Oxidative stress in sepsis: a redox redux
Full text
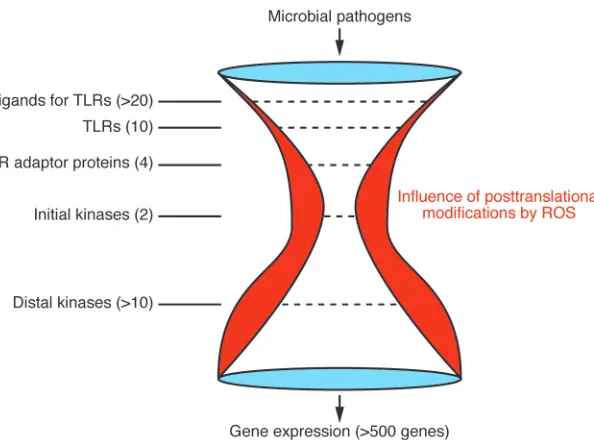
Figure
Related documents
Comparative results of EPCE using all three edge detection algorithms, shows that the proposed algorithm outperforms Sobel and Canny both on the basis of the measure and
After controlling for year of birth, season of birth, presence of siblings, and socioeconomic sta- tus, the inverse association between breastfeeding and risk of neonatal
An ion exchange experiment was also conducted to remove nitrate from synthetic water similar to water in the EJH wellfield and from a nitrate solution by performing
Often the forage diet will contain nutrient levels considered adequate, but the bioavailability of some minerals may be reduced because of interactions between S and Se; Cu and Zn;
Although the spirit of philanthropy governs the provision of health services in disasters, lack of ethical knowledge is conducive to a performance contrary to professional
Generally, (see Table 6 and Figure 7) when any clay, exemplified by Cl 25A, is added to the resin containing a conventional flame retardant, a slight increase in TTI and
Based on the gathered information, a data bank was created and using the generalized physical vulnerability developed for typical Iranian buildings, a series of scenario based,
An Interactive Qualifying Project submitted to the faculty of Worcester Polytechnic Institute in partial fulfillment of the requirements for the Degree of Bachelor of
