The inhibition of vascular smooth muscle cell
migration by peptide and antibody antagonists
of the alphavbeta3 integrin complex is reversed
by activated calcium/calmodulin- dependent
protein kinase II.
C Bilato, … , A J White, M T Crow
J Clin Invest.
1997;
100(3)
:693-704.
https://doi.org/10.1172/JCI119582
.
The migration of vascular smooth muscle cells (VSMCs) is thought to play a key role in the
pathogenesis of many vascular diseases and is regulated by soluble growth factors/
chemoattractants as well as interactions with the extracellular matrix. We have studied the
effects of antibodies to rat beta3 and human alphavbeta3 integrins on the migration of
VSMCs. Both integrin antibodies as well as cyclic RGD peptides that bind to the vitronectin
receptors alphavbeta3 and alphavbeta5 significantly inhibited PDGF-directed migration.
This resulted in a reduction in the accumulation of inositol (1,4,5) trisphosphate and the
activation of calcium/calmodulin-dependent protein kinase II (CamKII), an important
regulatory event in VSMC migration identified previously. PDGF-directed VSMC migration
in the presence of the anti-integrin antibodies and cyclic RGD peptides was restored when
intracellular CamKII activity was elevated by either raising intracellular calcium levels with
the ionophore, ionomycin, or infecting with a replication-defective recombinant adenovirus
expressing a constitutively activated CamKII cDNA (AdCMV.CKIID3). Rescue of rat VSMCs
was also observed in stably transfected cell lines expressing constitutively activated but not
wild-type CamKII. These observations identify a key intermediate in the regulation of VSMC
migration by outside-in signaling from the integrin alphavbeta3.
Research Article
Find the latest version:
The Journal of Clinical Investigation Volume 100, Number 3, August 1997, 693–704 http://www.jci.org
The Inhibition of Vascular Smooth Muscle Cell Migration by Peptide and Antibody
Antagonists of the
a
vb
3Integrin Complex Is Reversed by Activated
Calcium/Calmodulin–dependent Protein Kinase II
Claudio Bilato, Karen A. Curto, Robert E. Monticone, Rebecca R. Pauly, Angela J. White, and Michael T. Crow
Vascular Biology Unit, Laboratory of Cardiovascular Science, Gerontology Research Center, National Institute on Aging-NIH, Baltimore, Maryland 21224
Abstract
The migration of vascular smooth muscle cells (VSMCs) is thought to play a key role in the pathogenesis of many vas-cular diseases and is regulated by soluble growth factors/ chemoattractants as well as interactions with the extracellu-lar matrix. We have studied the effects of antibodies to rat
b3 and human avb3 integrins on the migration of VSMCs.
Both integrin antibodies as well as cyclic RGD peptides that bind to the vitronectin receptors avb3 and avb5 significantly inhibited PDGF-directed migration. This resulted in a re-duction in the accumulation of inositol (1,4,5) trisphosphate and the activation of calcium/calmodulin–dependent protein kinase II (CamKII), an important regulatory event in VSMC migration identified previously. PDGF-directed VSMC mi-gration in the presence of the anti-integrin antibodies and cyclic RGD peptides was restored when intracellular CamKII activity was elevated by either raising intracellular calcium levels with the ionophore, ionomycin, or infecting with a repli-cation-defective recombinant adenovirus expressing a consti-tutively activated CamKII cDNA (AdCMV.CKIID3). Rescue of rat VSMCs was also observed in stably transfected cell lines expressing constitutively activated but not wild-type CamKII. These observations identify a key intermediate in the regulation of VSMC migration by outside–in signaling from the integrin avb3. (J. Clin. Invest. 1997. 100:693–704.)
Key words: cell migration • vascular smooth muscle cells •
integrins • calcium • calcium/calmodulin–dependent
pro-tein kinase II
Introduction
The migration of vascular smooth muscle cells (VSMCs)1 from the tunica media to the intima is considered an important
pathogenic event in many vascular disorders such as athero-sclerosis and restenosis after balloon angioplasty (1, 2). The majority of VSMCs in the normal arterial wall reside in the tu-nica media, where they are surrounded by and embedded in a variety of extracellular matrix (ECM) molecules. VSMCs in-teract with these matrix components, which are likely to serve not only as anchorage sites and barriers to cell movement, but also as regulators of VSMC gene expression and modulators of intracellular signaling pathways initiated by growth factors and/or chemoattractants. For example, the composition of the ECM has been shown to promote the modulation of VSMCs from a contractile to a synthetic phenotype (3, 4), a process re-quired for VSMC migration (5).
Cell–matrix interactions are mediated, in part, by the ubiq-uitously expressed class of cell surface receptors known as in-tegrins (6). These molecules assemble as heterodimers through the noncovalent association of a and b subunits (7, 8). Both subunits are membrane glycoproteins composed of a large ex-tracellular domain, a single membrane-spanning segment and, with the exception of the b4 subunit, a short cytoplasmic tail. In general, the extracellular region contains the binding site for ECM ligands, while the intracellular portion associates with cytoskeletal elements (9). On the basis of the different b subunit types, the integrins are currently classified into eight subgroups (10). Integrins containing either the b1 or b3 sub-units are primarily involved in cell–ECM interactions, while other b subclasses mediate cell–cell interactions. Integrins not only serve as transmembrane links between the ECM and the cell surface resulting in increased adhesion, but they also can trigger important intracellular events (11). The role of integ-rins in controlling cell migration is likely, therefore, to be mul-tifaceted and involve not only the regulation of adhesion and spreading, but also intracellular signaling events triggered by various chemoattractants. Previous studies have established the presence of various integrins on the cell surface of VSMCs that are responsible for adhesion to the ECM and regulation of cell movement (4, 12–19). Among these, the integrin com-plex avb3 is of special interest. It is the most promiscuous of all integrin complexes in terms of its ability to recognize a wide variety of Arg-Gly-Asp (RGD)-containing ECM components (20–23). An important role for this complex in the pathogene-sis of various diseases is supported by several observations. First, avb3 expression is upregulated in several invasive tumors (24, 25). In human melanoma cells, its expression is correlated with malignancy grade (26), collagenase production (27), and cell growth (28). Second, blocking the avb3 receptor with pep-tide antagonists or function-blocking antibodies prevents the vascularization of tissues and tumors (29) and reduces tumor size (30). Third, avb3 expression is upregulated in many intimal lesions associated with the various stages of atherosclerosis (31). In animal models of restenosis, peptide antagonists of avb3 effectively block the fibroproliferative response seen in
C. Bilato is a Research Fellow of the Glenn Foundation.
Address correspondence to Michael T. Crow, Ph.D., Laboratory of Cardiovascular Science, Gerontology Research Center, National Insti-tute on Aging-NIH, 4940 Eastern Ave., Baltimore, MD 21224. Phone: 410-558-8207; FAX: 410-558-8150; E-mail: [email protected]
Received for publication 25 February 1997 and accepted in revised form 24 April 1997.
blood vessels after balloon catheter injury (32, 33). Moreover, there is growing evidence that the long term beneficial effects of a humanized b3 integrin antibody on survival after balloon angioplasty in humans (34) might be due, in part, to its ability to inhibit both aIIbb3 present on platelets and the avb3 complex present on other vascular cells.
Multiple intracellular signaling events control the directed migration of VSMCs toward the chemoattractant, platelet-derived growth factor (PDGF) (35, 36). We have demon-strated previously that the activation of the multifunctional protein kinase, calcium/calmodulin–dependent protein kinase II (CamKII), is a critical signaling event for VSMC migration. This enzyme is regulated by the phenotypic state of the cell (37) and the cooperation between PDGF and basic fibroblast growth factor (bFGF or FGF-2), which is synthesized by VSMCs in response to PDGF (38). Here we report that occu-pancy of avb3 is also required for the PDGF-induced migration of both rat and human VSMCs and that the inhibition of mi-gration caused by cyclic RGD (cRGD) peptides or b3/avb3 in-tegrin antibodies is associated with a failure of PDGF to fully activate CamKII. When CamKII was activated independently of PDGF either by elevating intracellular calcium with an ion-ophore or through forced expression of a recombinant-engi-neered constitutively activated CamKII, the antagonists were no longer effective in suppressing migration. Our results dem-onstrate that PDGF-directed migration in VSMCs requires integrin-mediated intracellular events that regulate chemo-attractant-activation of the multifunctional protein kinase, CamKII.
Methods
Materials.Recombinant human PDGF-BB (Collaborative Research Inc., Lexington/Waltham, MA or Upstate Biotechnology, Inc., Lake Placid, NY) was dissolved in Dulbecco’s modified MEM (high glu-cose; GIBCO BRL, Gaithersburg, MD) containing 0.1% BSA, ali-quoted, and stored at 2708C. Calmodulin (Upstate Biotechnology, Inc.) and autocamtide (Peninsula Laboratories Inc., Belmont, CA) were dissolved in water, aliquoted, and stored at 2208C. All reagents were used only once after thawing. cRGD (GpenGRGDSPCA), RGD (GRGDSP), and RGE (GRGESP) peptides were purchased from GIBCO BRL. Cyclo-RGDFV was purchased from Peptides In-ternational (Lexington, KY). All peptides were freshly prepared for each experiment by reconstitution in water. cRGD specifically recog-nizes the vitronectin receptors avb3 and avb5 and has no effect on
other integrins (12, 39). Monoclonal antibody F11 (40), directed against rat b3–containing integrins, was generously provided by Dr. Michael Horton (University College, London) or purchased from PharMingen (San Diego, CA). Monoclonal antibodies LM609 and P1F6, directed against the human avb3 and avb5 integrins,
respec-tively, were provided generously by Dr. David Cheresh (Scripps In-stitute, La Jolla, CA) or purchased from Chemicon International, Inc. (Temecula, CA). Stably transfected cell lines of rat aortic medial VSMCs expressing full-length wild type (WT) and constitutively acti-vated (D3 mutation) for rat brain CamKII a-subunit have been de-scribed previously and characterized (37).
Cell cultures. Medial VSMCs were enzymatically dissociated from the thoracic aorta of 3–6-mo-old Wistar rats as described previ-ously (5). Cells were maintained in culture with Dulbecco’s modified MEM (high glucose) supplemented with 10% heat-inactivated FBS, 1 mmol/liter nonessential amino acids, 20 mmol/liter L-glutamine, 50
mg/ml streptomycin, and 10 mg/ml neomycin (GIBCO BRL) in a hu-midified 5% CO2 atmosphere at 378C. Human aortic VSMCs were
purchased from Clonetics (San Diego, CA) and grown in smooth muscle growth medium-2 (SMGM-2) supplemented with 5% FBS
and 5 mg/ml insulin. 2 mg/ml human FGF, 0.5 ng/ml human EGF, 0.5
mg/ml gentamicin, and 0.5 mg/ml amphotericin (all purchased from Clonetics). For rat VSMCs, proliferating cells were used between the 7th and 14th passages and for human VSMCs between the 5th and 7th passages. Cells were harvested for the migration/chemotaxis assay or biochemical analyses between 50 and 80% confluence. The trans-formed human embryonic kidney cell line 293 (ATCC CRL 1573) (American Type Culture Collection, Rockville, MD) was grown in improved minimal essential medium (I-MEM) (Biofluids, Inc., Rock-ville, MD) containing 10% FBS, 2 mmol/liter glutamine, 50 U/ml pen-icillin, and 50 mg/ml streptomycin.
Chemotaxis/migration assay. Chemotaxis/migration assays were performed in a modified Boyden chamber as described previously (5, 41). Polycarbonate filters (Nucleopore polycarbonate, 8 mm pores; Costar Sci. Corp., Pleasanton, CA) were coated overnight with 50 mg/ ml of either human or rat fibronectin (GIBCO BRL). A solution of 0.4 nmol/liter PDGF-BB diluted in Dulbecco’s modified MEM (high glucose) and 0.1% BSA was placed in the bottom chamber of the Boy-den apparatus. Cells (200,000) suspended in Dulbecco’s modified MEM (high glucose) and 0.1% containing 0.1% BSA were then added to the upper chamber in a final volume of 0.8 ml. Other reagents were used at the concentrations indicated and either added to the upper chamber at the time of cell seeding or preincubated with the cells for 30 min before cell seeding of the chambers. The distinction between chemotaxis and chemokinesis was evaluated as described previously (38). When adhesion of the cells on the coated filter was assessed, only 50,000 VSMCs were plated and the number of cells on the top surface of the filter was calculated at the end of the 4 h experiment. In all cases, four fields per filter were counted and all experiments were run in triplicate. Each triplicate assay was repeated at least three times on separate occasions with different VSMC preparations.
Cell adhesion assay. The individual wells of a 96-well microtiter plate (Nunc, Inc., Naperville, IL) were coated overnight at 48C with 10–50 mg/ml of purified fibronectin, vitronectin, or thrombospondin dissolved in Dulbecco’s PBS. Nonspecific binding sites were blocked with 5 mg/ml denatured BSA for 1 h at 378C. Peptides and antibody antagonists were added to the cell suspension before plating. Cells re-suspended in Dulbecco’s modified MEM and 1% BSA were added to the coated wells (30,000 cells per well) and adhesion at 378C for 1 h was measured by first rinsing away nonadherent cells with Dulbecco’s PBS followed by fixation of adherent cells with 4% formaldehyde in PBS. The plates were then stained with 0.5% toluidine blue in 4% formaldehyde for 5 min and then rinsed extensively with water. The stain was eluted from the adherent cells by incubating in 1% SDS for 15 min at room temperature and then quantified at 595 nm.
CamKinase II activation and activity assays. Since CamKII under-goes regulatory autophosphorylation (42), the activation of the en-zyme in response to PDGF-BB can be evaluated by following the in-corporation of radioactive phosphate into the molecule. For this measurement, proliferating VSMCs at 70–80% confluency were incu-bated in phosphate-free medium containing 0.3 mCi/ml [32
P]ortho-phosphoric acid for 3–5 h and then stimulated with PDGF-BB in the presence or absence of cRGD peptide or antibody, F11. The cells were then extracted and prepared for immunoprecipitation as de-scribed previously (37). Immunoprecipitation was carried out using an antibody (generous gift of Dr. Harold Singer, Weis Center for Re-search, Danville, PA) against the d-subunit of CamKII, which is the predominant isoform expressed by rat VSMCs (37, 43). Equivalent amounts of TCA-precipitable counts were separated by SDS-PAGE on a 10% gel, which was then dried and exposed for autoradiography. The incorporation of 32P into CamKII was quantified using a
Recombinant adenovirus vector construction. A replication-deficient adenovirus in which the majority of the E1 region had been deleted and replaced with a cassette containing a site for gene insertion and the cytomegalovirus (CMV) early promoter was used. A cDNA for a constitutively active mutant of CamKII (known as CKIID3), gener-ated by site-directed mutagenesis of amino acids 286 and 287 to as-partic acid, was placed downstream of the CMV promoter. The muta-tions mimic the negative charge introduced in this region by the phosphorylation of the wild-type molecule, resulting in a constitutively activated enzyme (44). AdCMV.CKIID3 was produced by first insert-ing the CKIID3 cDNA into the pS5 shuttle vector to generate pS5CMV.CKIID3. This was then transfected into 293 cells along with the plasmid pJM17 (Microbix Inc., Toronto, Canada) that contains the full sequence of E1-deleted Ad 5 DNA (45) and individual plaques arising from homologous recombination were isolated. Ad-CMV.CKIID3 was propagated in 293 cell cultures, purified by CsCl density centrifugation (46), dialyzed, and stored at 2708C in 10 mM Tris-HCl, pH 7.4/1 mM MgCl2 buffer. The titer of the viral stock was
determined by plaque assay using 293 cells (47). An adenovirus ex-pressing the Escherichia colib-galactosidase gene (AdCMV.gal) was used as a control for infection.
AdCMV.CKIID3 and AdCMV.gal infection of VSMCs. Cultured VSMCs at 70–80% confluency were exposed to recombinant adeno-virus at an moi of 100 in a minimal volume of Dulbecco’s modified MEM (high glucose) for 1 h. Additional media containing 10% FBS was then added and the culture incubated at 378C in 5% CO2
atmo-sphere. After 24 h, the virus-containing medium was replaced with vi-rus-free medium and the cells collected for analysis of expression or for migration assays. Under the conditions described above, no cyto-pathic or cytotoxic effects were observed and . 90% of the cells were infected, as demonstrated by staining the AdCMV.gal–infected VSMCs with X-Gal (5-bromo-4-chloro-3-indolyl-D -galactopyrano-side; Gold Biotechnology Inc., St. Louis, MO) or the AdCMV. CKIID3–infected VSMCs by immunocytochemistry using an a -Cam-kinase II antibody (Boehringer Mannheim Biochemicals, India-napolis, IN). Expression of CKIID3 was also monitored by Northern and Western blotting. Total RNA was isolated by the guanidinium isothiocyanate procedure (48) from cultures of VSMCs at different times after adenoviral infection and probed by Northern blotting analysis as described previously (7). A full-length CamKII cDNA probe generated by PCR and cloned into the pCRII vector (Invitro-gen Corp., La Jolla, CA) was verified by dideoxy DNA sequenc-ing. The rat glyceraldehyde 3-phosphate dehydrogenase (GAPDH) probe has been described elsewhere (38). For Western blotting analy-sis, infected cells were extracted in RIPA buffer (150 mmol/liter NaCl, 1% NP40, 0.5% deoxycholic acid, 0.1% SDS, 50 mmol/liter Tris HCl, pH 8.0, 10 mg/ml aprotinin, 10 mg/ml leupeptin, and 0.1 mM phenylmethylsulfonylfluoride (PMSF) and clarified by centrifugation and assayed for protein content. 20 mg of protein per sample was sep-arated by SDS-PAGE on a 10% gel and transferred to a polyvi-nylidene difluoride (PVDF) membrane with a semidry transfer appa-ratus (Bio-Rad Laboratories, Hercules, CA) at 170 mA for 2 h. The membrane was blocked for 1 h in 5% nonfat milk/PBS (blocking so-lution) and then incubated overnight at 48C with a CamKII a -iso-form–specific mouse monoclonal antibody (10 mg/ml) (Boehringer Mannheim Biochemicals). The membrane was incubated with a horseradish peroxidase–conjugated anti–mouse antibody (1 mg/ml) for 4 h at 48C. The membrane was rinsed with PBS and antigen–anti-body complexes were detected using enhanced chemiluminescence (ECL) (Amersham Corp., Arlington Heights, IL) according to the manufacturer’s instructions.
IP3 levels. IP3 levels in VSMCs were determined with a commer-cially available IP3 receptor binding assay kit (Amersham Corp.). VSMCs were plated on 100 mm tissue culture plates so that the final cell density after an overnight incubation in serum-containing media followed by 48 h of serum starvation was between 0.7 and 1.3 3 106
cells per plate. Cells were exposed to 1 mg/ml cRGD peptide for 30 min before PDGF-BB stimulation. Stimulation was terminated by
placing the plates on ice, removing the serum-free media, and adding 0.5 ml of 4% perchloric acid. Each sample was collected by scraping the cells and precipitated proteins into a 1.5 ml centrifuge tube. The extract was placed on ice for 20 min and then clarified by centrifuga-tion at 48C. The supernatant was neutralized to approximately pH 7.5 with 1.5 N KOH/60 mM Hepes buffer, pH 8. The pellet was dissolved in 0.3 N NaOH and protein content determined using the BCA pro-tein assay reagent (Pierce Chemical Co., Rockford, IL). IP3 levels in 0.1-ml aliquots of the neutralized supernatants were assayed with a competitive IP3 receptor binding radioassay according to the manu-facturer’s instructions. Nonspecific binding was determined by the addition of excess unlabeled IP3 to the assay, while total binding for each assay was determined as the amount of labeled IP3 bound in the absence of competitor. A standard curve was established using serial dilutions of purified IP3. IP3 content in the extracts was determined by interpolation of the standard curve using the curve fitting pro-gram, AssayZap (Biosoft Co., Ferguson, IL) and was expressed rela-tive to total protein content.
Tyrosine phosphorylation of PLCg. After pretreatment and stim-ulation, VSMCs were rinsed in ice-cold Dulbecco’s PBS and then extracted by scraping the cells into 0.5 ml/100 mm plate of ice-cold ly-sis buffer (0.5 M Tris, pH 8.0, 137 mM NaCl, 1 mM Na3VO4, 1%
NP-40, 1 mM PMSF, 10 mg/ml aprotinin, and 10% glycerol). The extract was placed on ice for 30 min and then clarified by centrifugation at 48C. Supernatants containing equal amounts of protein were adjusted to a final volume of 1 ml using wash buffer (20 mM Tris, pH 8, 137 mM NaCl, 1 mM PMSF, and 10 mg/ml aprotinin). Immunoprecipita-tion of PLCg was performed overnight at 48C with 1 mg/ml of mixed monoclonal antibodies to bovine PLCg (Upstate Biotechnology Inc.). Protein A-Sepharose (50 ml of a 50% slurry) (Sigma Chemical Co.) was added to each sample, which was then incubated with mix-ing at 48C for another hour. Immunocomplexes were pelleted by 30 s centrifugation in a microfuge and the pellet washed with 1 ml of each of the following solutions: (a) 1% NP40, 150 mM NaCl, 20 mM Tris, pH 8.0, 10% glycerol, 2 mM EDTA, 10 mg/ml aprotinin, 10 mg/ml leu-peptin, 0.1 mM PMSF; (b) 0.5% NP40, 0.5 M LiCl, 50 mM Tris HCl, pH 7.5; (c) 0.5 M LiCl, 50 mM Tris HCl, pH 7.5; and (d) 10 mM Tris HCl, pH 7.5. The pellet was resuspended in 50 ml 23 sample buffer (63 mM Tris, pH 6.8, 2% SDS, 10% glycerol, 0.05% 2-mercaptoetha-nol, and 0.02% glycerol), boiled for 5 min, and then separated by SDS-PAGE on a 6% gel. After separation, proteins were electro-phoretically transferred to a PVDF membrane for 4 h at 170 mA us-ing a semidry blottus-ing apparatus. The membrane was blocked over-night at 48C in PBS containing 0.1% Tween 20 and 1% BSA and then washed three times for 5 min each at room temperature in Western buffer (50 mM NaCl, 10 mM Tris, pH 7.0, 1 mM EDTA, and 0.1% Tween 20). This was followed by incubation of the membrane in 0.5 mg/ml HRP-conjugated antiphosphotyrosine (ICN Biomedicals, Inc., Irvine, CA) for 2 h at room temperature. The membrane was washed in Western buffer at room temperature and the reactive bands on the membrane visualized by chemiluminescence according to the manu-facturer’s instructions (Amersham Corp.). Band intensity was quanti-tated using a densitometer (Molecular Dynamics, Sunnyvale, CA).
Statistical evaluations and comparisons. All the data are expressed as the mean6SEM. The comparison of the mean values among the different groups was made by ANOVA with P value corrected by the Bonferroni method (49).
Results
power field [HPF] PDGF-BB, 81.364.9 cells per HPF). When the rat b3 integrin antibody F11 was added to the upper cham-ber, there was a dose-dependent reduction in PDGF-mediated migration, with a half-maximal effect at , 5 mg/ml (Fig. 1 A). In contrast, equivalent amounts of mouse IgG had no effect on PDGF-directed chemotaxis.
Likewise, when the cRGD peptides, GPenGRGDSPCA and cyclo-RGDFV, were each added to the upper chamber, there was also a dose-dependent reduction in PDGF-directed migration. As shown in Fig. 1 B, there was an z 50% reduction
in PDGF-directed chemotaxis seen with 0.5 mg/ml of either cRGD peptide. In contrast, the addition of equivalent amounts of the control RGE peptide, GRGESP, did not affect the PDGF-directed migration of rat VSMCs. Although further in-hibition of chemotaxis was observed when the dose of the GPenGRGDSPCA peptide was increased above 2 mg/ml, this high dose inhibition was not seen with cyclo-RGDFV. Even at these high doses, the cRGD peptides did not interfere with ei-ther cell adhesion or spreading on the substrates used to coat the filters. When the cells on the top surface of the filter were counted after the 4 h incubation period, there was no signifi-cant difference in cell attachment between cRGD peptide-treated and unpeptide-treated cells (68.1613.1 versus 69.6613.9). Ad-hesion was also measured using a microplate assay in which different ECM components were absorbed to polyvinylchlo-ride plates (Fig. 2). These results show no change in attach-ment of cells to fibronectin as a result of preincubation with
cRGD, RGE, or the b3 integrin antibody, F11. As expected, attachment of cRGD-treated but not RGE-treated cells was inhibited on vitronectin-coated plates, indicating that the cy-clic peptide does interfere with ECM-vitronectin receptor in-teractions. F11 also did not affect attachment to fibronectin. Unlike the cRGD peptides, F11 also did not alter adhesion to vitronectin, probably because vitronectin-binding integrin complexes, such as avb5, are unaffected by F11. These results show that while both F11 and the cRGD peptides interfere with PDGF-directed chemotaxis, this is not a consequence of the inability of the treated cells to attach to the ECM-coated membrane used in the Boyden apparatus. The fact that both the cRGD peptide, which engages av-containing integrins, and the F11 antibody, which recognizes only b3-containing integrin complexes, inhibit PDGF-directed chemotaxis to the same ex-tent in these experiments suggests that the target of these re-agents is probably avb3. This can be directly tested in human VSMCs where antibodies to specific human integrin com-plexes are available.
[image:5.612.54.265.55.316.2]As was the case for rat VSMCs, the number of human VSMCs migrating to the underside of the membrane was markedly higher when the cells were exposed to a PDGF-BB gradient compared to BSA alone (85.562.5 versus 8.360.5; data not shown). Preincubation with 1 mg/ml cRGD peptide reduced the migration of these cells by . 50% relative to cells exposed to PDGF-BB alone. Monoclonal antibodies directed against specific integrins were then used to identify which integ-rins were responsible for the inhibitory effect of the cRGD peptide. The monoclonal antibody LM609 is specifically di-rected against the integrin, avb3. Treatment of migrating cells with this antibody resulted in a dose-dependent inhibition of migration of up to 60%. Treatment with either a monoclonal antibody directed against avb5 (PIF6) or with an isotype-matched nonimmune mouse IgG (IgG1) had no effect on PDGF-directed migration (Fig. 3 A). The combination of LM609 and P1F6 produced no additional effect other than that Figure 1. b3 integrin antibody and cyclic RGD peptides inhibit
PDGF-directed chemotaxis in rat VSMCs. Boyden chamber chemo-taxis assays of cultured rat medial VSMCs toward BSA or 0.4 nM PDGF-BB in the presence of varying doses of the b3 integrin anti-body, F11 (A) or the cRGD peptides, GPenGRGDSPCA and cyclo-RGDFV (B). Results represent at least three independent experi-ments and are expressed relative to chemotaxis/migration in the absence of antibody or peptide after subtraction of BSA background. (BSA: 5.2561.3 cells per HPF; PDGF: 81.364.9 cells per HPF).
Figure 2. Effect of the b3 antibody F11, cRGD, and RGE peptides on rat VSMC adhesion to fibronectin and vitronectin. Adhesion of cultured rat medial VSMCs was measured using a microtiter plate as-say with the individual wells of the plate coated with 50 mg/ml of each of the indicated substrates. Adhesion was allowed to occur for 1 h at 378C. Nonspecific binding was blocked with 5 mg/ml denatured BSA and was subtracted from all adhesion data. Data are expressed rela-tive to that observed in the absence of either peptide. FN: fibronectin;
[image:5.612.314.535.490.643.2]seen with LM609 alone (data not shown). These results dem-onstrate that the integrin complex regulating PDGF-directed migration in human VSMCs was likely to be avb3. As was the case for rat VSMCs, none of the antibodies or peptides used to block human VSMC chemotaxis affected the ability of human VSMCs to attach to fibronectin. While cRGD was effective in blocking binding of human VSMCs to vitronectin, only a com-bination of the monoclonal antibodies, LM609 and P1F6, pro-duced any significant blockade of adhesion to vitronectin alone (Fig. 3 B). This is consistent with results published previ-ously which indicate that attachment of cells to vitronectin can occur through both avb3 and avb5 (50, 51). However, LM609 alone was able to block attachment of the human VSMCs to thrombospondin (TSP) while P1F6 was ineffective (Fig. 3 B).
PDGF-stimulated CamKinase II activation is suppressed in VSMCs treated with cRGD peptides and b3-integrin antibod-ies. While numerous intracellular signaling pathways have been implicated in the control of cell migration, a critical role for the multifunctional protein kinase CamKII in VSMC mi-gration has been demonstrated recently (37, 38). To determine if antagonists of the vitronectin receptor affect the ability of VSMCs to activate CamKII in response to PDGF-BB, we measured its activation in treated cells. Activation of CamKII by calcium and calmodulin occurs as a result of autophosphor-ylation, resulting in an enzyme whose catalytic activity is no longer dependent on calcium and calmodulin. The activity of this enzyme can, therefore, be measured by either monitoring
the incorporation of radiolabeled phosphate into the molecule or by measuring the ability of the enzyme to phosphorylate a synthetic peptide substrate in permeabilized cells. Fig. 4 A shows the results of monitoring autophosphorylation of the enzyme. After 1 min of incubation with 10 ng/ml PDGF-BB, phosphorylation of CamKII was increased significantly. When VSMCs were pretreated with 1 mg/ml of cRGD peptide for 30 min before the addition of PDGF, there was an z 50%
de-crease in 32P incorporation. Similar results were obtained when we tested the activity of the enzyme using a synthetic substrate in permeabilized cells (Fig. 4 B). In this case, autonomous ac-tivity refers to enzyme that is active in the presence of EGTA and in the absence of additional calmodulin. It is reported as the percentage of total activity, which is obtained in parallel cultures in which excess calcium and calmodulin was added to the permeabilization solution. Unstimulated VSMC cultures showed a basal level of autonomous activity of 8.961.4%. The addition of PDGF-BB resulted in a significant increase of CamKinase II–independent activity within the first minute (58.561.8% autonomous activity). Blocking vitronectin recep-tors with 1 mg/ml cRGD peptide significantly suppressed PDGF-stimulated CamKII activation by z 53% (Fig. 4 B).
Similar results were observed when VSMCs were pretreated with the b3-integrin antibody, F11 (data not shown).
Ionomycin and basic FGF restore PDGF-directed chemo-taxis in cRGD peptide- and b3 integrin antibody–treated VSMCs. We have shown previously that elevating intracellu-Figure 3. Inhibition of PDGF-directed chemotaxis/migration of human VSMCs by cRGD peptide or monoclonal antibody, LM609. (A) The ef-fects of 1 mg/ml cRGD (GPenGRGDSPCA), the avb3 neutralizing monoclonal antibody, LM609, the avb5 neutralizing monoclonal antibody,
[image:6.612.62.514.63.313.2]lar free calcium levels with ionomycin activates CamKII inde-pendently of PDGF and restores migration in differentiated cells (37) or in cells with sequestered autocrine factors (38). Given that the cRGD peptide and b3 integrin antibody sup-presses CamKII activation in response to PDGF, we deter-mined whether ionomycin could restore migration in antago-nist-treated cells (Fig. 5). Proliferating rat VSMCs were placed in the upper chamber of the Boyden apparatus and their abil-ity to migrate was tested in presence or absence of cRGD pep-tide and ionomycin. The data in Fig. 5 shows that the inhibi-tion of migrainhibi-tion resulting from incubainhibi-tion with 1 mg/ml of cRGD peptide was reversed completely by the inclusion of 1 mM ionomycin. KN62, a specific inhibitor of CamKII activa-tion (52), completely blocked the ability of ionomycin to re-verse the effects of cRGD. Together, these results demon-strate that elevating intracellular calcium levels independently of PDGF-BB can fully restore the migration of proliferating VSMCs that are inhibited by cRGD and that this restoration is due, at least in part, to the activation of CamKII.
In addition to ionomycin, we have shown previously that addition of bFGF (FGF-2) can restore PDGF-directed chemo-taxis to cells when CamKII activity is suppressed by growth ar-rest/differentiation (38). FGF-2 alone is neither a chemoattrac-tant for VSMCs nor a stimulus for CamKII activation or activity (38). To determine whether the effect of FGF-2 is up-stream or downup-stream of the b3-integrin effect on PDGF-directed chemotaxis, the migration of F11-treated rat VSMCs or LM609-treated human VSMCs was measured in the pres-ence and abspres-ence of FGF-2. Fig. 6 shows that FGF-2 fully re-stored PDGF-directed migration to both rat and human
VSMCs treated with cRGD peptides or the integrin antibodies and that this restoration was inhibited by KN62, consistent with our previous study on bFGF and migration (38). These results demonstrate that the rescue of migration by bFGF oc-curs downstream of the integrin effect and upstream of CamKII activation.
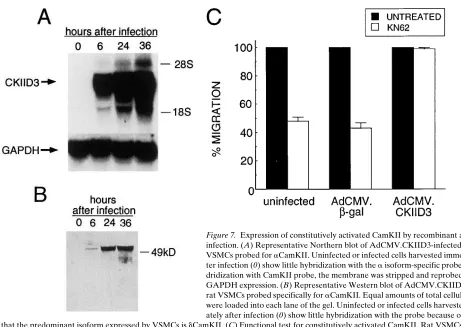
[image:7.612.58.298.59.314.2]Forced expression of constitutively activated CamKII by-passes the effect of cRGD peptides and b3 integrin antibodies on VSMC chemotaxis. Both ionomycin and FGF-2 are likely to activate other signaling pathways in the cell. To determine if the activation of CamKII alone is sufficient to restore migra-tion in VSMCs treated with cRGD peptides and b3 integrin antibodies, VSMCs were infected with a recombinant rep-lication-defective adenovirus (AdCMV.CKIID3) expressing CamKII that is locked into the activated state by site-directed mutagenesis. When used at an moi of 100, it was routinely possi-ble to demonstrate successful infection with AdCMV.CKIID3 into at least 90% of VSMCs without any accompanying cyto-toxic effects (data not shown). Increased steady levels of mRNA expression for the exogenous CamKII in rat VSMCs after infection are shown in Fig. 7 A. Since the majority of the endogenous CamKII in VSMCs is d-isoform, the expression of the exogenous CamKII mutant protein, which was constructed from an a-isoform cDNA, can be monitored with an a-iso-form–specific antibody (Fig. 7 B). Expression of the mutant CamKII protein was seen as early as 6 h after infection and continued to increase over the next 24 h. Similar results were Figure 4. CamKII
acti-vation and activity by PDGF-BB is inhibited by cRGD. (A) Activa-tion of CamKII mea-sured by 32P
incorpora-tion into cellular CamKII. Rat VSMCs were incubated with
32PO
4, treated, and then
extracted with RIPA buffer 1 min after PDGF-BB stimulation. The extract was then in-cubated with an anti-sera against dCamKII and immunoprecipi-tated with protein A–Sepharose. The im-munoprecipitate was separated by SDS-PAGE and the gel treated for autoradiog-raphy to detect 32P
in-corporation. The arrow
indicates the position of a purified CamKII standard, migrating at z 53 kD. (B) CamKII
activity was measured 1 min after 10 ng/ml PDGF-BB stimulation. The activity is expressed as percent autonomous activity, which de-scribes the percent of total activity present in the permeabilized cell extract that is independent of calcium and calmodulin.
Figure 5. Ionomycin prevents the inhibition of PDGF-directed mi-gration by cRGD peptide. PDGF-directed mimi-gration of rat VSMCs. Bar 1: migration in the absence of a PDGF gradient (6.560.4 cells per HPF); bar 2, migration toward 10 ng/ml PDGF-BB in the lower chamber (78.263.1 cells per HPF); bar 3, migration toward PDGF-BB in the bottom chamber and 1 mg/ml of the cRGD peptide, GPen-GRGDSPCA, in the upper chamber (32.762.6 cells per HPF); bar 4, same conditions as in bar 3 but with 1 mM ionomycin also in the up-per chamber (65.663.6 cells per HPF); bar 5, same conditions as in bar 4 with 10 mM KN62 also in the upper chamber (2562.5 cells per HPF). Results are averaged data from four independent experi-ments. *Indicates statistically significant from PDGF alone (bar 2),
[image:7.612.316.553.59.244.2]Figure 6. Exogenous bFGF prevents the inhibition of PDGF-directed migration by cRGD peptide. (A) PDGF-directed migration of rat VSMCs. Migration expressed relative to that toward 0.4 nM PDGF-BB in the absence of any additional treatments. F11 antibody was added to the upper chamber at a final concentration of 25 mg/ml, while the cRGD peptide, GPenGRGDSPCA, was added at a final concentration of 1 mg/ml. Basic FGF (FGF-2) and KN62 was added to the upper chamber at final concentrations of 10 ng/ml and 10 mM, respectively. Results are averaged data from four independent experiments. *Statistically significant from PDGF alone, P , 0.001; **not statistically significant from PDGF alone). (B) PDGF-directed migration of human VSMCs. Migration expressed relative to that toward 0.4 nM PDGF-BB in the absence of any additional treatments. LM609 antibody was added to the upper chmaber at a final concentration of 50 mg/ml. Basic FGF (FGF-2) and KN62 was added to the upper chamber at final concentrations of 10 ng/ml and 10 mM, respectively. Results are averaged data from four independent experiments. *Statistically significant from PDGF alone, P , 0.001; **not statistically significant from PDGF alone.
[image:8.612.56.517.379.706.2]obtained when human VSMCs were infected with the recom-binant virus (data not shown).
To evaluate whether the AdCMV.CKIID3 infection of VSMCs resulted in production of a functionally active mole-cule, we examined the migration of infected VSMCs in pres-ence of KN62. As shown in Fig. 7 C, the addition of 10 mM KN62 to the upper chamber of the Boyden apparatus sig-nificantly reduced the migration of both uninfected and AdCMV.bgal-infected VSMCs. In contrast, VSMCs infected with AdCMV.CKIID3 were completely insensitive to the ef-fects of KN62 (percentage of migration compared to KN62-untreated cells 5 99.1).
The ability of infected rat VSMCs to migrate toward PDGF in presence or absence of 1 mg/ml of cRGD peptide is shown on Fig. 8 A. Whereas infection with either AdCMV.-CKIID3 or AdCMV.bgal caused a small overall reduction in the number of migrating cells, more than an eightfold increase in migration occurred in response to PDGF-BB. The migra-tion of cells infected with AdCMV.CKIID3, however, was no longer sensitive to the inhibitory effects of the cRGD peptide, whereas the migration of cells infected with AdCMV.bgal and treated with the cRGD peptide was inhibited by . 50%. The magnitude of this inhibition was similar to that seen in the ab-sence of infection (Fig. 1).
Similar results were obtained with rat VSMC lines that sta-bly expressed constitutively activated CamKII (D3 VSMCs)
(Fig. 8 B). The control for these experiments were cells stably expressing the wild-type cDNA for the rat brain CamKII a-sub-unit (WT VSMCs). PDGF-BB was a strong chemoattractant for these cells and induced a . 10-fold increase in migration compared to BSA (D3: 8765.4 cells per field versus 862.7 cells per field; WT: 8862.2 cells per field versus 762.3 cells per field). D3 VSMCs, however, were completely insensitive to the effects of cRGD peptide (8663.6 cells per field), while block-ing the vitronectin receptors in WT VSMCs reduced the num-ber of the cells that migrated toward PDGF-BB by more than 50% (4362.9 cells per field). These experiments show that the transfected CamKII needed to be activated to block the effects of cRDG peptide and exclude the possibility that overexpres-sion of CamKII was affecting cell migration by acting as a calmodulin sink.
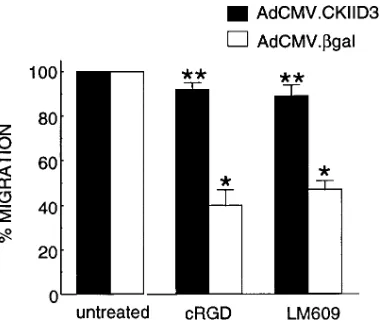
Overexpression of CamKII D3 gene not only restored the ability of human VSMCs to migrate toward PDGF in presence of the cRGD peptide, but also after treatment with the avb3 -specific monoclonal antibody LM609 (Fig. 9). While human VSMCs infected with AdCMV.bGal (white bars) showed an
z 55–60% reduction in migration in the presence of the cRGD
peptide (1 mg/ml) or LM609 (50 mg/ml), these same antago-nists were ineffective in human VSMCs infected with AdCMV.-CKIID3.
[image:9.612.57.300.367.670.2]cRGD peptides suppress PDGF-induced changes in cellular IP3 content. To determine how the cRGD peptides could sup-press CamKII activation in response to PDGF-BB, we exam-ined the effect of this antagonist on PDGF-stimulated IP3 for-mation. Changes in IP3 content are likely to be responsible for the initial rapid phase of intracellular calcium release in VSMCs after PDGF stimulation (38). Fig. 10 shows that in un-treated cells, PDGF BB leads to an approximately twofold in-crease in IP3 content at 2 min after stimulation (the time at which the maximum increase occurs; Curto, K., unpublished data). Pretreatment of the cells with 1 mg/ml cRGD signifi-cantly blunted this PDGF-stimulated increase.
Figure 8. Expression of constitutively activated CamKII restores PDGF-directed migra-tion to cRGD peptide-treated rat VSMCs. (A) PDGF-BB–directed mi-gration of rat VSMCs infected with AdCMV.- CKIID3 or
AdCMV.-bgal in the absence or presence of cRGD pep-tide. Absolute values for untreated and cRGD-treated Ad-CMV.CKIID3 cells: 6565.2 versus 6167.2 cells per HPF; for un-treated and un-treated cRGD-treated Ad-CMV.bgal cells: 6462.3 versus 3062.9 cells per HPF. *Statistically sig-nificant from infected cells in the absence of cRGD, P , 0.001; **not statistically significant from infected cells in the absence of cRGD. (B) PDGF-BB–directed migration of rat VSMC lines stably expressing either wild-type (WT) or D3 mutant aCamKII (CamKIID3). The characterization of these lines have been described previously (37). *Statistically significant from WT-CamKII–express-ing VSMCs in the absence of cRGD, P , 0.001; **not statistically sig-nificant from CamKIID3-expressing VSMCs in the absence of cRGD.
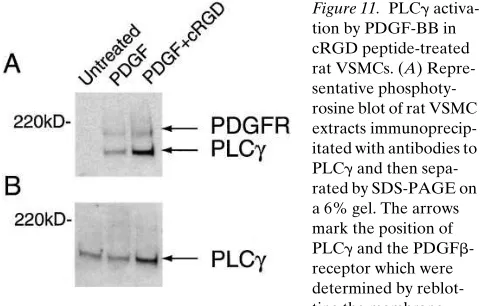
[image:9.612.317.507.490.653.2]In PDGF-stimulated cells, IP3 is produced from phosphati-dylinositol (4,5) bisphosphate [PI(4,5)P2] through the action of PLCg, which requires tyrosine phosphorylation after its associ-ation with the PDGF receptor. To determine whether the blunting of the IP3 response in cRGD-treated rat VSMCs was a consequence of the suppression of PLCg activation in re-sponse to PDGF-receptor, the relative level of PLCg tyrosine phosphorylation was measured by immunoprecipitating PLCg from cell extracts and then Western blotting the immunopre-cipitate with a phosphotyrosine-specific antibody. The results of this determination are shown in Fig. 11. The relative level of PLCg tyrosine phosphorylation increased dramatically upon the addition of PDGF BB and was unaffected by preincubat-ing the cells with 1 mg/ml cRGD peptide.
Discussion
Our results demonstrate that occupancy of the integrin com-plex, avb3, by either cRGD peptide, b3 integrin antibodies, or complex-specific antibodies, effectively suppresses both rat and human VSMC chemotaxis in response to PDGF-BB. This inhibition occurs without affecting overall cell attachment to the fibronectin coating used in the assay chamber. In addition,
the inhibition of chemotaxis caused by the peptides and anti-bodies was associated with a reduction in the activation of the multifunctional protein kinase, CamKII, an enzyme that previ-ously has been shown to play an important role in regulating VSMC migration (37, 38). Inhibition of chemotaxis after occu-pancy of avb3, was overcome through exogenous expression of a mutant CamKII cDNA that had been locked in a constitu-tively activated state. Migration was also restored when intra-cellular calcium levels were elevated independently of growth factor-mediated pathways using the calcium ionophore, iono-mycin. The restoration in this case was sensitive to the specific inhibitor of CamKII, KN62, suggesting that the beneficial effects of elevating intracellular calcium were largely due to the chemoattractant-independent activation of endogenous CamKII.
In human VSMCs, the integrin complex responsible for modulating PDGF-directed migration was directly identified as avb3 through the use of the avb3-specific monoclonal anti-body, LM609. This antibody blocked the adhesion of VSMCs to thrombospondin but not fibronectin and inhibited VSMC chemotaxis toward PDGF. A function-blocking monoclonal antibody to avb5 had no effect on the PDGF-directed chemo-taxis of human VSMCs. For rat VSMCs, an avb3-specific mono-clonal antibody is not available, but the fact that the inhibi-tory effects of either cRGD peptides, which apparently target av-containing integrin complexes, or b3 integrins are reversed by constitutitively activated CamKII, suggests that the respon-sible integrin complex in rat VSMCs is also avb3.
It is clear from studies in other cell types that integrins, in general, and avb3, in particular, not only mediate cell adhesion to various substrata, but also affect numerous cellular func-tions, including differentiation, gene expression, proliferation, movement, and apoptosis (53). For example, the viability and growth of human melanoma cells in three-dimensional dermal collagen requires avb3 receptor occupancy (28). Integrin-medi-ated ECM interactions necessary for cell survival have also been reported in human endothelial cells (53). Moreover, avb3 antagonists selectively induce apoptosis in vascular cells that have been stimulated to undergo angiogenesis (29). Recently, it has been observed that the systemic administration of mono-clonal antibodies directed to avb3 integrin significantly re-duced angiogenesis and growth of human breast carcinoma in a mouse/human chimeric model (30). Furthermore, occupancy of luminal avb3 in capillary endothelium modulates the perme-ability of the lung microvascular barrier (54) and elicits a signal leading to an enhanced invasiveness and production of type IV collagenase in human melanoma cells (27).
[image:10.612.56.304.57.183.2]A critical role for avb3 in the pathogenesis of vascular dis-ease is suggested by the following observations. While avb3 is absent in normal blood vessels, it is detectable in intimal le-sions associated with various stages of atherosclerotic lesion development (31). A number of extracellular matrix molecules that interact rather selectively with avb3 are upregulated after vessel injury and in atherosclerotic lesions. These include thrombospondin, tenascin, and osteopontin, which are often referred to as matricellular proteins, because they interact with both cell surface receptors (including avb3) and other extracel-lular matrix proteins (50, 55). Furthermore, cyclic peptide an-tagonists of the broad class of vitronectin receptors effectively inhibit neointimal formation after balloon catheter injury of rabbit and hamster arteries (32, 33). Indeed, it has been sug-gested that the long-term beneficial effects of humanized b3 Figure 10. PDGF
BB-stimulated IP3 produc-tion in rat VSMCs treated with cRGD. To-tal IP3 cellular content (pmol per mg of pro-tein) in unstimulated (control) and PDGF-stimulated VSMCs in absence (PDGF) or presence of cRGD pep-tide (PDGF1cRGD). VSMCs were serum starved for 48 h and then stimulated with PDGF for 2 min (50 ng/ml). cRGD peptide (1 mg/ml) was added to cell cultures 30 min before PDGF stimulation. IP3 assay was performed as described in Meth-ods. *Statistically significant from PDGF alone, P , 0.001.
Figure 11. PLCg activa-tion by PDGF-BB in cRGD peptide-treated rat VSMCs. (A) Repre-sentative phosphoty-rosine blot of rat VSMC extracts immunoprecip-itated with antibodies to PLCg and then sepa-rated by SDS-PAGE on a 6% gel. The arrows mark the position of PLCg and the PDGFb -receptor which were determined by reblot-ting the membrane with specific antibodies after stripping. (B) Same membrane shown in
[image:10.612.56.301.550.703.2]antibody on restenosis-related outcomes in humans may be re-lated to its ability to inhibit not only platelet activation and ag-gregation through the aIIbb3 receptors, but also later vascular events mediated by avb3 (34). Previous in vitro studies with isolated vascular smooth muscle cells have demonstrated that at least one of these effects acts on PDGF-directed migration, suggesting that occupancy of vitronectin receptors, such as avb3, modulates intracellular signaling initiated by soluble chemoattractants, such as PDGF-BB (32). The observations reported here suggest a general mechanism for integrating ECM-dependent and chemoattractant-mediated intracellular signaling pathways involved in the regulation of VSMC mi-gration.
This mechanism highlights a critical regulatory role for the multifunctional protein kinase, CamKII, in chemotaxis. Previ-ous studies have established that CamKII activation is crucial in other situations in which the PDGF-directed migration of VSMCs is blocked. These include growth arrest (37) and the application of antibodies or antisense oligonucleotides that ei-ther neutralize or inhibit endogenous bFGF expression (38). In both cases, there is also a significant or complete reduction in CamKII activation after the administration of PDGF-BB. Furthermore, chemotaxis in both circumstances is restored completely through either forced elevation of intracellular calcium (which causes PDGF-BB-independent activation of CamKII) or forced exogenous expression of a constitutively activated mutant CamKII cDNA. The blockade of PDGF-directed chemotaxis by neutralization of endogenously pro-duced bFGF could also be reversed if exogenous bFGF was supplied during the migration. We show here that exogenous bFGF also rescues VSMCs from the inhibitory effects of either the cRGD peptides or b3-integrin antibodies and that this res-cue is also dependent on CamKII activation (Fig. 6). These re-sults indicate that the requirement for bFGF is downstream of avb3 integrin regulation and suggest that occupancy of the avb3 regulates bFGF production/secretion.
How integrin occupancy influences the activation of CamKII is unknown. It is thought that the activation of CamKII is the result of the rapid release of calcium from intracellular stores triggered by an elevation in IP3 content (56, 57). Our data show that pretreatment of VSMCs with the cRGD peptide blocks the increase in IP3 content that normally occurs in re-sponse to PDGF. IP3 is generated along with diacylglycerol (DAG) by the enzyme, PLCg (58), which in response to PDGF associates with the PDGF receptor and becomes ty-rosine phosphorylated. Phosphorylation of PLCg at specific tyrosines has been shown to be necessary for enzyme activity (59). As we show here, however, tyrosine phosphorylation of PLCg in VSMCs in response to PDGF is unaffected by the cRGD peptides (Fig. 10). It is possible that, while tyrosine phosphorylation of PLCg is required, it is not sufficient for full in vivo activity. Indeed, evidence suggests that phosphorylation of PLCg on serine residues may negatively modulate its activ-ity (60). IP3 formation, however, could also be affected by sub-strate availability. For example, while intracellular calcium is rapidly mobilized in PDGF-stimulated fibroblast adhering to tissue culture plastic or immobilized ECM, the stimulation of suspended fibroblasts fails to do so (61, 62). In fact, IP3 forma-tion in response to PDGF is suppressed in suspended cells be-cause of a reduction in the substrate PI(4,5)P2 pool (63). The activity of the enzyme responsible for PI(4,5)P2 formation, namely phosphatidylinositol phosphate (PIP) 5-kinase (PI5-K),
is, in turn, regulated by integrin activation through a mecha-nism involving the small GTP-binding protein, rho, and is sup-pressed in suspended fibroblasts (64). Unlike cells in suspen-sion, however, the VSMCs used in this study that were treated with the cRGD peptide and integrin antibodies remained at-tached to the ECM, suggesting that the majority of integrin-associated focal contacts with the ECM persist in the presence of the cRGD peptides and b3 or avb3 antagonists. Therefore, unless PIP 5-K specifically associates with vitronectin recep-tors in these cells, its activity and the level of PI(4,5)P2 would be expected to remain unchanged.
In our experimental system, migration occurs toward a PDGF gradient that is established across a filter coated with fi-bronectin. Neither the antibody to avb3 nor the cRGD peptide interfered significantly with attachment to this substrate, rais-ing the question as to the identity of the extracellular matrix component(s) interacting with avb3 to regulate migration. It is possible that total avb3 levels are so low relative to that of the other cell surface binding sites for matrix proteins that their contribution to overall adhesion is minimal. On the other hand, avb3 may interact with endogenously produced ECM proteins. Giachelli et al. (55), for example, have reported that several growth factors, including PDGF-BB, increase the mRNA levels and protein expression of the matricellular pro-tein, osteopontin, which, in turn, interacts with avb3 to drive osteopontin-directed cell migration (50). Other extracellular matrix molecules produced by VSMCs that may interact with avb3 include TSP and tenascin. TSP is unique among other ECM binding proteins in that it not only binds to avb3 but also to the 50-kD integrin-associated protein (IAP) through a sepa-rate domain on the molecule (51). Because IAP tightly associ-ates with b3 integrins, it is possible that a single TSP molecule could engage both IAP and avb3. Such dual engagement may be necessary for signaling from the complex. As is the case for osteopontin, TSP expression is also upregulated by PDGF-BB in cell cultures and by injury to the vessel wall in vivo (65).
Our observations on the regulation of VSMC chemotaxis by integrin occupancy share many similarities to those re-ported by Blystone and colleagues (66, 67) for K562 cells transfected with cDNAs for both av and b3 integrins. These in-vestigators found that ligation of the resulting avb3 complex with either antibody or soluble vitronectin inhibited a5b1 -mediated phagocytosis, a process involving recognition of fi-bronectin coated particles, but had no effect on a5b1-mediated adhesion to fibronectin. We have shown here that antibodies to avb3 inhibit chemotaxis of VSMCs to PDGF on a fibronec-tin substrate without affecfibronec-ting VSMC adhesion to fibronecfibronec-tin. In the avb3-transfected K562 cells, the inhibition of a5b1 phagocytosis by avb3 was reversed by inhibiting a serine/threo-nine kinase, with an inhibitor profile consistent with, but not exclusive for, protein kinase C. On the other hand, we have shown that avb3 suppression of migration is accompanied by the failure of PDGF to stimulate another serine/threonine ki-nase, namely CamKII and that the effect of avb3 can be re-versed by expression of a constitutively activated CamKII. The relationship between the serine/threonine kinase which nega-tively regulates phagocytosis in K562 cells and CamKII, whose activity is required for VSMC migration, remains to be deter-mined.
67). In the case of the K562 cells, it is speculated that avb3 af-fects the affinity state of a5b1 so that low affinity functions such as adhesion are not affected, but higher affinity function, such as phagocytosis, are inhibited. It is possible that a similar phe-nomenon is occurring in anti-avb3 treated VSMCs, namely that the ligated avb3 integrin complex may affect the affinity state of other integrins. Indeed, recent experiments indicate that a decrease in the activation state of b1 integrins is seen in vivo after balloon catheter injury to vessels (68). Likewise, the treat-ment of human VSMCs with an antibody that induces the acti-vated or high affinity state for b1 integrins suppresses the mi-gration toward PDGF in cell culture (69).
The results presented here provide the outline of a mecha-nism by which traditional growth factor–signaling pathways are regulated through interactions of the cell with the ECM. In response to vessel injury, a gradient of PDGF is invariably es-tablished across the vessel wall, originating either from acti-vated platelets that adhere to the denuded endothelium or macrophages that are recruited to sites of developing athero-sclerotic lesions (1). The work of others has shown that the ex-pression of both avb3 integrin and thrombospondin are also upregulated in balloon-injured vessels and atherosclerotic le-sions (31, 55, 66). According to our data, outside–in signaling from the occupied avb3 integrin complex then enables PDGF to activate CamKII and for cell migration to proceed. The sig-naling events downstream of CamKII activation that are im-portant for cell movement are currently unknown and are the subject of continuing investigation. The results reported here provide the first demonstration of how ECM interactions with VSMCs regulate intracellular signaling pathways and possibly contribute to the pathogenesis of vascular lesions.
Acknowledgments
The authors acknowledge the generosity of Dr. Michael A. Horton (University College, London), Dr. David Cheresh (Scripps Research Institute, San Diego, CA), and Dr. Harold Singer (Geisinger Clinic, Danville, PA) for monoclonal antibody F11, monoclonal antibodies LM609 and P1F6, and polyclonal antibodies to dCamKII, respec-tively. We also thank Dr. Maurizio Capogrossi (Gene Therapy Unit, LCS, NIA, NIH) for use of the facilities for growing recombinant adenoviruses.
References
1. Ross, R. 1993. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature (Lond.). 362:801–809.
2. Clowes, A.W., M.A. Reidy, and M.M. Clowes. 1983. Kinetics of cellular proliferation after arterial injury. I. Smooth muscle growth in the absence of en-dothelium. Lab. Invest. 49:327–333.
3. Hedin, U., B.A. Bottger, E. Forsberg, S. Johansson, and J. Thyberg. 1988. Diverse effects of fibronectin and laminin on phenotypic properties of cultured arterial smooth muscle cells. J. Cell Biol. 107:307–319.
4. Yamamoto, M., K. Yamamoto, and T. Noumura. 1993. Type I collagen promotes modulation of cultured rabbit arterial smooth muscle cells from a contractile to a synthetic phenotype. Exp. Cell. Res. 204:121–129.
5. Pauly, R.R., A. Passaniti, C. Bilato, R. Monticone, L. Cheng, N. Papa-dopoulos, Y.A. Gluzband, L. Smith, C. Weinstein, E.G. Lakatta, and M.T. Crow. 1994. Migration of cultured vascular smooth muscle cells through a base-ment membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ. Res. 75:41–54.
6. Ruoslahti, E. 1991. Integrins. J. Clin. Invest. 87:1–5.
7. Hynes, R.O. 1987. Integrins, a family of cell surface receptors. Cell. 48: 549–555.
8. Ruoslahti, E., and M.D. Pierschbacher. 1987. New prospectives in cell ad-hesion: RGD and integrins. Science (Wash. DC). 238:491–497.
9. Schaller, M.D., and J.T. Parson. 1994. Focal adhesion kinase and associ-ated proteins. Curr. Opin. Cell Biol. 6:705–710.
10. Giancotti, F.G, and F. Mainiero. 1994. Integrin-mediated adhesion and signaling in tumorigenesis. Biochim. Biophys. Acta. 1198:47–64.
11. Hynes, R.O. 1992. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. 69:11–25.
12. Clyman, R.I., F. Mauray, and R.H. Kramer. 1992. b1 and b3 integrins have different roles in the adhesion and migration of vascular smooth muscle cells on extracellular matrix. Exp. Cell. Res. 200:272–284.
13. Bottger, B.A., U. Heldin, S. Johansson, and J. Thyberg. 1989. Integrin-type fibronectin receptors of rat arterial smooth muscle cells: isolation, partial characterization, and role in cytoskeletal organization and control of differenti-ated properties. Differentiation. 41:158–167.
14. Clyman, R.I., D.C. Turner, and R.H. Kramer. 1990. An a1b1-like
integ-rin receptor on rat aortic smooth muscle cells mediates adhesion to laminin and collagen types I and IV. Arteriosclerosis. 10:402–409.
15. Belkin, V.M., A.M. Belkin, and V.E. Koteliansky. 1990. Human smooth muscle VLA-1 integrin: purification, substrate specificity, localization in aorta, and expression during development. J. Cell Biol. 111:2159–2170.
16. Clyman, R.I., K.A. McDonald, and R.H. Kramer. 1990. Integrin recep-tors on aortic smooth muscle cells mediate adhesion to fibronectin, laminin, and collagen. Circ. Res. 67:175–186.
17. Janat, M.F., W.S. Agraves, and G. Liau. 1992. Regulation of vascular smooth muscle cell integrin expression by transforming growth factor b1 and platelet-derived growth factor-BB. J. Cell. Physiol. 151:588–595.
18. Skinner, M.P., E.W. Raines, and R. Ross. 1994. Dynamic expression of a1b1 and a2b1 integrin receptors by human vascular smooth muscle cells: a2b1
integrin is required for chemotaxis across type I collagen-coated membranes.
Am. J. Pathol. 145:1070–1081.
19. Lee, R.T., F. Berditchevski, G.C. Cheng, and M.E. Hemler. 1995. Integ-rin-mediated collagen matrix reorganization by cultured human vascular smooth muscle cells. Circ. Res. 76:209–214.
20. Cheresh, D.A., and R.C. Spiro. 1987. Biosynthetic and functional prop-erties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen and von Willebrand factor. J. Biol. Chem.
262:17703–17711.
21. Kramer, R.H., Y.-F. Cheng, and R. Clyman. 1990. Human microvascu-lar endothelial cells use b1 and b3 integrin receptor complexes to attach to laminin. J. Cell Biol. 111:1233–1243.
22. Chairo, I.F., L. Nannizzi, J.W. Smith , and D.A. Cheresh. 1991. The vit-ronectin receptor a5b3 binds fibronectin and acts in concert with avb3 in
pro-moting cellular attachment and spreading on fibronectin. J. Cell Biol. 111:2795– 2800.
23. Cheresh, D.A. 1991. Structure, function and biological properties of in-tegrin avb3 on human melanoma cells. Cancer Metastasis Rev. 10:3–10.
24. Albelda, S.M., S.A. Mette, D.E. Elder, R.M. Stewart, L. Damjanovich, M. Herlyn, and C.A. Buck. 1990. Integrin distribution in malignant melanoma: association of the b3 subunit with tumor progression. Cancer Res. 50:6757– 6764.
25. Gladson, C.L., and D.A. Cheresh. 1991. Glioblastoma expression of vi-tronectin and the avb3 integrin. Adhesion mechanism for transformed glial
cells. J. Clin. Invest. 88:1924–1932.
26. Felding-Habermann, B., B.M. Mueller, C.A. Romerdahl, and D.A. Cheresh. 1992. Involvment of integrin av gene expression in human melanoma
tumorigenicity. J. Clin. Invest. 89:2018–2022.
27. Seftor, R.E.B., E.A. Seftor, K.R. Gehlsen, W.G. Stetler-Stevenson, P.D. Brown, E. Ruoslahti, and M.J.C. Hendrix. 1992. Role of the avb3 integrin in
hu-man melanoma cell invasion. Proc. Natl. Acad. Sci. USA. 89:1557–1561. 28. Montgomery, A.M.P., R.A. Reisfeld, and D.A. Cheresh. 1994. Integrin avb3 rescues melanoma cell from apoptosis in three-dimensional dermal
col-lagen. Proc. Natl. Acad. Sci. USA. 91:8856–8860.
29. Brooks, P.C., A.M.P. Montgomery, M. Rosenfeld, R.A. Reisfeld, T. Hu, G. Klier, and D.A. Cheresh. 1994. Integrin avb3 antagonists promote tumor
re-gression by inducing apoptosis of angiogenic blood vessels. Cell. 79:1157–1164. 30. Brooks, P.C., S. Stromblad, R. Klemke, D. Visscher, F.H. Sarkar, and D.A. Cheresh. 1995. Anti-integrin avb3 blocks human breast cancer growth and
angiogenesis in human skin. J. Clin. Invest. 96:1815–1822.
31. Hoshiga, M., C.E. Alpers, L.L. Smith, C.M. Giachelli, and S.M. Schwartz. 1995. avb3 integrin expression in normal and atherosclerotic artery. Circ. Res. 77:1129–1135.
32. Choi, E.T., L. Engel, A.D. Callow, S. Sun, J. Trachtenberg, S. Santoro, and U.S. Ryan. 1994. Inhibition of neointimal hyperplasia by blocking avb3
in-tegrin with a small peptide antagonist GpenGRGDSPCA. J. Vasc. Surg. 19: 125–134.
33. Matsuno, H., J.M. Stassen, J. Vermylen, and H. Deckmyn. 1994. Inhibi-tion of integrin funcInhibi-tion by a cyclic RGD-containing peptide prevents neoin-tima formation. Circulation. 90:2203–2206.
34. Topol, E.J., R.M. Califf, H.F. Weisman, S.G. Ellis, J.E. Tcheng, S. Wor-ley, R. Ivanhoe, B.S. George, D. Fintel, M. Weston, et al. 1994. Randomized trial of coronary intervention with antibody against platelet IIb/IIIa integrin for reduction of clinical restenosis: results at six months. Lancet (N. Am. Ed.). 343: 881–886.
to-wards platelet derived growth factor. Proc. Natl. Acad. Sci. USA. 78:3669–3672. 36. Kundra, V., J.A. Escobedo, A. Kazlauskas, H.K. Kim, S.A.G. Rhee, L.T. Williams, and B.R. Zetter 1994. Regulation of chemotaxis by the platelet-derived growth factor receptor-b. Nature (Lond.). 367:474–476.
37. Pauly, R.R., C. Bilato, S.J. Sollott, R. Monticone, P.T. Kelly, E.G. Lakatta, and M.T. Crow. 1995. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circula-tion. 91:1107–1115.
38. Bilato, C., R.R. Pauly, G. Melillo, R. Monticone, D. Gorelick-Feldman, Y.A. Gluzband, S.J. Sollott, B. Ziman, E.G. Lakatta, and M.T. Crow. 1995. In-tracellular signaling pathways required for rat vascular smooth muscle cell mi-gration. J. Clin. Invest. 96:1905–1915.
39. Pierschbacher, M.D., and E. Ruoslathi. 1987. Influence of stereochemis-try of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J. Biol. Chem. 262:17294–17298.
40. Helfrich, M.H., S.A. Nesbitt, and M.A. Horton. 1992. Integrins on rat osteoclasts: characterization of two monoclonal antibodies (F4 and F11) to rat b3. J. Bone Miner. Res. 7:345–351.
41. Albini, A., Y. Iwamoto, H.K. Kleinman, G.R. Martin, S.A. Aaronson, J.M. Kozlowski, and R.N. McEwan. 1987. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 47:3239–3245.
42. Braun, A.P., and H. Schulman. 1995. The multifunctional calcium/cal-modulin-dependent protein kinase: from form to function. Annu. Rev. Bio-chem. 57:417–445.
43. Schworer, C.M., L.I. Rothblum, T.J. Thekkumkara, and H.J. Singer. 1993. Identification of novel isoforms of the d subunit of Ca21 /calmodulin-dependent protein kinase II. J. Biol. Chem. 268:14443–14449.
44. Waldmann, R., P.I. Hanson, and H. Schulman. 1990. Multifunctional Ca21/calmodulin-dependent protein kinase made Ca21-independent for func-tional studies. Biochemistry. 29:1679–1684.
45. McGroy, W.J., D.S. Bautista, and F.L. Graham. 1988. A simple tech-nique for the rescue of early region I mutations into infectious human adenovi-rus type 5. Virology. 163:614–617.
46. Graham, F.L., and A.J. Van Der Eb. 1973. A new technique for the as-say of infectivity of human adenovirus 5 DNA. Virology. 52:456–467.
47. Hersh, J., R.G. Crystal, and B. Bewig. 1995. Modulation of gene expres-sion after replication-deficient, recombinant adenovirus-mediated gene transfer by the product of a second adenovirus vector. Gene Therapy. 2:124–131.
48. Chirgwin, J.M., A.E. Przybyla, R.J. MacDonald, and W.J. Rutter. 1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonu-clease. Biochemistry. 18:5294–5299.
49. Devore, J. 1982. Probability and Statistics for Engineering and the Sci-ences. Brooks/Cole Publishing Co., Monterey, CA.
50. Liaw, L., M.P. Skinner, E.W. Raines, R. Ross, D.A. Cheresh, S.M. Schwartz, and C.M. Giachelli. 1995. The adhesive and migratory effects of os-teopontin are mediated via distinct cell surface integrins: role of avb3 in smooth
muscle migration to osteopontin in vitro. J. Clin. Invest. 95:713–724.
51. Gao, A.-G., F.P. Lindberg, M.B. Finn, S.D. Blystone, E.J . Brown, and W.A. Frazier. 1996. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J. Biol. Chem. 271:21–24.
52. Tokumitsu, H., T. Chijiwa, M. Hagiwara, A. Mizutani, M. Terasawa, and H. Hidaka. 1990. KN-62, 1-[N,O,-Bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenyl-piperizine, a specific inhibitor of Ca21
/calmodulin-depen-dent protein kinase II. J. Biol. Chem. 265:4315–4320.
53. Meredith, J.E., B. Fazeli, and M.A. Schwartz. 1993. The extracellular matrix as a cell survival factor. Mol. Biol. Cell. 4:953–961.
54. Tsukada, H., X. Ying, C. Fu, S. Ishikawa, P. McKeown-Longo, S. Al-belda, S. Bhattacharya, B. Anderson Bray, and J. Bhattacharya. 1995. Ligation of endothelial avb3 integrin increases capillary hydraulic conductivity of rat
lung. Circ. Res. 77:651–659.
55. Giachelli, C.M., N. Bae, M. Almeida, D.T. Denhardt, C.E. Alpers, and S.M. Schwartz. 1993. Osteopontin is elevated during neointima formation in rat arteries and is a novel component of human atherosclerotic plaques. J. Clin. In-vest. 92:1686–1696.
56. Berridge, M.J. 1993. Inositol trisphosphate and calcium signalling. Na-ture (Lond.). 361:315–325.
57. MacNicol, M., A.B. Jefferson, and H. Schulman. 1990. Calcium/calmod-ulin kinase is activated by the phosphatidylinositol signalling pathway and be-comes calcium-independent in PC12 cells. J. Biol. Chem. 265:18055–18058.
58. Nishizuka, Y. 1989. Turnover of inositol phospholipids and signal trans-duction. Science (Wash. DC). 244:546–550.
59. Kim, H.K., J.W. Kim, A. Zilberstein, B. Margolis, J.G. Kim, J. Schlessinger, and S.G. Rhee. 1991. PDGF stimulation of inositol phospholipid hydrolysis requires PLCg-1 phosphorylation on tyrosine residues 783 and 1254.
Cell. 65:435–441.
60. Park, D.J., H.K. Min, and S.G. Rhee. 1992. Inhibition of CD3-linked phospholipase C by phorbol ester and by cAMP is associated with decreased phosphotyrosine and increased phosphoserine contents of PLC-g1. J. Biol. Chem. 267:1496–1501.
61. Schwartz, M.A. 1993. Spreading of human endothelial cells on fibronec-tin or vitronecfibronec-tin triggers elevation of intracellular free calcium. J. Cell Biol.
120:1003–1010.
62. Tucker, R.W., K. Meade-Cobun, and D. Ferris. 1990. Cell shape and in-creased free cytosolic calcium by growth factors. Cell Calcium. 11:201–209.
63. McNamee, H.P., D.E. Ingber, and M.A. Schwartz. 1993. Adhesion to fi-bronectin stimulates inositol lipid synthesis and enhances PDGF-induced inosi-tol lipid breakdown. J. Cell Biol. 121:673–678.
64. Ching, L.D., A. Traynor-Kaplan, G.N. Bokoch, and M.A. Schwartz. 1994. The small GTP-binding protein rho regulates a phosphatidylinositol 4-phosphate 5-kinase in mammalian cells. Cell. 79:507–513.
65. Majack, R.A., J. Milbrandt, and V.M. Dixit. 1987. Induction of throm-bospondin messenger RNA levels occurs as an immediate promary response to platelet-derived growth factor. J. Biol. Chem. 262:8821–8825.
66. Blystone, S.D., I.L. Graham, F.P. Lindberg, and E.J. Brown. 1994. Integ-rin avb3 differentially regulates the adhesive and phagocytic functions of the
fi-bronectin receptor a5b1. J. Cell Biol. 127:1129–1137.
67. Blystone, S.D., F.P. Lindberg, S.E. LaFlamme, and E.J. Brown. 1995. Integrin b3 cytoplasmic tail is necessary and sufficient for regulation of a5b1
phagocytosis by avb3 and integrin associated protein. J. Cell Biol. 130:745–754.
68. Koyama, N., J. Seki, S. Vergel, E.J.R. Mattson, T. Yednock, N.L. Ko-vach, J.M. Harlan, and A.W. Clowes. 1996. Regulation and function of an acti-vation-dependent epitope of the b1 integrins in vascular cells after balloon in-jury in baboon arteries and in vitro. Am. J. Pathol. 148:749–761.