. Sifford, M. D. Cave, J. H. Bates, and J. T. Crawford. 1991. Detection of Mycobacterium tuberculosis in sputum samples using a polymerase chain reaction. Am. Rev. Respir. Dis. 144:1160–1163. 10. Ellner, J. J., A. R. Hinman, S. W. Dooley, M. A. Fischl, K. A. Sepkowitz, M. J.
Goldberger, T. M. Shinnick, M. D. Iseman, and W. R. Jacobs, Jr.1993.
Tuberculosis symposium: emerging problems and promise. J. Infect. Dis.
168:537–551.
11. Floyd, M. M., V. A. Silcox, W. D. Jones, Jr., W. R. Butler, and J. O. Kilburn. 1992. Separation of Mycobacterium bovis BCG from Mycobacterium
tubercu-losis and Mycobacterium bovis by using high-performance liquid
chromatog-raphy of mycolic acids. J. Clin. Microbiol. 30:1327–1330.
12. Ford, E. G., S. J. Snead, J. Todd, and N. G. Warren. 1993. Strains of
Mycobacterium terrae complex which react with DNA probes for M. tuber-culosis complex. J. Clin. Microbiol. 31:2805–2806.
13. Goodfellow, M., and L. G. Wayne. 1982. Taxonomy and nomenclature, p. 471–521. In R. Ratledge and J. Stanford (ed.), The biology of the mycobac-teria, vol. 1. Academic Press, London.
14. Goto, M., S. Oka, K. Okuzimi, S. Kimura, and K. Shimada. 1991. Evaluation of acridinium-ester-labeled DNA probes for identification of Mycobacterium
tuberculosis and Mycobacterium avium-Mycobacterium intracellulare complex
in culture. J. Clin. Microbiol. 29:2473–2476.
15. Groothuis, D. G., and M. D. Yates. 1991. European Society for Mycobacte-riology: manual of diagnostic and public health mycobacteriology. Bureau of Hygiene and Tropical Diseases, London.
16. Hampson, S. J., F. Portaels, J. Thompson, E. P. Green, M. T. Moss, J.
Hermon-Taylor, and J. J. McFadden.1989. DNA probes demonstrate a
single highly conserved strain of Mycobacterium avium infecting AIDS pa-tients. Lancet i:65–68.
17. Hermans, P. W. M., A. R. J. Schuitema, D. Van Soolingen, C. P. H. J.
Verstynen, E. M. Bik, J. E. R. Thole, A. H. J. Kolk, and J. D. A. van Embden.
Specific detection of Mycobacterium tuberculosis complex strains by poly-merase chain reaction. J. Clin. Microbiol. 28:1204–1213.
18. Hermans, P. W. M., D. van Soolingen, J. W. Dale, A. R. J. Schuitema, R. A.
McAdam, D. Catty, and J. D. A. van Embden.1990. Insertion element IS986
from Mycobacterium tuberculosis: a useful tool for diagnosis and epidemiol-ogy of tuberculosis. J. Clin. Microbiol. 28:2051–2058.
19. Hoffner, S. E. 1994. Pulmonary infections caused by less frequently encoun-tered slow-growing environmental mycobacteria. Eur. J. Clin. Microbiol. Infect. Dis. 13:937–941.
20. Hoffner, S. E., M. Haile, and G. Kållenius. 1992. A biphasic system for primary isolation of mycobacteria compared to solid medium and broth culture. J. Med. Microbiol. 37:332–334.
21. Horsburgh, C. R., Jr. 1991. Mycobacterium avium complex infection in the acquired immunodeficiency syndrome. N. Engl. J. Med. 324:1332–1338. 22. Hughes, M. S., R. A. Skuce, L. A. Beck, and S. D. Neil. 1993. Identification
of mycobacteria from animals by restriction enzyme analysis and direct DNA cycle sequencing of polymerase chain reaction-amplified 16S rRNA gene sequences. J. Clin. Microbiol. 31:3216–3222.
23. Jonas, V., M. J. Alden, J. I. Curry, K. Kamisango, C. A. Knott, R. Lankford,
J. M. Wolfe, and D. F. Moore.1993. Detection and identification of
Myco-bacterium tuberculosis directly from sputum sediments by amplification of
rRNA. J. Clin. Microbiol. 31:2410–2416.
24. Kirschner, P., B. Springer, U. Vogel, A. Meier, A. Wrede, M. Kiekenbeck,
F. C. Bange, and E. C. Bo¨ttger.1993. Genotypic identification of
mycobac-teria by nucleic acid sequence determination: report of a 2-year experience
on May 15, 2020 by guest
http://jcm.asm.org/
in a clinical laboratory. J. Clin. Microbiol. 31:2882–2889.
25. Kolk, A. H. J., G. T. Noordhoek, O. de Leeuw, S. Kuijper, and J. D. A. van
Embden.1994. Mycobacterium smegmatis strain for detection of
Mycobacte-rium tuberculosis by PCR used as internal control for inhibition of
amplifi-cation and for quantifiamplifi-cation of bacteria. J. Clin. Microbiol. 32:1354–1356. 26. Kolk, A. H. J., A. R. J. Schuitema, S. Kuijper, J. van Leeuwen, P. W. M.
Hermans, J. D. A. van Embden, and R. A. Hartskeerl.1992. Detection of
Mycobacterium tuberculosis in clinical samples by using polymerase chain
reaction and a nonradioactive detection system. J. Clin. Microbiol. 30:2567– 2575.
27. Kotloff, R. M. 1993. Infection caused by nontuberculous mycobacteria: clin-ical aspects. Semin. Roentgenol. 28:131–138.
28. Kox, L. F. F., S. Kuijper, and A. H. J. Kolk. Early diagnosis of tuberculous meningitis by polymerase chain reaction. Neurology, in press.
29. Kox, L. F. F., D. Rhienthong, A. Medo Miranda, N. Udomsantisuk, K. Ellis,
J. van Leeuwen, S. van Heusden, S. Kuijper, and A. H. J. Kolk.1994. A more
reliable PCR for detection of Mycobacterium tuberculosis in clinical samples. J. Clin. Microbiol. 32:672–678.
30. Kubica, G. P., W. E. Dye, M. L. Cohn, and G. Middlebrook. 1963. Sputum digestion and decontamination with N-acetyl-L-cysteine-sodium hydroxyde for culture of mycobacteria. Am. Rev. Respir. Dis. 87:775–779.
31. Kusunoki, S., and T. Ezaki. 1992. Proposal of Mycobacterium peregrinum sp. nov., nom. rev., and elevation of Mycobacterium chelonae subsp. abscessus (Kubica et al.) to species status: Mycobacterium abscessus comb. nov. Int. J. Syst. Bacteriol. 42:240–245.
32. Lebrun, L., F. Espinasse, J. D. Poveda, and V. Vincent-Levy-Frebault. 1992. Evaluation of nonradioactive DNA probes for identification of mycobacte-ria. J. Clin. Microbiol. 30:2476–2478.
33. Luquin, M., V. Ausina, F. Lo´pez Calahorra, F. Belda, M. Garcı´a Barcelo´, C.
Celma, and G. Prats.1991. Evaluation of practical chromatographic
proce-dures for identification of clinical isolates of mycobacteria. J. Clin. Microbiol.
29:120–130.
34. Mabilat, C., S. Desvarenne, G. Panteix, N. Machabert, M. H. Bernillon, G.
Guardiola, and P. Cros.1994. Routine identification of Mycobacterium
tu-berculosis complex isolates by automated hybridization. J. Clin. Microbiol.
32:2702–2705.
35. Masur, H., and the Public Health Service Task Force on Prophylaxis and
Therapy for Mycobacterium avium Complex. 1993. Recommendations on
prophylaxis and therapy for disseminated Mycobacterium avium complex disease in patients infected with the human immunodeficiency virus. N. Engl. J. Med. 329:898–904.
36. Miller, N., S. G. Hernandez, and T. J. Cleary. 1994. Evaluation of Gen-Probe Amplified Mycobacterium Tuberculosis Direct Test and PCR for direct detection of Mycobacterium tuberculosis in clinical specimens. J. Clin. Micro-biol. 32:393–397.
37. Miller, W. T., Jr., and W. T. Miller, Sr. 1993. Pulmonary infections with atypical mycobacteria in the normal host. Semin. Roentgenol. 28:139–149. 38. Nolte, F. S., B. Metchock, J. E. McGowan, Jr., A. Edwards, O. Okwumabua,
C. Thurmond, P. S. Mitchell, B. Plikaytis, and T. Shinnick.1993. Direct
detection of Mycobacterium tuberculosis in sputum by polymerase chain re-action and DNA hybridization. J. Clin. Microbiol. 31:1777–1782. 39. Pfyffer, G. E., P. Kissling, R. Wirth, and R. Weber. 1994. Direct detection of
Mycobacterium tuberculosis complex in respiratory specimens by a
target-amplified test system. J. Clin. Microbiol. 32:918–923.
40. Plikaytis, B. B., B. D. Plikaytis, M. A. Yakrus, W. R. Butler, C. L. Woodley,
V. A. Silcox, and T. M. Shinnick.1992. Differentiation of slowly growing
Mycobacterium species, including Mycobacterium tuberculosis, by gene
am-plification and restriction fragment length polymorphism analysis. J. Clin. Microbiol. 30:1815–1822.
41. Roberts, G. D., N. L. Goodman, L. Heifets, H. W. Larsh, T. H. Lindner, J. K.
McClatchy, M. R. McGinnis, S. H. Siddiqi, and P. Wright.1983. Evaluation
of the BACTEC radiometric method for recovery of mycobacteria and drug susceptibility testing of Mycobacterium tuberculosis from acid-fast smear-positive specimens. J. Clin. Microbiol. 18:689–696.
42. Rogall, T., J. Wolters, T. Flohr, and E. C. Bo¨ttger.1990. Towards a
phylog-eny and definition of species at the molecular level within the genus
Myco-bacterium. Int. J. Syst. Bacteriol. 40:323–330.
43. Saito, H., H. Tomioka, H. Tasaka, and D. J. D. Dawson. 1990. Identification of serovar strains of Mycobacterium avium complex by using DNA probes specific for Mycobacterium avium and Mycobacterium intracellulare. J. Clin. Microbiol. 28:1694–1697.
44. Scho¨ningh, R., C. P. H. J. Verstijnen, S. Kuijper, and A. H. J. Kolk.1990. Enzyme immunoassay for identification of heat-killed mycobacteria belong-ing to the Mycobacterium tuberculosis and Mycobacterium avium complexes and derived from early cultures. J. Clin. Microbiol. 28:708–713.
45. Sewell, D. L., A. L. Rashad, W. J. Rourke, Jr., S. L. Poor, J. A. C. McCarthy,
and M. A. Pfaller.1993. Comparison of the Septi-Chek AFB and BACTEC
systems and conventional culture for recovery of mycobacteria. J. Clin. Microbiol. 31:2689–2691.
46. Shafer, R. W., and M. F. Sierra. 1992. Mycobacterium xenopi, Mycobacterium
fortuitum, Mycobacterium kansasii, and other nontuberculous mycobacteria
in an area of endemicity for AIDS. Clin. Infect. Dis. 15:161–162. 47. Shinnick, T. M., and R. C. Good. 1994. Mycobacterial taxonomy. Eur. J. Clin.
Microbiol. Infect. Dis. 13:884–901.
48. Smithwick, R. W. 1976. Laboratory manual for acid-fast microscopy, 2nd ed. Centers for Disease Control, Atlanta.
49. Stager, C. E., J. P. Libonati, S. H. Siddiqi, J. R. Davis, N. M. Hooper, J. F.
Baker, and M. E. Carter.1991. Role of solid media when used in conjunction
with the BACTEC system for mycobacterial isolation and identification. J. Clin. Microbiol. 29:154–157.
50. Thierry, D., L. T. K. Tuyen, P. Chavarot, G. Marchal, H. M. Ly, N. N. Lan,
L. N. Van, S. Ledru, F. Fumoux, and J. L. Guesdon.1995. Mycobacterium
tuberculosis strains unidentified by using the IS6110 probe can be detected by
oligonucleotides derived from MT308 sequence, abstr. 456, p. 87. In Ab-stracts of the 7th European Congress of Clinical Microbiology and Infectious Diseases. European Society of Clinical Microbiology and Infectious Dis-eases, Vienna.
51. Thorel, M. F., M. Krichevsky, and V. Vincent Le´vy-Fre´bault. 1990. Numer-ical taxonomy of mycobactin-dependent mycobacteria, emended description of Mycobacterium avium, and description of Mycobacterium avium subsp.
avium subsp. nov., Mycobacterium avium subsp. paratuberculosis subsp. nov.,
and Mycobacterium avium subsp. silvaticum subsp. nov. 1990. Int. J. Syst. Bacteriol. 40:254–260.
52. Tsukamura, M. 1981. A review of the methods of identification and differ-entiation of mycobacteria. Rev. Infect. Dis. 3:841–861.
53. Van der Vliet, G. M. E., R. A. F. Schukkink, B. van Gemen, P. Schepers, and
P. R. Klatser.1993. Nucleic acid sequence-based amplification (NASBA) for
the identification of mycobacteria. J. Gen. Microbiol. 139:2423–2429. 54. Vaneechoutte, M., H. de Beenhouwer, G. Claeys, G. Verschraegen, A. de
Rouck, N. Paepe, A. Alachouni, and F. Portaels.1993. Identification of
Mycobacterium species by using amplified ribosomal DNA restriction
anal-ysis. J. Clin. Microbiol. 31:2061–2065.
55. Van Soolingen, D., P. E. W. de Haas, P. W. M. Hermans, P. M. A. Groenen,
and J. D. A. van Embden.1993. Comparison of various repetitive DNA
elements as genetic markers for strain differentiation and epidemiology of
Mycobacterium tuberculosis. J. Clin. Microbiol. 31:1987–1995.
56. Verstijnen, C. P. H. J., H. M. Ly, K. Polman, C. Richter, S. P. Smits, S. Y.
Maselle, P. Peerbooms, D. Rienthong, N. Montreewasuwat, S. Koanjanart,
D. D. Trach, S. Kuijper, and A. H. J. Kolk.1991. Enzyme-linked
immu-nosorbent assay using monoclonal antibodies for identification of mycobac-teria from early cultures. J. Clin. Microbiol. 29:1372–1375.
57. Vincent Le´vy-Fre´bault, V., and F. Portaels. 1992. Proposed minimal stan-dards for the genus Mycobacterium and for description of new slowly growing
Mycobacterium species. Int. J. Syst. Bacteriol. 42:315–323.
58. Wolinsky, E. 1992. Mycobacterial diseases other than tuberculosis. Clin. Infect. Dis. 15:1–12.
59. Woods, G. L., and J. A. Washington II. 1987. Mycobacteria other than
Mycobacterium tuberculosis; review of microbiologic and clinical aspects.
Rev. Infect. Dis. 9:275–294.