0095-1137/05/$08.00⫹0 doi:10.1128/JCM.43.10.5263–5271.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Use of Sequence Data Generated in the Bayer TruGene Genotyping
Assay To Recognize and Characterize Non-Subtype-B Human
Immunodeficiency Virus Type 1 Strains
Diane L. Hirigoyen
1and Charles P. Cartwright
1,2*
Department of Laboratory Medicine and Pathology, Hennepin County Medical Center, Minneapolis, Minnesota 55415,1and Department of Laboratory Medicine and Pathology, University
of Minnesota Medical School, Minneapolis, Minnesota 554552
Received 21 April 2005/Returned for modification 5 July 2005/Accepted 11 July 2005
Human immunodeficiency virus type 1 (HIV-1) protease (PR) and reverse transcriptase (RT) gene sequences obtained during antiretroviral resistance testing with a commercial genotyping assay (TruGene; Bayer Corp.) were analyzed to assess the utility of these data for detecting and characterizing non-subtype-B HIV-1 strains. A total of 125 viral sequences obtained from patients believed to have acquired their HIV-1 infection in Africa were analyzed, of which 121 were determined to belong to non-B subtypes. Utilizing TruGene sequence data alone, 92 (76%) of these viruses could be subtyped by conventional phylogenetic analysis. The addition of supplemental RT sequence data enabled a further 28 (23.1%) viruses to be classified, while one (0.9%) sample could not be classified conclusively. Two internet-accessible databases that generate HIV-1 subtypes from PR and RT sequences (HIV-SEQ and Geno2Pheno) were also evaluated, and both achieved 88% concordance (106/120) with phylogenetic analysis. Non-subtype-B and B-subtype HIV-1 sequences could be readily discrim-inated by tallying silent polymorphisms listed on the TruGene research report. The mean number of silent polymorphisms in the non-B HIV-1 sequences identified in this study was 58.3 (95% confidence interval [CI], 41.1 to 75.5), compared with 20.7 (95% CI, 9.9 to 31.5) for the four subtype B viruses in the study cohort and 118 case-matched B-subtype controls. Sequence data generated in the TruGene HIV-1 genotyping assay could, therefore, provide a ready means of tracking the prevalence and identity of non-B subtypes in HIV-1-infected populations undergoing routine antiretroviral resistance testing.
One of the hallmarks of the human immunodeficiency virus type 1 (HIV-1) is its enormous genetic diversity, a consequence of high rates of mutation and recombination during viral rep-lication, necessitating the development of an increasingly com-plex scheme for viral classification (30, 31, 35). The major (M) group of HIV-1 strains, responsible for the overwhelming ma-jority of infections, currently contains nine recognized subtypes (A to D, F to H, J, and K), two of which, A and F, are further divided into sub-subtypes (A1 and A2 and F1 and F2) (31). The circulation of multiple subtypes in certain geographic re-gions has led to further diversity since it has resulted in a subset of patients in these locales becoming simultaneously infected with viruses belonging to different subtypes, with subsequent intersubtype recombination events generating unique chimeric viruses (36). The epidemiologic importance of these viruses has been demonstrated in several studies conducted in sub-Saharan Africa, where unique chimeras have been shown to be present in 30% or more of HIV-1-infected individuals (10). Finally, certain specific intersubtype recombinant viruses have been found in multiple epidemiologically unrelated individu-als, indicating the establishment of entirely novel recombinant subtypes termed circulating recombinant forms (CRFs), of which 16 have been formally recognized to date (31, 36).
The impact of the bewildering genetic variability of HIV-1
on therapeutic response and antiretroviral resistance develop-ment remains uncertain (11, 33, 34). This lack of knowledge is due both to the comparatively limited use of antiretroviral drugs in those areas of the world where HIV-1 diversity is greatest and to the overwhelming predominance of a single subtype, namely B, in those areas of the world where antiret-roviral use is heaviest (33). Conflicting reports have been pub-lished on the relative rates of de novo primary resistance mu-tations to antiretroviral drugs in B versus non-B subtypes (2, 3, 8, 9, 22, 29), and insufficient data are currently available to definitively determine whether the genetic background of a viral isolate (as indicated by subtype) influences the phenotypic significance of resistance-associated mutations (11, 22, 33). Given both burgeoning efforts to implement antiretroviral treatment programs in the developing world, such as the WHO “3 million by 2005” initiative (14), and the emerging signifi-cance of non-B HIV-1 subtypes in the developed world (7, 21, 27, 32), it would seem prudent to perform viral subtype sur-veillance in patients prior to commencing antiretroviral ther-apy in order to better elucidate relationships between viral subtype and therapeutic outcome.
Phylogenetic analysis of nucleotide sequences of one or more regions of the HIV-1 genome remains the primary ap-proach for determining viral subtype (31). Definitive assigna-tion of a viral isolate to a particular subtype or CRF and optimal identification of unique chimeras require sequencing of multiple regions of the genome, a costly and laborious exercise. Although such extensive evaluation is clearly neces-sary in investigations seeking either to thoroughly assess viral
* Corresponding author. Mailing address: Clinical Laboratories P4, Hennepin County Medical Center, 701 Park Ave., Minneapolis, MN 55415. Phone: (612) 873-3026. Fax: (612) 904-4229. E-mail: charles [email protected].
5263
on May 15, 2020 by guest
http://jcm.asm.org/
diversity in a given population or to determine local and global patterns of viral transmission, it may not be required if the scope of the investigation is more limited, such as investigating the influence of subtype on therapeutic outcome. With the exception of the recently licensed fusion inhibitor enfuvirtide (16), the target site for all antiretroviral agents is either the protease (PR) or reverse transcriptase (RT) of HIV-1, proteins encoded by contiguous genes within thepolregion of the viral genome. Determination of resistance to these agents is primar-ily accomplished by RT-PCR amplification of circulating viral RNA followed by nucleotide sequencing to detect the presence of resistance-conferring mutations, a technique commonly re-ferred to as HIV genotyping. To assess the influence of genetic variation on parameters of therapeutic effectiveness, it would seem reasonable to determine subtype using the genomic re-gion targeted by antiretroviral agents, and thus the sequence information generated by commercial HIV genotyping sys-tems, such as the ViroSeq HIV-1 genotyping system (Celera Diagnostics) and the TruGene HIV-1 genotyping test (Bayer Corp.), seems ideally suited for this purpose. Preliminary eval-uations of this approach with the ViroSeq system have been encouraging, although determination of subtype in all studies to date has been accomplished using complicated, time-con-suming, phylogenetic analysis of data (4, 13). Interestingly, however, a number of web-based algorithms are now available that offer a real-time determination of viral subtype from PR and RT. HIV-SEQ is one of several applications available at the Stanford University HIV Drug Resistance Database (http://hivdb.stanford.edu) and includes an algorithm for as-signing subtypes that involves determining comparative nucle-otide similarity scores between submitted sequences and a panel of reference sequences representative of the main sub-types and CRFs of group M HIV-1 strains (18). Geno2Pheno (http://195.37.60.133/cgi-bin/geno2pheno.pl) is an application developed by Genafor (Bonn, Germany) ostensibly for predict-ing phenotypic resistance to antiretroviral agents from geno-typic data using decision tree analysis (6); however, a supple-mentary algorithm simultaneously performs a BLAST-type comparison of submitted sequences with a database containing several representative sequences of each subtype and CRF. Utilization of these services requires comparatively little effort by the end user; HIV-SEQ will accept sequence files directly from the TruGene system, obviating even the need to convert them into file formats compatible with FASTA/BLAST anal-yses (28), and no knowledge of phylogenetic analysis is needed. If such simplified analyses can provide a reasonably accurate determination of subtype, it offers the possibility that most, if not all, laboratories performing HIV genotyping could deter-mine viral subtype relatively routinely and, ultimately, that algorithmic approaches similar to those currently offered by third parties could be incorporated into future iterations of the sequence analysis software provided by the genotyping assay manufacturers.
The present study was undertaken both to establish the utility of sequence data generated by the TruGene HIV-1 genotyping system for determining viral subtype using conven-tional phylogenetic analysis and to examine the accuracy of web-based algorithms for elucidating subtypes from the same data. In addition, information provided in the TruGene geno-typing report was scrutinized in order to determine whether
some simple screening metric could be used to demarcate B from non-B viruses prior to subjecting sequences to further interrogation.
MATERIALS AND METHODS
Study institution and patients.Hennepin County Medical Center is a 450-bed public teaching facility located in Minneapolis, Minnesota. The institution aver-ages 20,000 patient admissions and 400,000 outpatient clinic and emergency room visits annually and provides continuing care for approximately 1,500 per-sons with HIV infection. Individuals who acquired their infection in Africa and are thus highly likely to be infected with a non-B HIV-1 subtype have become an increasingly significant component of those receiving care in the HIV clinic at Hennepin County Medical Center, comprising approximately 15% of the current patient population (1). From a database maintained in the HIV clinic, a total of 125 patients were identified who were believed to have acquired their HIV infection in Africa and had either had a TruGene HIV genotype test performed
(n⫽95) or had an archived plasma sample available (n⫽30) that contained at
least 50 copies of HIV RNA/ml. Additional demographic, clinical, and virologic characteristics of this cohort of patients have been described elsewhere (1).
HIV-1 genotyping.HIV-1 PR and RT sequences were generated from archived plasma samples using the TruGene HIV-1 genotyping assay (v1.5) (Bayer Diag-nostics) essentially as recommended by the manufacturer. The only significant modification was the addition of an initial viral concentration step on samples with HIV RNA concentrations of 50 to 1,000 copies/ml in which 1 ml of plasma
was centrifuged at 28,000⫻gfor 60 min at 4°C and 860l of supernatant was
removed prior to resuspension of the resultant pellet. The TruGene HIV-1 genotyping assay consists of an RT reaction performed on HIV RNA extracted
from plasma followed by PCR amplification of a 1,318-bp fragment of thepol
gene. Cycle sequencing of two noncontiguous regions of this fragment is then performed, generating a 288-bp sequence of the PR gene (codons 4 to 99) and a 630-bp sequence of the RT gene (codons 36 to 247). The OpenGene (Bayer Diagnostics) software system edits and aligns the resultant data, generates a protein translation of the sequence, and determines the presence of antiretro-viral resistance-conferring polymorphisms (24).
Phylogenetic analysis of sequence data.PR and RT sequences generated using the TruGene HIV genotyping assay were converted from the native format of the Unix-based OpenGene program (.glc files) to FASTA-formatted text files using the Exporter utility and then exported to a desktop personal computer running the Windows operating system. Since the TruGene system generates noncontig-uous sequences for the protease and RT genes, sequences for each gene were independently aligned with 59 representative sequences of HIV-1 group M subtypes and CRFs available from the Los Alamos database (http://www.hiv.lanl .gov/content/hiv-db) using BioEdit v. 5.0.9. Sequences used for alignment were as follows: A1-Q23-17 (AF004885), A1-SE7253 (AF069670), A1-U455 (M62320), A1-UG037 (U51190), A2–97CDKFE4 (AF286240), A2–97CDKS10 (AF286241), A2– 94CY017–41 (AF286237), B-HXB2 (K03455), B-RF (M17451), B-JRFL (U63632),
B-WEAU160 (U211135), C-92BR025 (U52953), C-96BW0502 (AF110967),
C-ETH2220 (U46016), C-99ET3 (AY255827), C-SM145 (AF447850), C-95IN21068 (AF067155), D-ELI (K03454), D-NDK (M27323), D-84ZR085 (U88822), D-94UG114 (U88824), F1-VI850 (AF077336), F1–93BR020.1 (AF005494), F1-FIN9363 (AF07503), F1-MP411 (AJ249238), F2-MP255 (AJ249236), F2-MP257 (AJ249237), G-DRCBL (AF084936), G-92NG083 (U88826), G-SE6165 (AF061642), VI991 (AF190127), H-VI997 (AF190128), H-90CF056 (AF005496), J-SE7887 (AF082394), J-SE7022 (AF082395), K-EQTB11C (AJ249235), K-MP535 (AJ249239), CRF01_AE-90CF11697 (AF197340), CRF01_AE-90CF402 (U51188), CRF01_AE-90CF4071 (AF197341), CRF01_AE-CM240 (U54771), CRF02_AG-LB12 (AF212281), CRF02_AG-LB-POC44951 (AF447831), CRF02_AG-97CM-MP807 (AJ286133), CRF02_AG-DJ264 (AF063224), CRF02_AG-IBNG (L39106), CRF02_AG-SE7812 (AF107770), CRF03_ AB-KAL153-2 (AF193276), CRF04_cpx-CY032 (AF049337), CRF05_DF-VI1310 (AF193253), CRF06_cpx-BFP90 (AF064669), CRF07_BC-97CN001 (AF286226), CRF08_BC-97CNGX-7F (AY008716), CRF10_CD-96TZ-BF061 (AF289548), CRF11_cpx-MP818 (AJ291718), CRF12_BF-ARMA159 (AF385936), CRF13_cpx-1849 (AF460972), CRF14_BG-X475 (AF423759), SIV-CPZGAB (X52154). Phylogenetic distances were then elucidated by constructing neighbor-joining trees based on Kimura’s two-parameter matrix, and the robustness of these relationships was tested by the bootstrap method using 100 replications. These analyses were conducted using applications provided in the MEGA 2.1 software suite (www.megasoftware.net) (23). The simian immunodeficiency virus se-quence CPZGAB was used as the outgroup in all phylogenetic trees. Determi-nation of subtype or CRF was considered definitive only if the bootstrap value linking a given sample exceeded 70% for both gene regions analyzed. Samples
on May 15, 2020 by guest
http://jcm.asm.org/
that could not be unambiguously assigned to a subtype using this approach were subjected to further analysis, consisting of the performance of an additional amplification and sequencing reaction to fill the 111-bp gap between codon 99 of the PR gene and codon 36 of the RT gene. Using TruGene RT-PCR products as
templates, PCR was performed using primers polfp2 (5⬘-GGTACAGTATTAG
TAGGACCTACA-3⬘) and polrp2 (5⬘-CCCACATCTAGTACTGTCACT-3⬘),
generating a 415-bp amplicon spanning the 3⬘end of the PR gene and the 5⬘end
of the RT gene. PCR products were purified, and sequencing was performed on an automated DNA sequencer (ABI model 3730; Applied Biosystems) using the ABI Prism Big Dye Terminator Cycle Sequencing kit v3.1 (Applied Biosystems).
Contiguous 1,029-bppolsequences were then assembled, and phylogenetic
anal-ysis was repeated as described above. For samples still yielding ambiguous results, the possibility that these sequences consisted of intersubtype mosaics was initially investigated using the recombination identification program (RIP v2.0; http://hivweb.lanl.gov/RIP/RIPsubmit.html) and then further analyzed by bootscanning using the SimPlot v2.5 software package (http://sray.med.som.jhmi .edu/SCRoftware).
Subtype determination using web-based applications.The subtype of the 125 samples from African-born individuals was also determined by uploading FASTA-formatted files of TruGene sequence data to two internet-accessible, public domain computer programs, namely, HIV-SEQ (Stanford University, Palo Alto, Calif.; www.hivdb.stanford.edu), and Geno2Pheno (Genafor, Bonn, Germany; http://195.37.60.133/cgi-bin/geno2pheno.pl). Since HIV-SEQ indepen-dently analyzes RT and protease sequence data, use of this program resulted in the generation of a subtype for each of the genomic regions analyzed, whereas Geno2Pheno provided only a single subtype based on the best fit of the entire sequence to the reference sequences in its database.
Nucleotide sequence accession numbers.GenBank accession numbers for the
contiguouspolsequences determined in the study are DQ009495 to DQ009523.
RESULTS
Viral quantitation of non-B-subtype samples used for HIV-1 genotyping. The viral loads of all plasma samples used for genotyping that contained non-B-subtype HIV-1 strains were ascertained using either the Quantiplex bDNA v3.0 assay (Bayer Diagnostics, Tarrytown, NY) or the Roche COBAS Monitor v1.5 UltraSensitive RT-PCR assay (Roche Molecular Diagnostics, Indianapolis, Ind.), both of which have been dem-onstrated to provide accurate determinations of non-sub-type-B HIV-1 RNA levels (12). Viral loads of plasma samples (n⫽28) collected prior to August 2002 were determined using the bDNA assay and ranged from 2.70 to 5.74 log10copies/ml
with a mean viral load of 4.13 log10copies/ml. The viral loads
of the remaining samples (n⫽93) were determined using the UltraSensitive RT-PCR assay and ranged from 2.20 to ⬎5.0 log10copies/ml. Thirty-two of these samples (34.4%) had viral
loads in excess of the upper limit of quantitation of this assay (5.0 log10copies/ml); the mean viral load of the remaining 61
samples was 3.96 log10copies/ml.
Performance of the TruGene HIV-1 genotype assay.Version 1.5 of the TruGene HIV genotyping assay successfully gener-ated double-stranded sequence in both the protease and RT regions for all non-B viruses tested. This included nine samples with viral loads of⬍3.0 log10copies/ml, the stated lower limit
of sensitivity of the TruGene assay, that were subjected to an ultracentrifugation procedure prior to isolation of HIV RNA. The TruGene assay contains four sets of sequencing primers, two of which (PR and P2) cover a virtually identical region of the protease gene and two (RT beginning and RT middle) that between them cover codons 36 to 247 of the RT gene. No difference was observed in the performance of the RT se-quencing primers when non-B and B-subtype viruses were compared (data not shown), with excellent double-stranded sequence being obtained in this region irrespective of the viral
subtype. Similarly, excellent sequence data were obtained us-ing the P2 protease primer pair; however, the PR protease primer pair performed relatively poorly with non-B-subtype sequences. For only 41/121 (33.9%) of the non-B-subtype se-quences analyzed was the PR sequence of sufficient quality in both directions to be usable. The success rate of the PR primer pair was somewhat subtype specific, with the highest success rate being observed with viruses belonging to CRF02_AG (20/ 26; 76.9%) and subtype D (9/12; 75.0%). In contrast, only 12.5% (6/48) of subtype C viruses and 13.0% (3/23) of subtype A viruses yielded usable sequence with the PR primer set.
[image:3.585.299.541.89.367.2]Phylogenetic determination of HIV-1 subtype. Since the TruGene assay yields two noncontiguous sequences, one for the PR gene and one for the RT gene, phylogenetic analysis initially was performed independently for each of the genes. Using the criterion of a bootstrap value in excess of 70% (based on 100 replicates) to assign each unknown sequence to a viral subtype, 96/125 (76.8%) samples gave unambiguous subtyping results for both the PR and RT regions using the TruGene sequencing data (Table 1). Four of the patients in the initial cohort were determined by this analysis to be infected with subtype B viruses; thus, the success rate for non-B viruses using this approach was 76.0% (92/121). Ninety-one of the 92 non-B viruses whose subtype could be resolved using phyloge-netic analysis of only the TruGene sequence data belonged to pure subtypes or CRFs, while a single sample contained an unequivocally chimeric virus that clustered with subtype C
TABLE 1. Results of phylogenetic analysis of 121 non-B HIV-1 viruses using TruGene sequencing data
Analysisa(n) Subtype
No. of samples
classified bypolregion
PR RT PR-RT
Primary analysis (121) A 15 17 12
C 47 46 45
D 10 12 10
F 1 0 0
G 1 2 1
CRF02_AG 25 24 23
Chimeric (C/D) 1 1 1
NDb 22 20 29
Secondary analysis (29) A 11
C 3
D 2
G 2
CRF02_AG 3
CRF06_cpx 1
Chimeric 6
ND 1
Final designation (121) A 23
C 48
D 12
G 3
CRF02_AG 26
CRF06_cpx 1
Chimeric 7
ND 1
aPrimary analysis shows the results obtained using TruGene data alone;
sec-ondary analysis shows the results obtained after supplemental sequencing al-lowed analysis of contiguous 1,029-bp PR-RT sequence.
bND, not determinable.
on May 15, 2020 by guest
http://jcm.asm.org/
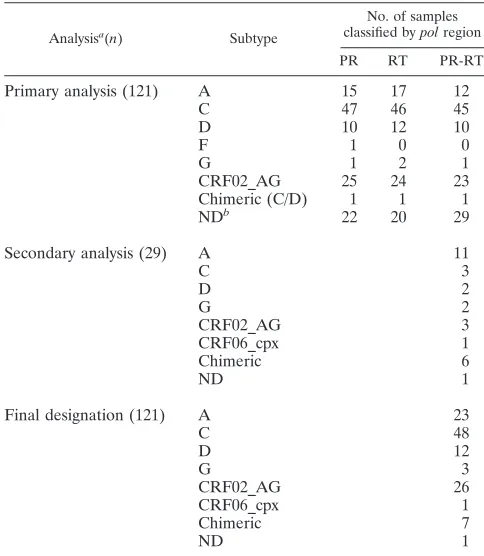
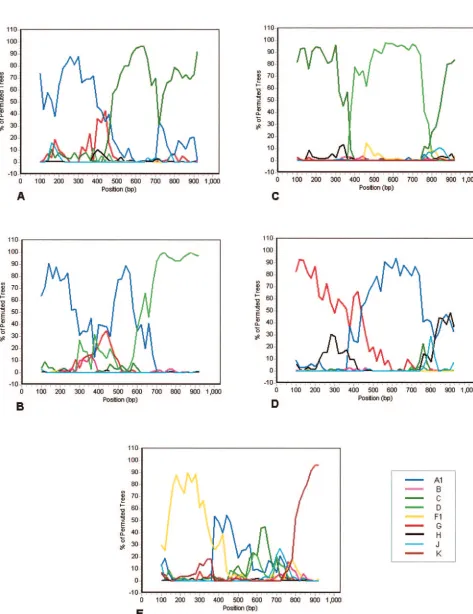
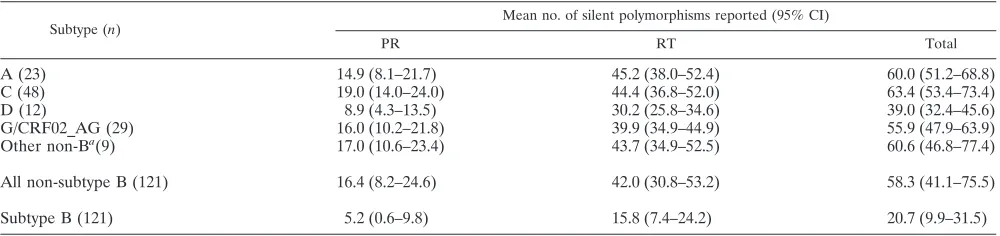
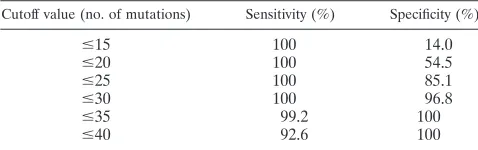
viruses in the protease region and with subtype D viruses in the RT region. Of the 29 viruses whose subtype could not be conclusively ascertained using purely TruGene data, 12 (41.4%) yielded ambiguous results for both gene regions, 10 (34.5%) could be assigned a subtype based on analysis of the RT but not the PR gene, and 7 (24.1%) could be assigned based on analysis of the PR but not the RT gene. The gener-ation of additional RT sequence enabled phylogenetic analysis of a contiguous 1,029-bppolfragment to be performed on the 29 problematic viruses (Table 1). Twenty-eight of these 29 samples could then be subtyped with an acceptable level of confidence, with 22 belonging to single subtypes or CRFs and six being chimeric in this region (three G/A, one A/D, one A/C, and one F/K). Representative examples of the bootscanning plots obtained with these intersubtype recombinants are shown in Fig. 1. The subtype composition of a single sample could not be conclusively determined, although it appeared to be most likely an A/C recombinant. The subtype designation of the remaining 120 non-B viruses in the sample cohort is listed in Table 1.
Subtype designation using HIV-SEQ (Stanford).HIV-SEQ interrogates PR and RT sequence data independently; thus, two viral subtypes were generated for each TruGene sequence uploaded to the database. For 106 of the 120 samples (88.3%) in our cohort that were determined to be non-B and could be assigned to a subtype by phylogenetic analysis, the results of the HIV-SEQ analysis were entirely in concordance with the ultimate result of the phylogenetic analysis (Table 2). Of the 14 samples that were assigned incorrectly using HIV-SEQ, four (28.6%) were assigned incorrectly for the PR gene but cor-rectly for the RT gene, three (21.4%) were assigned incorcor-rectly for the RT gene but correctly for the PR gene, and seven (50.0%) were assigned incorrectly for both regions. The most common error was the designation of a viral sequence as be-longing to CRF01_AE when the virus was subtype A. This type of error accounted for 56.5% (13/23) of miscalls in either gene (Table 2). The remaining misidentifications consisted of three failures to detect intersubtype chimeras (all of which were G/A), one misidentification of a subtype C virus as subtype K in the PR region (and therefore as a K/C chimera), one mis-identification of a subtype G virus as CRF02_AG in the PR region, and one failure to identify a CRF (CRF06_cpx) in either region (Table 2). Although HIV-SEQ reports a percent similarity between the submitted sequence and the closest match in its database, we found no correlation between this value and the likelihood of the HIV-SEQ subtype determina-tion being in agreement with the phylogenetic analysis (data not shown).
Subtype designation using Geno2Pheno (Genafor).Subtype determination by the Geno2Pheno program was concordant with phylogenetic analysis for 106/120 (88.3%; Table 2) se-quences in our non-B HIV-1 cohort. Not surprisingly, given that Geno2Pheno generated only a single “best-fit” subtype for each PR-RT sequence submitted, it misclassified all seven of the chimeric sequences in the cohort. Of the remaining seven errors made by Geno2Pheno, five were incorrect designations of pure subtype viruses as CRFs, one was an incorrect desig-nation of a CRF as a pure subtype, and one was a misidenti-fication of a subtype C virus as subtype K (Table 2).
Interest-ingly, this was not the same subtype C virus that was misclassified as a K/C chimera by HIV-SEQ.
Validation of a screening algorithm for identifying non-B-subtype sequences. Once the determination of forward and reverse PR and RT sequences has been completed in the TruGene assay, the OpenGene software program combines these sequences and compares them with an archetypal sub-type B viral sequence, HIV-LAI (A04321). A final report is then generated which, in addition to listing significant muta-tions at known resistance codons, also contains a line listing of silent polymorphisms. Given that the comparator sequence is a subtype B virus, we reasoned that the number of silent poly-morphisms listed should be considerably higher if the sample sequence belongs to a non-subtype-B virus. Table 3 shows the mean number and 95% confidence interval (CI) of silent poly-morphisms for the 121 non-B viruses analyzed in this study and for 121 subtype B viruses. The subtype B cohort includes the viruses from the four patients originally included in our study cohort and 118 additional sequences retrieved from our ar-chives. These patients were selected to match the non-B cohort with respect to viral load, CD4⫹cell count, and exposure to antiretroviral drugs, and their subtype was confirmed as B by phylogenetic analysis of TruGene sequences. As expected, sig-nificantly more silent polymorphisms were detected in the non-subtype-B viral sequences than in the B sequences (Table 3). The mean number of silent polymorphisms in the PR and RT regions combined was 20.7 for subtype B viruses (95% CI, 9.9 to 31.5) versus 58.3 (95% CI, 41.1 to 75.5) for non-subtype-B viruses (Table 3). When the individual gene regions were an-alyzed independently, the mean number of silent polymor-phisms was significantly higher in non-B than in B viruses for the RT region but not for the PR region (Table 3). We also examined the number of silent polymorphisms reported by subtype to establish that all the non-B subtypes detected in our cohort could be effectively differentiated from subtype B vi-ruses using an algorithm based on this metric (Table 3). All of the subtypes identified in our non-B cohort could be differen-tiated from subtype B viruses, at the 95% confidence level, based on either the number of mutations in the RT sequence alone or that in the PR and RT regions combined (Table 3); however, neither subtype A nor subtype D viruses had signif-icantly higher numbers of mutations in the PR region than subtype B viruses did (Table 3). Subtype D viruses also had significantly lower numbers of mutations in both the PR and RT regions than did any of the other non-B-subtype viruses identified in our cohort (Table 3).
To determine an appropriate cutoff for screening TruGene assay results for non-B viral sequences, we determined the sensitivity and specificity of using various total numbers of silent mutations for this purpose (Table 4). A cutoff value of 30 or fewer total silent mutations appeared to result in an optimal combination of sensitivity (100%, with no non-B viruses having a total mutation count of ⬍35) and specificity (96.8%), en-abling detection of all non-B clade viruses with exclusion of all but four subtype B viruses in our cohort (Table 4). Although using a cutoff value of 35 or fewer mutations would have eliminated all subtype B viral sequences from further analysis, one subtype D virus in our cohort had 35 silent mutations, and a further six of the 12 subtype D viruses had silent mutation counts between 35 and 38, suggesting that the negative
on May 15, 2020 by guest
http://jcm.asm.org/
FIG. 1. Representative bootscanning plots of putative chimeric viral sequences compared to representative isolates of HIV-1 subtypes A1 (U455), B (LAI), C (ETH220), D (UG114), F1 (VI850), G (NG083), H (VI991), J (SE7887), and K (EQTB). (A) A/C chimera; (B) A/D chimera; (C) C/D chimera; (D) G/A chimera; (E) F/K chimera. Plots were obtained using a sliding window of 200 bp moving in 20-bp steps.
on May 15, 2020 by guest
http://jcm.asm.org/
tive value of such an aggressive cutoff value might be less than ideal in populations with higher frequencies of subtype D in-fection.
DISCUSSION
The results of this study demonstrate that the TruGene HIV-1 genotyping assay can be used to generate sequence data for determining antiretroviral resistance in phylogenetically diverse non-B-subtype viruses, even those with viral loads sub-stantially below the claimed lower limit of the assay, and that the sequence information thus generated can be used to de-termine with a reasonable degree of accuracy the subtype of such viruses.
Our finding that version 1.5 of the TruGene assay enables non-B HIV-1 subtype viruses to be sequenced with a high success rate is consistent with previous studies examining the performance characteristics of this version of the assay (5, 15, 20). The primary difference between versions 1.0 and 1.5 of the TruGene HIV-1 genotyping assay is a modification to the RT-PCR primers (20). This change resulted in a more efficient and robust RT-PCR amplification, which in turn improved the per-formance of the assay on both low-viral-load samples and sam-ples containing non-B-subtype viruses (20). Our finding of a significant failure rate of the PR sequencing primer pair of the TruGene assay for most non-B subtypes is also in agreement with previous studies. In our sample cohort, only 34% of pro-tease sequences could be elucidated using the PR primer pair,
a result comparable to the 33% reported by Fontaine et al. (15) and 22% determined by Jagodzinski and coworkers (20). In-terestingly, we obtained a significantly higher rate of success with the PR primers for determining the protease sequence of both subtype D (75%) and CRF02_AG (77%) viruses com-pared with other non-B viruses. The relatively small phyloge-netic distance between subtypes B and D, and the resultant lower overall frequency of mismatches in both the RT-PCR and sequencing primer regions, is a logical explanation for the relative success of this primer pair with subtype D. Presumably, the absence of certain mismatches in the primer regions also accounts for the relative success of the PR primer pair with CRF02_AG viruses compared with viruses belonging to other subtypes whose overall sequence composition is equally diver-gent from subtype B. The ultimate importance of the PR primer sequencing failures was negligible, since excellent dou-ble-stranded sequence was typically obtained with the alternate protease sequencing primers (P2). Nevertheless, the observed subtype-specific variation in the performance of the PR primer pair highlights the need for continued surveillance of the per-formance of systems such as the TruGene assay on novel CRFs and unique viral recombinants as they are discovered to ensure that these assays continue to perform effectively on all group M HIV-1 strains.
[image:6.585.44.542.89.233.2]The need to perform epidemiologic surveillance of HIV-1 diversity in the United States has been recognized by the Cen-ters for Disease Control and Prevention (37), although the
TABLE 2. Comparison of subtyping results obtained with Geno2Pheno and HIV-SEQ analysis of TruGene PR and RT sequences with results obtained using conventional phylogenetic analysis
Phylogenetic result
(n)
Geno2Pheno HIV-SEQ
PR-RT No. of
errors PR RT
No. of errors
A (23) A (20), CRF01_AE (3) 3 A (17), CRF01_AE (6) A (16), CRF01_AE (7) 8
C (48) C (47), K (1) 1 C (47), K (1) C (48) 1
D (12) D (10), CRF10_CD (2) 2 D (12) D (12) 0
G (3) G (3) 0 G (2), CRF02_AG (1) G (3) 1
CRF02_AG (26) CRF02_AG (25), G (1) 1 CRF02_AG (26) CRF02_AG (26) 0
CRF06_cpx (1) CRF06_cpx (1) 0 CRF02_AG (1) G (1) 1
A/C (1) C (1) 1 A (1) C (1) 0
A/D (1) D (1) 1 A (1) D (1) 0
G/A (3) G (1), CRF01_AE (2) 3 G (1), CRF02_AG (2) G (1), CRF01_AE (2) 3
C/D (1) D (1) 1 C (1) D (1) 0
F/K (1) CRF09_cpx (1) 1 F (1) K (1) 0
TABLE 3. Comparison of the mean numbers of silent polymorphisms listed on the TruGene report by individual subtype
Subtype (n)
Mean no. of silent polymorphisms reported (95% CI)
PR RT Total
A (23) 14.9 (8.1–21.7) 45.2 (38.0–52.4) 60.0 (51.2–68.8)
C (48) 19.0 (14.0–24.0) 44.4 (36.8–52.0) 63.4 (53.4–73.4)
D (12) 8.9 (4.3–13.5) 30.2 (25.8–34.6) 39.0 (32.4–45.6)
G/CRF02_AG (29) 16.0 (10.2–21.8) 39.9 (34.9–44.9) 55.9 (47.9–63.9)
Other non-Ba(9) 17.0 (10.6–23.4) 43.7 (34.9–52.5) 60.6 (46.8–77.4)
All non-subtype B (121) 16.4 (8.2–24.6) 42.0 (30.8–53.2) 58.3 (41.1–75.5)
Subtype B (121) 5.2 (0.6–9.8) 15.8 (7.4–24.2) 20.7 (9.9–31.5)
a“Other” includes CRF06_cpx (1), A/C (1), A/D (1), G/A (3), C/D (1), F/K (1), and unclassified (1).
on May 15, 2020 by guest
http://jcm.asm.org/
[image:6.585.43.543.598.716.2]precise mechanism by which such surveillance should or could take place remains to be determined. Given the relative rarity of non-B viruses in most populations of HIV-infected persons in the United States, conducting active surveillance for viral variants by collecting samples for sequence analysis on a rep-resentative sampling of newly diagnosed individuals would be a highly onerous, expensive, and largely unrevealing exercise. Emerging evidence of increasing rates of transmission of vi-ruses harboring antiretroviral resistance mutations has re-sulted in the publication of more expansive guidelines for using genotypic resistance assays (19), however, making passive sur-veillance of viral diversity by screening genotyping data an increasingly viable option. The possibility of using the list of silent (synonymous) mutations documented on the TruGene report for differentiating B from non-B viral sequences was first intimated in the study by Jagodzinski and colleagues (20), although the relatively small sample size of their study pre-cluded them from definitively establishing the validity of this technique. We analyzed the number of silent mutations re-ported by TruGene for each non-B virus in our cohort and compared this group with a matched group of subtype B vi-ruses that had been sequenced in our laboratory (Table 4). The use of a simple cutoff value of 35 silent polymorphisms enabled detection of all non-B viral sequences while eliminating 117 of the 121 subtype B viral sequences (Table 4). These data con-vincingly establish that non-B viruses can be detected in a population that contains predominantly subtype B viruses without any expenditure of effort beyond that necessary to perform the TruGene assay for its intended clinical purpose of detecting antiretroviral resistance mutations. Screening Tru-Gene assay reports, with samples whose analysis resulted in a listing of⬎30 silent polymorphisms being retained for further investigation, could provide a highly cost-effective means of monitoring the incidence of non-B viral subtypes in settings where genotyping assays are commonly performed at the time of HIV diagnosis.
The primary purpose of this investigation was to assess the utility of the sequence data generated in the TruGene assay for determining viral subtype, either via conventional phylogenetic analysis or by using public-domain, internet-based software programs. Since the TruGene assay does not currently gener-ate sequence for the entire RT-PCR amplicon, independent phylogenetic analyses were initially performed for the PR and RT sequences. Using comparatively stringent criteria for sub-type classification (see Materials and Methods), only 92 of the 121 (76%) non-B-subtype viruses in our cohort could be de-finitively assigned to a given subtype or CRF. This is not surprising given the relatively short sequence lengths, 288 bp
and 630 bp for the PR and RT genes, respectively, generated by the TruGene assay, and the high degree of sequence con-servation in thepolregion of HIV-1 (4, 31). The encouraging findings of a number of other investigators regarding the use of thepolregion, rather than the more diverse and widely utilized
envandgagregions, for subtype designation of HIV-1 viruses (4, 13, 18, 32) led us to believe that it was likely the limitation of having only two short, noncontiguous sequences for analysis that was responsible for our overall lack of initial success. To resolve this problem, we performed additional PCR amplifica-tions on TruGene RT-PCR amplicons generated from those samples that could not be subtyped definitively using the Tru-Gene sequence data alone, sequenced the products, and then generated contiguous PR-RT sequences encompassing 1,029 bp of thepolgene. Phylogenetic analysis of these sequences enabled us to conclusively identify the subtype of 28/29 (96.6%) viruses that had previously yielded ambiguous results, resulting in an extremely high overall success rate of 99.2% (120/121).
Not surprisingly, viral sequences that proved problematic for subtype determination were not evenly distributed among the different subtypes identified in our study. Subtypes G and A were significantly overrepresented in the problematic viral group with two of three subtype G viruses (66%) and 11 of 23 (48%) subtype A viruses requiring additional sequencing for classification. This is not unexpected given that differentiation of subtype A and CRF01_AE, which has a subtype A-derived
pol sequence, and to a lesser extent subtype G and CRF02_AG, which has a subtype G-derivedpolsequence, can be problematic if only a limited amount of sequence informa-tion is available for analysis (31). Subtype C and D viruses, in contrast, were readily identified without the need for addi-tional sequencing, with only 3 of 48 (6%) subtype C and 1 of 12 (8%) subtype D viruses yielding ambiguous results upon anal-ysis of TruGene-generated sequence data.
Among the viruses in our non-B cohort were seven that could be identified as intersubtype chimeras with recombina-tion breakpoints within the PR-RT genes and one virus that was almost certainly chimeric, although the precise subtype composition of the PR-RT region of that virus could not be elucidated. Numerous previous studies have demonstrated that, in those parts of the world where multiple subtypes occur with some frequency, unique recombinant viruses are not un-common and may even constitute a substantial minority of the total viral population (4, 10, 25, 38). The fact that a number of intersubtype recombinant viruses were detected in our uns-elected cohort further demonstrates the value of determining subtype composition within the PR-RT region ofpol. Although more conventional envsequence-based subtyping analysis of HIV-1 is highly probative for differentiating pure subtypes and CRFs, the very sequence heterogeneity in this region that makes it valuable for phylogenetic differentiation makes it a relatively poor location for viral recombination (26, 35), and chimeric viruses are likely to remain undetected if only env
sequences are analyzed. Sequence analysis ofpolis essential, therefore, if a definitive determination is to be made of the significance of viral heterogeneity for antiretroviral response and antiretroviral resistance development.
[image:7.585.43.282.99.174.2]In addition to performing phylogenetic analysis on TruGene sequences, we also examined the ability of two public-domain,
TABLE 4. Sensitivity and specificity of using various numbers of total silent mutations listed on the TruGene report as cutoff values
for differentiating non-subtype-B from subtype B viruses
Cutoff value (no. of mutations) Sensitivity (%) Specificity (%)
ⱕ15 100 14.0
ⱕ20 100 54.5
ⱕ25 100 85.1
ⱕ30 100 96.8
ⱕ35 99.2 100
ⱕ40 92.6 100
on May 15, 2020 by guest
http://jcm.asm.org/
internet-accessible computer algorithms, namely, the HIV-SEQ program offered at the Stanford University HIV Drug Resistance Database and the Geno2Pheno program of Genafor, to assign subtypes to the non-B viruses in our cohort. Both algorithms performed identically overall, achieving an 88.3% concordance (106/120) with the phylogenetic determi-nation. The nature of the erroneous calls made by the two programs was, however, somewhat different. Since the HIV-SEQ program analyzes the PR and RT gene sequences inde-pendently, it was reasonably successful in recognizing chimeric viruses with recombination breakpoints at, or close to, the PR-RT junction, with four of seven (57.1%) of such recombi-nants being correctly identified. The Geno2Pheno program, in contrast, determines a single “best fit” genotype for the entire sequence submitted and not surprisingly failed to identify a single chimeric sequence. Both algorithms made only a single complete misclassification of a pure subtype: Geno2Pheno identified a subtype C virus as subtype K while HIV-SEQ identified a different subtype C virus as a K/C chimera. The majority of the errors made by the HIV-SEQ program resulted in it identifying subtype A viruses as belonging to CRF01_AE (Table 2). It is not surprising that a relatively simple algorithm for designating subtype usingpolsequence data such as that employed by HIV-SEQ should experience difficulty in differ-entiating these two subtypes; indeed, one could question whether attempting to identify CRFs in this manner should even be attempted. The only other miscalls made by the HIV-SEQ program were the failure to identify two chimeric viral sequences and one CRF (CRF06_cpx) that is not currently included in the program’s database. Although the two com-puter algorithms performed comparably overall, our prefer-ence would be to use the HIV-SEQ program for TruGene sequences both because files generated by OpenGene can be uploaded directly to the website without manipulation and because the independent analysis of PR and RT sequences allows for the detection of at least some chimeric viral se-quences.
In conclusion, this study demonstrates that sequence infor-mation generated by the TruGene HIV-1 genotyping assay can be effectively used both to detect non-B viruses and to eluci-date which subtype such viruses belong to. Certain limitations to this analysis exist at the moment, the primary one being the lack of a single contiguous sequence covering most, if not all, of the RT-PCR amplicon. This deficiency hampered conven-tional phylogenetic analysis of the samples in our cohort, ne-cessitating the generation of additional sequence information for a considerable number of samples. The performance of the simple web-based algorithms as a surrogate for phylogenetic analysis was generally acceptable, although our results suggest that their identification of CRFs other than CRF02-AG should be routinely scrutinized by other means. Intriguingly, Gale and colleagues (17) recently described a rapid, automated, and potentially powerful computational approach for generating subtype information frompolsequences generated for antiret-roviral resistance testing, to which they gave the acronym STAR. Perhaps the inclusion of algorithms for identifying non-B subtypes along with analysis tools similar to the STAR program in the software packages provided with applications such as the TruGene genotyping assay could help facilitate the important task of monitoring HIV-1 subtype movement and
divergence using the increasingly available resource of se-quence information generated for determination of antiretro-viral resistance.
REFERENCES
1.Akinsete, O., T. Sides, D. Hirigoyen, C. P. Cartwright, C. Boraas, C. Davey, L. Pessoa-Brandao, and K. Henry.Unpublished data.
2.Apetrei, C., D. Descamps, G. Collin, I. Loussert-Ajaka, F. Damond, M. Duca, F. Simon, and F. Brun-Vezinet.1998. Human immunodeficiency virus type F
reverse transcriptase sequence and drug susceptibility. J. Virol.72:3534–
3538.
3.Barlow, K. L., I. D. Tatt, P. A. Cane, D. Pillay, and J. P. Clewley.2001. Recombinant strains of HIV type 1 in the United Kingdom. AIDS Res.
Hum. Retrovir.17:467–474.
4.Becker-Pergola, G., P. Kataaha, L. Johnston-Dow, S. Fung, J. B. Jackson, and S. H. Eshleman.2000. Analysis of HIV type 1 protease and reverse transcriptase in antiretroviral drug-naive Ugandan adults. AIDS Res. Hum.
Retrovir.16:807–813.
5.Beddows, S., S. Galpin, S. H. Kazmi, A. Ashraf, A. Johargy, A. J. Frater, N. White, R. Braganza, J. Clarke, M. McClure, and J. N. Weber.2003. Perfor-mance of two commercially available sequence-based HIV-1 genotyping systems for the detection of drug resistance against HIV type 1 group M
subtypes. J. Med. Virol.70:337–342.
6.Beerenwinkel, N., B. Schmidt, H. Walter, R. Kaiser, T. Lengauer, D. Hoff-mann, K. Korn, and J. Selbig.2002. Diversity and complexity of HIV-1 drug resistance: a bioinformatics approach to predicting phenotype from
geno-type. Proc. Natl. Acad. Sci. USA99:8271–8276.
7.Chaix, M. L., D. Descamps, M. Harzic, V. Schneider, C. Deveau, C. Tamalet, I. Pellegrin, J. Izopet, A. Ruffault, B. Masquelier, L. Meyer, C. Rouzioux, F. Brun-Vezinet, and D. Costagliola.2003. Stable prevalence of genotypic drug resistance mutations but increase in non-B virus among patients with
pri-mary HIV-1 infection in France. AIDS17:2635–2643.
8.Cornelissen, M., R. van den Burg, F. Zorgdrager, V. Lukashov, and J. Goudsmit.1997.polgene diversity of five human immunodeficiency virus type 1 subtypes: evidence for naturally occurring mutations that contribute to drug resistance, limited recombination patterns, and common ancestry for
subtypes B and D. J. Virol.71:6348–6358.
9.Descamps, D., C. Apetrei, G. Collin, F. Damond, F. Simon, and F. Brun-Vezinet.1998. Naturally occurring decreased susceptibility of HIV-1 subtype
G to protease inhibitors. AIDS12:1109–1111.
10.Dowling, W. E., B. Kim, C. J. Mason, K. M. Wasunna, U. Alam, L. Elson, D. L. Birx, M. L. Robb, F. E. McCutcheon, and J. K. Carr.2002. Forty-one near full-length HIV-1 sequences from Kenya reveal an epidemic of subtype
A and A-containing recombinants. AIDS16:1809–1820.
11.Dumans, A. T., M. A. Soares, E. S. Machado, S. Hue, R. M. Brindeiro, D. Pillay, and A. Tanuri.2004. Synonymous genetic polymorphisms within Brazilian human immunodeficiency virus type 1 subtypes may influence
mutational routes to drug resistance. J. Infect. Dis.189:1232–1238.
12.Elbeik, T., W. G. Alvord, R. Trichavaroj, M. de Souza, R. Dewar, A. Brown, D. Chernoff, N. L. Michael, P. Nassos, K. Hadley, and V. L. Ng.2002. Comparative analysis of HIV-1 viral load assays on subtype quantification: Bayer Versant HIV-1 RNA 3.0 versus Roche Amplicor HIV-1 Monitor
version 1.5. J. Acquir. Immune Defic. Syndr.29:330–339.
13.Eshelman, S. H., J. Hackett, P. Swanson, S. P. Cunningham, B. Drews, C. Brennan, S. G. Devare, L. Zekeng, L. Kaptue, and N. Marlowe.2004. Per-formance of the Celera Diagnostics ViroSeq HIV-1 genotyping system for sequence-based analysis of diverse human immunodeficiency virus type 1
strains. J. Clin. Microbiol.42:2711–2717.
14.Fleck, F.2004. WHO hopes 3-by-5 plan will reverse Africa’s HIV/AIDS
epidemic. Bull. W. H. O.82:77–78.
15.Fontaine, E., C. Riva, M. Peeters, J.-C. Schmit, E. Delaporte, K. van Laet-hem, K. van Vaerenbergh, J. Snoeck, E. van Wijngaerden, E. de Clercq, M. van Ranst, and A.-M. Vandamme.2001. Evaluation of two commercial kits for the detection of genotypic drug resistance on a panel of HIV type 1
subtypes A through J. J. Acquir. Immune Defic. Syndr.28:254–258.
16.Fung, H. B., and Y. Guo.2004. Enfuvirtide: a fusion inhibitor for the
treat-ment of HIV infection. Clin. Ther.26:352–378.
17.Gale, C. V., R. Myers, R. S. Tedder, I. G. Williams, and P. Kellam.2004. Development of a novel human immunodeficiency virus type 1 subtyping tool, subtype analyzer (STAR): analysis of subtype distribution in London.
AIDS Res. Hum. Retrovir.20:457–464.
18.Gonzales, M. J., R. N. Machekano, and R. W. Shafer.2001. Human immu-nodeficiency virus type 1 reverse-transcriptase and protease subtypes: clas-sification, amino acid mutation patterns, and prevalence in a northern
Cal-ifornia clinic-based population. J. Infect. Dis.184:998–1006.
19.Hirsch, M. S., F. Brun-Vezinet, B. Clotet, B. Conway, D. R. Kuritzkes, R. T. D’Aquila, L. M. Demeter, S. M. Hammer, V. A. Johnson, C. Loveday, J. W. Mellors, D. M. Jacobsen, and D. D. Richman. 2003. Antiretroviral drug resistance testing in adults infected with human immunodeficiency virus type 1: 2003 recommendations of an International AIDS Society-USA panel.
Clin. Infect. Dis.37:113–128.
on May 15, 2020 by guest
http://jcm.asm.org/
20.Jagodzinski, L. L., J. D. Cooley, M. Weber, and N. L. Michael.2003. Per-formance characteristics of human immunodeficiency virus type 1 (HIV-1) genotyping systems in sequence-based analysis of subtypes other than HIV-1
subtype B. J. Clin. Microbiol.41:998–1003.
21.Jorgensen, L. B., M. B. Christensen, J. Gerstoft, L. R. Mathiesen, N. Obel, C. Pedersen, H. Nielsen, and C. Nielsen.2003. Prevalence of drug resistance mutations and non-B subtypes in newly diagnosed HIV-1 patients in
Den-mark. Scand. J. Infect. Dis.35:800–807.
22.Kantor, R., and D. Katzenstein.2004. Drug resistance in non-subtype B
HIV-1. J. Clin. Virol.29:152–159.
23.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei.2001. MEGA2:
molec-ular evolutionary genetics analysis software. Bioinformatics17:1244–1245.
24.Kuritzkes, D. R., R. M. Grant, P. Feorino, M. Griswold, M. Hoover, R. Young, S. Day, R. M. Lloyd, C. Reid, G. F. Morgan, and D. L. Winslow.2003.
Performance characteristics of the TRUGENEHIV-1genotyping kit and the
Opengene DNA sequencing system. J. Clin. Microbiol.41:1594–1599.
25.Pandrea, I., D. L. Robertson, R. Onanga, F. Gao, M. Makuwa, P. Ngari, I. Bedjabaga, P. Roques, F. Simon, and C. Apetrei.2002. Analysis of partial pol and env sequences indicates a high prevalence of HIV type 1 recombinant
strains circulating in Gabon. AIDS Res. Hum. Retrovir.18:1103–1116.
26.Paraskevis, D., E. Magiorkinis, C. Anastassopoulou, M. Lazanas, G. Chry-sos, A. M. Vandamme, and A. Hatzakis.2001. Molecular characterization of a complex, recombinant human immunodeficiency virus type 1 (HIV-1) isolate (A/G/J/K/?): evidence to support the existence of a novel HIV-1
subtype. J. Gen. Virol.82:2509–2514.
27.Parry, J. V., G. Murphy, K. L. Barlow, K. Lewis, P. A. Rogers, F. J. Belda, A. Nicoll, C. McGarrigle, S. Cliffe, P. P. Mortimer, and J. P. Clewley.2001. National surveillance of HIV-1 subtypes for England and Wales: design,
methods, and initial findings. J. Acquir. Immune Defic. Syndr.26:381–388.
28.Pearson, W. R., and D. J. Lipman.1988. Improved tools for biological
sequence comparison. Proc. Natl. Acad. Sci. USA85:2444–2448.
29.Perez-Alvarez, L., R. Carmona, M. Munoz, E. Delgado, M. M. Thomson, G. Contreras, J. D. Pedreira, R. Rodriguez Real, E. Vazquez de Parga, L. Medrano, J. A. Taboada, R. Najera, et al.2003. High incidence of non-B and
recombinant HIV-1 strains in newly diagnosed patients in Galicia, Spain:
study of genotypic resistance. Antivir. Ther.8:355–360.
30.Quinones-Mateu, M. E., and E. J. Arts.1999. Recombination in HIV-1:
update and implications. AIDS Rev.1:89–100.
31.Robertson, D. L., J. P. Anderson, J. A. Bradac, J. K. Carr, B. Foley, R. K. Funkhouser, F. Gao, B. H. Hahn, M. L. Kalish, C. Kuiken, G. H. Learn, T. Leitner, F. McCutchan, S. Osmanov, M. Peeters, D. Pieniazek, M. Salminen, P. M. Sharp, S. Wolinksky, and B. Korber. 2000. HIV-1 nomenclature
proposal. Science288:55–56.
32.Snoeck, J., K. Van Laethem, P. Hermans, E. Van Wijngaerden, I. Der-delinckx, Y. Schrooten, D. A. van de Vijver, S. De Wit, N. Clumeck, and A. M. Vandamme.2004. Rising prevalence of HIV-1 non-B subtypes in Belgium:
1983–2001. J. Acquir. Immune Defic. Syndr.35:279–285.
33.Spira, S., M. A. Wainberg, H. Loemba, D. Turner, and B. G. Brenner.2003. Impact of clade diversity on HIV-1 virulence, antiretroviral drug sensitivity
and drug resistance. J. Antimicrob. Chemother.51:229–240.
34.Tatt, I. D., K. L. Barlow, A. Nicoll, and J. P. Clewley.2001. The public health
significance of HIV-1 subtypes. AIDS15(Suppl. 5):S59–S71.
35.Temin, H. M.1993. Retrovirus variation and reverse transcription: abnormal strand transfers result in retrovirus genetic variation. Proc. Natl. Acad. Sci.
USA90:6900–6903.
36.Thomson, M. M., L. Perez-Alvarez, and R. Najera.2002. Molecular epide-miology of HIV-1 genetic forms and its significance for vaccine development
and therapy. Lancet Infect. Dis.2:461–471.
37.Weidle, P. J., C. E. Ganea, K. L. Irwin, D. Pieniazek, J. P. McGowan, N. Olivo, A. Ramos, C. Schable, R. B. Lal, S. D. Holmberg, and J. A. Ernst.
2000. Presence of human immunodeficiency virus (HIV) type 1, group M, non-B subtypes, Bronx, New York: a sentinel site for monitoring HIV
ge-netic diversity in the United States. J. Infect. Dis.181:470–475.
38.Yang, C., M. Li, Y. P. Shi, J. Winter, A. M. van Eijk, J. Ayisi, D. J. Hu, R. Steketee, B. L. Nahlen, and R. B. Lal.2004. Genetic diversity and high proportion of intersubtype recombinants among HIV type 1-infected
preg-nant women in Kisumu, western Kenya. AIDS Res. Hum. Retrovir.20:565–
574.