Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Novel Diagnostic Algorithm for Identification of Mycobacteria Using
Genus-Specific Amplification of the 16S-23S rRNA Gene
Spacer and Restriction Endonucleases
ANDREAS ROTH,1* UDO REISCHL,2ANNA STREUBEL,1LUDMILA NAUMANN,2
REINER M. KROPPENSTEDT,3MARION HABICHT,1MARGA FISCHER,1
ANDHARALD MAUCH1
Institut fu¨r Mikrobiologie und Immunologie, Lungenklinik Heckeshorn, 14109 Berlin,1Institut fu¨r Medizinische Mikrobiologie und Hygiene, Universita¨t Regensburg, 93053 Regensburg,2and Deutsche Sammlung
von Mikroorganismen und Zellkulturen, 38124 Braunschweig,3Germany Received 11 August 1999/Returned for modification 22 October 1999/Accepted 8 December 1999
A novel genus-specific PCR for mycobacteria with simple identification to the species level by restriction fragment length polymorphism (RFLP) was established using the 16S-23S ribosomal RNA gene (rDNA) spacer as a target. Panspecificity of primers was demonstrated on the genus level by testing 811 bacterial strains (122 species in 37 genera from 286 reference strains and 525 clinical isolates). All mycobacterial isolates (678 strains among 48 defined species and 5 indeterminate taxons) were amplified by the new primers. Among nonmycobacterial isolates, onlyGordonia terraewas amplified. The RFLP scheme devised involves estimation of variable PCR product sizes together withHaeIII andCfoI restriction analysis. It yielded 58HaeIII patterns, of which 49 (84%) were unique on the species level. Hence, HaeIII digestion together withCfoI results was sufficient for correct identification of 39 of 54 mycobacterial taxons and one of three or four of seven RFLP genotypes found inMycobacterium intracellulareandMycobacterium kansasii, respectively. Following a clearly laid out diagnostic algorithm, the remaining unidentified organisms fell into five clusters of closely related species (i.e., theMycobacterium aviumcomplex orMycobacterium chelonae-Mycobacterium abscessus) that were successfully separated using additional enzymes (TaqI,MspI,DdeI, orAvaII). Thus, next to slowly growing mycobacteria, all rapidly growing species studied, includingM. abscessus,M. chelonae,Mycobacterium farcino-genes,Mycobacterium fortuitum,Mycobacterium peregrinum, andMycobacterium senegalense(with a very high 16S rDNA sequence similarity) were correctly identified. A high intraspecies sequence stability and the good discriminative power of patterns indicate that this method is very suitable for rapid and cost-effective identi-fication of a wide variety of mycobacterial species without the need for sequencing. Phylogenetically, spacer sequence data stand in good agreement with 16S rDNA sequencing results, as was shown by including strains with unsettled taxonomy. Since this approach recognized significant subspecific genotypes while identification of a broad spectrum of mycobacteria rested on identification of one specific RFLP pattern within a species, this method can be used by both reference (or research) and routine laboratories.
The genusMycobacteriumis represented by a wide range of species. They form a heterogenous group in terms of occur-rence in clinical or environmental material, complex pheno-typic and genetic data, and disease association (25, 33). Cur-rently, identification of mycobacteria grown in culture is achieved by standard culture and biochemical methods, and for a few species, probes are commercially available ( Mycobac-terium tuberculosis, Mycobacterium avium, Mycobacterium in-tracellulare, Mycobacterium gordonae,Mycobacterium kansasii, andMycobacterium fortuitum) (10, 12). Determination of phe-notypic features is time-consuming, difficult to assimilate into a precise diagnosis concerning closely related taxa, and not al-ways highly reproducible (26). The majority of clinically iso-lated nontuberculous bacteria, such asM. gordonaeor rapidly growing species, are not pathogenic or are of doubtful clinical relevance (3, 7). On the other hand, a rise in incidence of nontuberculous bacteria, including newly described species or subspecific phylogenetic lineages of potential clinical signifi-cance, and the crucial role of the laboratory in establishing the
diagnosis demand methods that provide accurate results in a more timely fashion (7, 26). Therefore, efforts for rapid and accurate molecular identification have been undertaken in re-cent years (13–17, 21–23, 26, 29, 30; J. L. Miller, training manual, MIDI Inc., Newark, N.J., 1997). Today, sequencing of the 16S RNA gene (rDNA) is regarded as the most suitable method for identification of mycobacteria (14, 32). Even so, the high expense, together with a lack of clinical relevance for most species identified in routine laboratory practice, renders sequencing unacceptable for general use. Limitations of the 16S RNA gene are evident because the number of polymor-phic sites within the genus Mycobacteriumis rather low (13, 22). Some species have the same sequence or a very high degree of similarity (22). This leads to problems in develop-ment of simpler sequence analysis methods, such as restriction fragment length polymorphism (RFLP) analysis or hybridiza-tion with probes (3, 6, 15). To meet this need, alternative genetic targets have been studied (13, 16, 22, 27, 28). Of these, thehsp65gene has so far been best investigated, and the data were recently improved by inclusion of more species, especially some rapidly growing mycobacteria (e.g.,Mycobacterium che-lonae andMycobacterium abscessus) (5, 21). However,hsp65
gene-based PCR-RFLP analysis has been impeded by difficul-ties, such as minor differences of band sizes between some
* Corresponding author. Mailing address: Institut fu¨r Mikrobiolo-gie und ImmunoloMikrobiolo-gie, Lungenklinik Heckeshorn-Zehlendorf, Zum Heckeshorn 33, D 14109 Berlin, Germany. Phone: 49-30-8002 2254. Fax: 49-30-8002 2299. E-mail: [email protected].
1094
on May 15, 2020 by guest
http://jcm.asm.org/
species and the occurrence of new patterns not previously reported (20, 34; B. A. Forbes, K. S. Hicks, and D. L. Kiska, Abstr. 9th Eur. Cong. Clin. Microbiol. Infect. Dis., Clin. Mi-crobiol. Infect.5:(Suppl. 3), abstr. O37, p. 62, 1999). Moreover, all current molecular approaches to detect mycobacteria have the common disadvantage that the primers used for amplifi-cation are not specific for mycobacteria. Thus, undesirable amplification of other gram-positive bacterial contaminants, such as corynebacteria, represents a potential issue of concern in most clinical settings.
In view of this, we aimed to investigate the 16S-23S ribo-somal DNA (rDNA) internal transcribed spacers of a larger number of mycobacterial species for their suitability to estab-lish a PCR-RFLP-based identification scheme (16, 23). The first goal was to develop and evaluate novel primers for genus-specific amplification of mycobacteria and, secondly, to estab-lish a reliable diagnostic algorithm for identification of a broad spectrum of mycobacterial species with one to three endo-nucleases. The interspecies discriminatory power and the de-gree of intraspecies divergence of patterns of such a new RFLP-based approach were investigated by using 678 myco-bacterial strains within 48 species.
MATERIALS AND METHODS
Bacterial strains, identification, and sequencing.The bacteria used in this study comprised 811 strains listed in Table 1. They constituted 122 species within
37 genera of 286 reference strains and 525 clinical isolates. SixNocardiaclinical
isolates were identified to the genus level only. Species other than mycobacteria were chosen mostly from taxa of actinomycetes closely related to mycobacteria. Mycobacteria were represented by a total of 678 strains (179 reference strains
and 499 clinical isolates) of 48 defined species and 5Mycobacteriumspp. that
failed to match either biochemical species patterns or known 16S rDNA signa-ture sequences (26).
All clinical isolates were identified to the species level by standard biochemical methods and/or AccuProbes (12, 25). Most of the reference strains and all
clinical isolates—with the exception ofM. avium,M. tuberculosis, 23M. gordonae
isolates, and 14Mycobacterium xenopiisolates—were sequenced in the variable
regions A and B within the 16S RNA gene (14, 22, 25). To allow for a better understanding, we sequenced the nearly 1.5-kbp 16S rDNA in a few strains with
unique or discordant RFLP patterns (one clinical isolate each ofM. kansasii,
Mycobacterium phlei, andMycobacterium triviale, and the reference strains My-cobacterium flavescensS526 and S318 andMycobacterium parafortuitumDSM 43526) and some of those with unsettled taxonomic status (strains M511, S245, S279, S369, and S504) using a method described elsewhere (24). To obtain more 16S-23S spacer sequence data, a selection of strains were also sequenced within the 16S-23S spacer (22). A few DSM reference strains with apparently wrong designation according to the RFLP results were subsequently reclassified after partial 16S rDNA sequencing and analyses of fatty acids by gas chromatography and of mycolic acids by high-performance liquid chromatography (17; Miller, 1997).
PCR amplification.Chromosomal DNA was released from bacterial suspen-sions by sonication with glass beads according to methods described elsewhere (22). Amplification of a part of the 16S-23S spacer was performed with primers
Sp1 (5⬘-ACC TCC TTT CTA AGG AGC ACC-3⬘) (AAGGA corresponds to the
beginning of the spacer sequence) and Sp2 (5⬘-GAT GCT CGC AAC CAC TAT
CCA-3⬘) (positions 210 to 190 of theM. tuberculosisspacer sequence; EMBL
accession number L15623). The amplification was done with a 50-l reaction
mixture containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 1.5 mM MgCl2, 0.1%
Triton X-100, 200 M (each) deoxynucleoside triphosphate (dATP, dGTP,
dCTP, and dUTP), 75 ng of each primer, 1 U ofThermus aquaticus DNA
polymerase (all reagents were from Pharmacia Biotech, Freiburg, Germany), and
5l of DNA. The thermal profile involved initial denaturation for 5 min at 96°C
and 38 cycles with the following steps: 1-min denaturation at 94°C, annealing at 59°C, and extension at 72°C.
RFLP analysis.The amplified products were digested separately with 2 U of
restriction enzymeHaeIII,CfoI,TaqI,MspI (Sigma, Diesenhofen, Germany),
DdeI (Promega, Madison, Wis.), orAvaII andHinfI (Amersham, Braunschweig,
Germany) according to the recommendations of the manufacturers and electro-phoresed in 4% Small agarose (Biozym, Oldendorf, Germany) in the presence of
ethidium bromide at 65 V for 2.0 to 3.0 h. For restriction withHinfI, dUTP in the
PCR mixture was replaced by dTTP. Fragment band sizes were estimated
visu-ally by comparison with appropriate controls run in parallel (type strains ofM.
avium,M. intracellulare, andM. kansasii) and a 100-bp ladder. All restriction fragment sizes of patterns shown for slowly growing mycobacteria rely on se-quence data (fragments smaller than 30 bp are not shown). Due to unavailable sequence data for most rapidly growing representatives, their RFLP fragment
sizes were estimated visually without computerized help and rounded to the nearest 5 bp.
Nucleotide sequence accession numbers.The 16S RNA gene sequences of
Mycobacteriumspp. strains S245 (MCRO 33;scrofulaceum), S318 (M. flavescens),
and S369 (M. xenopi) were deposited in the GenBank database under the
acces-sion numbers AF152559, AF174289, and AF174290, respectively. Cultures of S245, S279, S369, S318, and S522 were deposited in the strain collection of the Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany under the numbers 44427, 44429, 44428, 44430, and 44431.
RESULTS
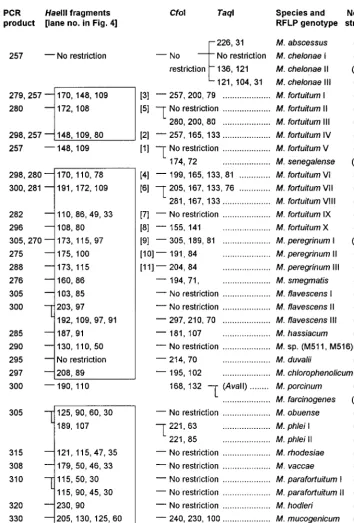
Specificity of primers and RFLP patterns.With the excep-tion ofGordonia terrae, none of the bacteria other than myco-bacteria were amplifiable. Amplification products of varying sizes were obtained from all mycobacteria tested. Amplicon sizes varied from 200 bp (M. xenopi) to 330 bp (Mycobacterium neoaurum). Mycobacterium nonchromogenicum, Mycobacte-rium terrae, M. triviale, and rapidly growing species showed fragments larger than 250 bp.HaeIII was selected as the first-line enzyme that, together with the knowledge about amplicon sizes, would produce the most discriminative RFLP patterns. Of 58 discernibleHaeIII patterns, 49 (84%) were unique and thus indicative and sufficient for identification to the species level.HaeIII species-specific patterns are highlighted in Fig. 1 and 2. TheHaeIII patterns of slowly growing mycobacteria,M. fortuitum, andMycobacterium peregrinumare displayed in Fig. 3 and 4. Except for the patterns shown in Fig. 3, lanes 3 and 4, 6 and 7, 13 and 16, 18 and 19, or 24 and 25 exhibiting minor differences of 3 to 9 bp,HaeIII restriction produced mostly two to three DNA fragments whose sizes could be easily estimated by visual inspection of the gels. Since primer-dimer formation was never noticed, fragments as small as 30 bp were also used for classification of RFLP patterns.
The remaining nineHaeIII patterns that did not give a final species assignment needed further analysis with additional en-donucleases. Therefore, all test organisms were subjected to
CfoI digestion. AlthoughCfoI patterns were not necessary for final identification of most species (in particular, rapidly grow-ing ones), as a practical routine, immediate restriction with bothHaeIII andCfoI may be advisable (and thus it is shown for all species) because the reliability of results for one enzyme is augmented if confirmed by a second endonuclease analysis. Following the established algorithm for slowly growing myco-bacteria (Fig. 1), precise estimates of fragments after CfoI restriction were not generally required. Rather, the distin-guishing feature of this enzyme was mostly confined to the question of whether the amplicon was cut or not. Owing to the high sequence similarity of some species such asM. avium,M. chelonae,M. kansasii, andMycobacterium simiaeto their near-est relatives, these species formed groups that needed further analysis byDdeI,TaqI, orAvaII for accurate identification as shown in Fig. 1, 2, and 3. Four of these clusters (Fig. 3, lanes 2, 7, 9, and 22) were readily resolved using DdeI, and three were resolved using TaqI, but the latter enzyme could not distinguish Mycobacterium genavense, Mycobacterium lentifla-vum, and Mycobacterium triplex. If necessary, these species, which have a high spacer sequence similarity of 95%, can be separated by restriction withMspI:M. simiaeand M. lentifla-vumstrains were cut once (139 and 86 bp), andM. genavense
andM. triplexwere cut twice (114, 86, and 25 and 86, 79, and 60 bp). The endonucleases AvaII andHinfI may be of use in exceptional cases, notably, in research or reference facilities, for separation of M. kansasii V from Mycobacterium leprae
(HinfI), orMycobacterium porcinumfromMycobacterium far-cinogenes(AvaII). One isolate of this specific M. kansasii ge-notype and theM. porcinumtype strain (Table 1) tested were cut, with resulting fragments of 116 and 105 bp and 215 and 85
on May 15, 2020 by guest
http://jcm.asm.org/
TABLE 1. Reference strains and clinical isolates of bacteria used in the present study
Genus Species No. ofstrains Reference strain(s)a No. of clinical
isolates
Actinomadura madurae 1 DSM 43067T
Actinomadura pelletieri 1 DSM 43383T
Actinomyces israelii 3 DSM 43320T, S293 1
Actinomyces meyeri 1 1
Actinomyces naeslundii 2 DSM 43013T 1
Actinomyces neuii 1 DSM 8577T
Actinomyces viscosus 2 DSM 43327T 1
Amycolatopsis orientalis 2 DSM 40040T, 43387
Arcanobacterium haemolyticum 3 DSM 20595T, S333 1
Arcanobacterium pyogenes 1 DSM 20630T
Prevotella buccae 1 1
Prevotella disiens 1 1
Bacteroides fragilis 1 DSM 2151T
Cellulomonas cellulans 1 DSM 43879T
Cellulomonas turbata 2 DSM 20577T, S330
Clostridium glycolicum 1 1
Clostridium perfringens 1 1
Corynebacterium diphtheriae 2 DSM 44123T 1
Corynebacterium jeikeium 2 DSM 7171T 1
Corynebacterium pseudodiphthericum 1 1
Corynebacterium renale 1 DSM 20688T
Corynebacterium striatum 2 DSM 20668T 1
Corynebacterium urealyticum 2 DSM 7109T 1
Corynebacterium xerosis 5 DSM 20743T, 20170 3
Dermatophilus congolensis 1 DSM 44180T
Dietzia maris 1 DSM 43672T
Escherichia coli 3 DSM 1103, 5923, ATCC 9637
Enterococcus faecalis 1 DSM 2570
Fusobacterium nucleatum 1 DSM 20482T
Gordonia aichensis 1 DSM 43978T
Gordonia amarae 2 DSM 43392T, 43391
Gordonia bronchialis 1 DSM 43247T
Gordonia hirsuta 1 DSM 44140T
Gordonia hydrophobica 1 DSM 44015T
Gordonia rubropertinctus 2 DSM 43197T, 10347
Gordonia sputi 2 DSM 43896T, 43979
Gordonia terrae 5 DSM 43249T, 46040, 43342, 43568, 43569
Haemophilus influenzae 1 ATCC 49247
Lactobacillus rhamnosus 1 S331
Listeria ivanovii 1 S302
Listeria monocytogenes 3 S303, 305, 306
Mycobacterium abscessus 9 DSM 44196T, 43492, 43493, S322–324 3
Mycobacterium asiaticum 3 DSM 44056, 44292, 44297
Mycobacterium aurum 2 DSM 43999T, S283
Mycobacterium avium 111 DSM 44156T, DSM 44133T 109
Mycobacterium bohemicum 2 DSM 44277T 1
Mycobacterium celatum 15 DSM 44243T, S274, 275 12
Mycobacterium chelonae 14 DSM 43804T, 43217, 43276, 43483, 43484, 43487, 43488–43490, 46626, S268 3
Mycobacterium chlorophenolicum 1 DSM 43826T
Mycobacterium conspicuum 1 DSM 44136T
Mycobacterium duvalii 1 DSM 44244T
Mycobacterium farcinogenes 20 DSM 43637T, M9, 15, 16, 39, 52, 57, 191, 217, 269, 274, 275, 281, 285, 612, 687, 785, N710, 715, 725
Mycobacterium flavescens 7 DSM 43991T, S318, 523–527
Mycobacterium fortuitum 26 DSM 46621T, 44220T, M205, 368, 390, N723, S113, S485, 487–493 11
Mycobacterium gastri 5 DSM 43505T, 43506, S227–229
Mycobacterium genavense 11 11
Mycobacterium gordonae 48 DSM 44160T 47
Mycobacterium haemophilum 3 3
Mycobacterium hassiacum 1 DSM 44199T
Mycobacterium hodleri 1 DSM 44183T
Mycobacterium interjectum 1 DSM 44064T
Mycobacterium intermedium 4 DSM 44049T 3
Mycobacterium intracellulare 34 DSM 43223T, S138, 347 31
Mycobacterium kansasii 52 DSM 44162T 51
Mycobacterium lentiflavum 5 DSM 44195T, 44194, S136, 360 1
Mycobacterium malmoense 11 DSM 44163T, S217 9
Mycobacterium marinum 16 DSM 44344T, 43518, 43519, 43824, 44345, S287 10
Mycobacterium mucogenicum 1 DSM 44124
Mycobacterium neoaurum 1 DSM 44074T
Mycobacterium nonchromogenicum 4 DSM 44164T, S264–266
Mycobacterium obuense 1 DSM 44075T
Continued on following page
on May 15, 2020 by guest
http://jcm.asm.org/
bp, respectively.M. farcinogenesisolates were not cut, nor was
M. lepraeaccording to the database sequence. Finally,AvaII is shown in Fig. 1 for optional application in connection withM. avium,M. kansasiiIV, andMycobacterium bohemicumbecause these showedHaeIII patterns with the highest degree of
[image:4.612.58.552.92.608.2]sim-ilarity. The first was digested by AvaII (144 and 75 bp); the latter two taxons were not. Mycobacterium marinumand My-cobacterium ulceranspossess identical spacer sequences (1, 22). Therefore, these organisms could not be separated by this method.
TABLE 1—Continued
Genus Species No. ofstrains Reference strain(s)a No. of clinical
isolates
Mycobacterium parafortuitum 4 DSM 43528T, 43526, 43527, S528
Mycobacterium peregrinum 17 DSM 43271T, M418–420, S254, 486, 494–496 8
Mycobacterium phlei 5 DSM 43239T, 43214, 44018 2
Mycobacterium porcinum 1 DSM 44242T
Mycobacterium rhodesiae 1 DSM 44223T
Mycobacterium scrofulaceum 11 DSM 43992T, 43226, 43512, 43513, S343, 244 5
Mycobacterium senegalense 10 DSM 43656T, 43658, M266, 555, N714, 717–718, 721, 728, S114
Mycobacterium shimoidei 3 DSM 44152T, S234 1
Mycobacterium simiae 15 DSM 44165T, S137, 140, 141, 146, 148, 149 8
Mycobacterium smegmatis 2 DSM 43756T, 43299
Mycobacterium sp. 2 M511, 516
Mycobacterium sp. (gastri)b 10 DSM 43221, 43507, S230–233, R230–233
Mycobacterium sp. (malmoense) 2 S222, 279 2
Mycobacterium sp. (scrofulaceum) 5 S245, 313–314, 316, R39 5
Mycobacterium sp. (xenopi) 2 S369, 504 2
Mycobacterium szulgai 8 DSM 44166T, S97 6
Mycobacterium terrae 9 DSM 43227T, S280–281, 353 5
Mycobacterium triplex 2 S139 1
Mycobacterium triviale 2 DSM 44153T 1
Mycobacterium tuberculosis 90 DSM 44156 89
Mycobacterium ulcerans 3 ATCC 19423T, S219 1
Mycobacterium vaccae 4 DSM 43292T, 43229, 43514, S345
Mycobacterium xenopi 59 DSM 43995T 58
Nocardia asteroides 12 DSM 43757T, 43003–43005, 43208, 43244, 43289, S308, 328, ATCC 23824 2
Nocardia brasiliensis 2 DSM 43758T, 43009
Nocardia brevicatena 1 DSM 43024T
Nocardia carnea 2 DSM 43397T, 40840
Nocardia corynebacterioides 1 DSM 20151T
Nocardia farcinica 4 DSM 43665T, 43666, S304, 327
Nocardia nova 1 ATCC 33726
Nocardia otitidiscaviarum 3 DSM 43242T, 43010, 43398
Nocardia pseudobrasiliensis 1 DSM 44290T
Nocardia seriolae 1 DSM 44129T
Nocardia sp. 6 6
Nocardioides albus 1 DSM 43109T
Nocardiopsis dassonvillei 1 DSM 43111T
Peptostreptococcus anaerobius 1 DSM 2949
Pseudomonas aeruginosa 1 DSM 1117
Pseudonocardia autotrophica 2 DSM 535T, 43082
Rhodococcus equi 2 DSM 20307T, S307
Rhodococcus rhodochrous 1 DSM 43241T
Rhodococcus ruber 1 DSM 43338T
Rothia dentocariosa 3 DSM 43762T, S325, 326
Saccharomonospora glauca 1 DSM 43769T
Saccharomonospora viridis 1 DSM 43017T
Saccharopolyspora rectivirgula 1 DSM 43747T
Saccharothrix aerocolonigenes 1 DSM 40034T
Skermania piniformis 1 DSM 43998T
Staphylococcus aureus 2 DSM 2569, 1104
Streptococcus pneumoniae 2 ATCC 6303 1
Streptomyces albus 1 DSM 40313T
Streptomyces somaliensis 1 DSM 40738T
Thermoactinomyces vulgaris 1 DSM 43016T
Tsukamurella paurometabolum 3 DSM 20162T, 44119, S329
Tsukamurella pulmonis 1 DSM 44142T
Tsukamurella tyrosinosolvens 1 DSM 44234T
Turicella otitidis 1 DSM 8821T
Total 811 525
aATCC, American Type Culture Collection, Manassas, Va.; DSM, Deutsche Sammlung von Mikroorganismen und Zellkulturen, Braunschweig, Germany; M and
N, strain collection, Department of Microbiology, The Medical School, University of Newcastle, Newcastle upon Tyne, United Kingdom; R, strain collection, Institut fu¨r Mikrobiologie und Hygiene, Universita¨tsklinikum Regensburg, Regensburg, Germany; S, strain collection, Institut fu¨r Mikrobiologie und Immunologie, Kranken-haus Zehlendorf, Berlin, Germany.
bProvisionally termedMycobacteriumsp. on the basis of phenotypic and genotypic data (for details, see the text).
on May 15, 2020 by guest
http://jcm.asm.org/
According to the spacer RFLP method, four reference strains deposited as M. fortuitum (DSM 43276 and 46626),
Nocardia farcinica(DSM 43231), andMycobacterium thermore-sistibile(DSM 43644) were diagnosed asM. chelonae(the first
[image:5.612.131.484.71.603.2]two isolates),Mycobacterium senegalense, andM. phlei, respec-tively. Partial 16S rDNA sequencing and analysis of fatty and mycolic acids were in full agreement with these findings, and these strains were thereafter reclassified (the names used in FIG. 1. Algorithm of RFLP patterns of 28 slowly growing mycobacterial species and 4Mycobacteriumspp. of uncertain taxonomic status from PCR-amplified 16S-23S rDNA spacer sequences (547 strains). PCR products and restriction fragments are designated by molecular sizes in base pairs.HaeIII species-specific patterns are highlighted by boxes.CfoI patterns A to D are as follows: A, 126 to 144 and 91 to 96 (digest size varies depending on the PCR product size); B, 129 to 146 and 83; C, 126, 63, and 30; D, 160 and 62.DdeI patterns A-E are as follows: A, 120 and 90; B, 120 and 80; C, 120 and 70; D, 120 and 100; E, 214.TaqI pattern A is 155 and 70; 0, no restriction. Type strains were assigned to genotype I if more than one pattern occurred in a species. Genotypes Ia and Ib or IIa and IIb indicate that the strains are genetically very similar but new RFLP genotypes have occurred after loss or acquisition of oneHaeIII restriction site due to allelic microheterogeneity.M. lepraeandM. kansasiiIII RFLP patterns were deduced from nucleotide sequence accession no. X56657 (EMBL) and theM. kansasiigenotype III sequence published by Alcaide et al. (1). For details concerningAvaII,HinfI, andMspI patterns and descriptions ofMycobacteriumspp., see the text.
on May 15, 2020 by guest
http://jcm.asm.org/
Table 1 are those after reclassification). Of five G. terrae
strains, three showed a 315-bp and two showed a 330-bp PCR product, and the respectiveHaeIII RFLP patterns were 200, 170, and 130 and 185 and 160 bp, respectively.
Taxonomically uncertain strains. Two rapidly photochro-mogenic strains deposited as Mycobacterium sp. (reference strains M511 and M516) had a unique RFLP pattern com-pared to other rapidly growing mycobacteria. Data on the exact phenotype were not available, but complete 16S rDNA
sequencing revealed three substitutions compared to Mycobac-terium smegmatis: ACA3ATA, TAG3TGG, and TTA3 TGA at positions 137, 162, and 1075 of the reference sequence (EMBL X52922). Four groups comprising slowly growing strains with uncertain taxonomic status were formed and ten-tatively named Mycobacterium sp. (Table 1). Although this nomenclature has no taxonomic standing (the names of the most closely related species are provisionally added in paren-theses), phenotypic and genotypic data together with the find-FIG. 2. Algorithm of RFLP patterns of 21 rapidly growing mycobacterial species and one rapidly growingMycobacteriumsp. of unknown taxonomic status from PCR-amplified 16S-23S rDNA spacer sequences. Details are given in the legend to Fig. 1.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:6.612.128.482.66.589.2]ing that all showed unique spacer RFLP patterns obviated the need to separate these groups from established species. The following is a detailed description of the results.
(i) Mycobacterium sp. (gastri). Ten reference strains origi-nally deposited as either M. kansasiiorMycobacterium gastri
clustered in one RFLP genotype (Fig. 3, lane 21). All these strains were sequenced in the spacer except for strains S230 to
S233, which had previously been characterized as M. gastri
[image:7.612.130.472.71.329.2]spacer genotype Mga B (22). This revealed that the fourM. kansasii strains and two M. gastri strains (DSM 43221 and 43507) were attributable to spacer M. kansasii genotype IV described by Alcaide et al. (1) (Table 2 gathers available data together with the findings of this study). The spacer genotype sequences IV and Mga B have a similarity value of 99.9% due
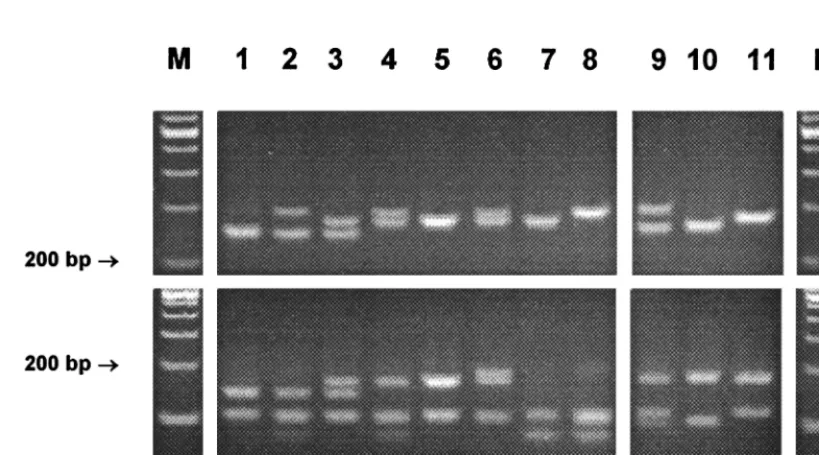
FIG. 3. Gel electrophoresis andHaeIII RFLP patterns of slowly growing mycobacteria from PCR-amplified 16S-23S rDNA spacer sequences (the upper panel
shows PCR products without restriction). The molecular sizes of the fragments are given in Fig. 1. The patterns are displayed in order of increasing size of the biggest
fragment. M, molecular size marker (100-bp ladder). MAIS,M. avium-M. intracellulare-M. scrofulaceum.
FIG. 4. Gel electrophoresis andHaeIII RFLP patterns ofM. fortuitum(lanes 1 to 8) andM. peregrinum(lanes 9 to 11) from PCR-amplified 16S-23S rDNA spacer
sequences (the upper panel shows PCR products without restriction). The patterns are described in the legend to Fig. 2. M, molecular size marker (100-bp ladder).
on May 15, 2020 by guest
http://jcm.asm.org/
[image:7.612.99.509.481.709.2]to a 1-nucleotide substitution at position 223 (ACT3AAT), and sequences Mga B and Mga A (M. gastri type strain se-quence) display a similarity value of 98% (4-nucleotide differ-ence). Careful reassessment of biochemical tests showed that these strains were incapable of hydrolyzing Tween 80 (both
M. gastriandM. kansasiihydrolyze Tween), while tests clearly positive for M. kansasii(nitrate reduction, catalase, and pho-tochromogenicity) were weakly positive (delayed colony pig-ment formation). Thus, these strains were assumed to be an indeterminate (or new) taxon near to the speciesM. gastri, as evidenced by their 16S-23S rDNA sequences. In contrast, all
M. kansasii strains hydrolyzed Tween, and their spacer se-quences showed sufficient diversity to emerge as three distinct subgroups (dendrogram not shown): sequence genotypes (se-quevars) I together with II, and III together with Mka B and Mka C as separate clusters, while type V showed the highest degree of similarity toM. kansasiitype IV (90%). Resolution of 16S rDNA sequences within these entities was poor (Table 2). We found and thus confirmed minor variants in the variable region B recently described by Richter et al. (20). Full-length 16S rDNA sequencing of one strain of concern because of its negative AccuProbe result (strain S522 with spacer genotype Mka C) flawed the probability that this strain could be an unrecognized new species, since it showed complete identity with the M. kansasii type strain sequence downstream from variable region B.
(ii)Mycobacteriumsp. (malmoense).Two clinical scotochro-mogenic isolates phenotypically resembling Mycobacterium malmoense exhibited a 16S rDNA sequence with nine substi-tutions compared to that ofM. malmoense: CCC CGA3CCA
CTT, GGG3GTG, ACG3ATG, TGG3TAG, CCT TGT
3CCC CGT, and TCG3TTG at positions 141, 159, 220, 601, 1062, and 1403 of the reference sequence (EMBL X52930). These strains could represent subspecies ofM. malmoenseand, interestingly, they emerged as a distinct RFLP genotype in the vicinity ofM. malmoense.
(iii)Mycobacteriumsp. (scrofulaceum).Similarly to the form-er case, five clinical isolates phenotypically vform-ery closely related toMycobacterium scrofulaceum (the only physiological differ-ence was a lack of growth at 25°C) showed an RFLP genotype near to but distinguishable from that ofM. scrofulaceum, and in good correlation with this, possessed a distinctive 16S rDNA sequence. The latter was identical to the MCRO 33 sequence published previously (26), which typically shows identity with
M. scrofulaceum in variable region A and identity withM. si-miaein region B (deletion of 12 nucleotides).
(iv)Mycobacteriumsp. (xenopi).Two strains, S369 and S504, isolated from the sputa of two patients with lung disease, dis-played identical complete 16S rDNA sequences, with the high-est similarity to that of the M. xenopi type strain (97%). A missing arylsulfatase activity (2 weeks) and negative nicotin-amidase and pyrazinnicotin-amidase were reactions in discordance with M. xenopi. The RFLP results were somewhat different from those ofM. xenopi, exhibiting a uniqueHaeIII pattern.
Intraspecies stability of spacer sequences.Intraspecies spacer sequence polymorphisms seemed to be more frequent in rap-idly growing mycobacteria than in slowly growing species. In fact, many of the rapidly growing representatives for which multiple strains within a species were studied presented more than one RFLP pattern. As expected from the known variabil-ity found in the 16S rDNA,M. fortuitumwas associated with a considerable variability leading to eight different HaeIII pat-terns (Fig. 4). They all shared a 108- to 110-bp band, and some of them showed typical PCR products of two different sizes due to interoperon variability. These features, together with the
CfoI result, gave the correct species identification in all cases.
M. fortuitum subsp. acetamidolyticum (DSM 44220) was as-signed to RFLP genotype II, and one strain with a 16S signa-ture ofM. fortuitumbiovariant 3 was assigned to RFLP geno-type VII.
The occurrence of RFLP genotypes different from type strain genotypes inM. flavescensand M. parafortuitum refer-ence strains (Fig. 2) was found to be associated with hitherto unknown sequence polymorphisms in the 16S RNA gene.M. flavescensII (S526) and III (S318) had identical 16S sequences, but they differed by as many as 23 nucleotides from the type strain sequence, which raises the question of the species integ-rity of these reference strains. M. parafortuitumRFLP geno-type II (DSM 43526) displayed six base substitutions compared to the type strain 16S rDNA sequence: five differences in the signature sequence of variable region A (AAT AGG ATC
ACT GGC TTC ATG GTC) and one mismatch (GAA 3
GGA) at position 882 of the reference sequence (EMBL X93183). Full-length 16S rDNA sequences ofM. phleiandM. triviale RFLP genotypes II (one strain each) showed 100% identity with the respective type strain sequences.
[image:8.612.51.552.83.231.2]Of slowly growing mycobacteria, onlyM. kansasii(as noted), theM. terraecomplex, and, to a lesser extent,M. intracellulare TABLE 2. Spacer genotyping results forM. kansasiiandM. gastriicompared to 16S RNA andhsp65gene
Species
Results 16S-23S spacer
16S rDNA sequence variants
(nt 461–469)b
AccuProbe (new version)
hsp65genec
RFLP geno-type No. of strains analyzed Sequence sequevara No. of strains
analyzed EMBL no.
Geno-type
Restriction fragments (bp)
BstEII HaeIII
M. kansasii I 23 I/Mka A 6 X97632/L42262 M. kansasii/gastri
CGG GTT CTC Positive I 231, 212 127, 103, 78
II 15 II 7 CGG GTT GTC Positive II 231, 133, 79 127, 103 (70)
IV 9 Mka B 2 L42263 CGG GTT TTC Positive III 231, 133, 79 127, 94, 69 V 1 Mka C 1 L42264 CGG GTT TCC Negative VI 231, 133, 79 127, 103, 69
VI 4 V 1 M. kansasii/gastri Positive V 325, 125 140, 100, 80
Mycobacteriumsp.
(gastri) 10 IVMga B 64 Y14182 M. kansasii/gastriM. kansasii/gastri PositivePositive IV Unknown231, 118, 79 127, 112, 69Unknown
M. gastri 5 Mga A 4 X97633 M. kansasii/gastri Negative 231, 133, 79 127, 103, 69
aGenotypes I to IV and Mga A and B in accordance with references 1 and 22, respectively. Sequevar Mka B and C sequences were deposited in the EMBL database
in 1995 (unpublished data) and were later confirmed by Richter et al. (20). Sequence sequevar III (1) is not shown because the genotype was not found in this study.
bRefers to partial 16S rDNA sequencing within the variable region B only. Underlined bases indicate positions of substitutions.
cAccording to references 1, 18, and 20. Boldface numbers indicate patterns that are identical. TheHaeIII 70-bp fragment was not present in all isolates (1).
on May 15, 2020 by guest
http://jcm.asm.org/
andM. scrofulaceumwere characterized by a genetic hetero-geneity leading to more than one RFLP pattern in a species. Intraspecies sequence variations for many slowly growing my-cobacteria, such as theM. aviumcomplex,M. simiae,M. gor-donae(the last deposited as EMBL accession numbers L42258 to 42261), andM. xenopihave been described (8, 22). Despite this, allelic heterogeneities had no detrimental impact on the clearly arranged RFLP algorithm. Even so, we sequenced a selection of strains by way of example to obtain a better esti-mate of the occurrence of sequence diversity within the spacer. Species were chosen in which a diversity might be expected because sequence variations occur both in the 16S rDNA and the hsp65gene. The results are shown in an alignment with published sequences in Fig. 5. The reproducibility (and thus the degree of stability) of spacer sequences was confirmed because the sequences found were in full agreement with pub-lished data (8, 9), albeit some new sequevars were found (Mac J to L and Mgo E and F). Of 81M. aviumstrains examined, 44 and 37 fell into the Mav A and B sequevars, respectively. The spacer sequences of sixM. celatumisolates were all identical. The positions of restriction sites that generate species-specific or subspecific RFLP genotypes are located in the more con-served stem-loop regions. For example, in the case ofM. in-tracellulare these contain distinct sequence motifs found in subspecific groups related to either Min or Mac sequevars, which in turn have led to the formation of two RFLP geno-types. These clusters consist of a larger number of sequevars, which are characterized by a high rate of substitutions in vari-able regions, such as the antitermination elements (position 130 to 160) or within helices 2, 5, and 6. The last two lie beyond
the part of the spacer amplified by primers Sp1 and Sp2, and a single sporadic mutation with generation of a new restriction site within helix 2 was observed only once (Mac K with RFLP genotypeM. intracellulare IIb). Although substitutions in the stem regions can be expected to occur rarely, we found sub-stitutions at the transition from helix 3 to the stem sequence (position 76 [Fig. 5]). Hence,M. kansasiiII andM. scrofula-ceumwere split into two RFLP genotypes attributable to the same mutational event. Ultimately, the high number of strains studied within some species associated with only one RFLP genotype despite sequence microheterogeneities, such asM. gordonaeorM. xenopi, provide firm evidence that the degree of stability of the RFLP patterns is very high.
DISCUSSION
We sought to establish a new molecular method for identi-fication of mycobacteria that on the one hand would be capa-ble of identifying all taxons to the species level with high accuracy and reliability and on the other hand would be simple enough for application even in routine laboratories. In view of this, emphasis was laid on inclusion of a broad spectrum of species and, even more important, on examination of a larger number of reference and clinical isolates within a species. First, this was important in order to determine the reliability of new primers chosen within a genetic target with a tendency to show more frequent sequence rearrangements due to a higher evo-lutionary rate (11, 22). Second, the occurrence of additional RFLP patterns due to sequence polymorphisms not presently recognized due to unavailable clinical isolates may later seri-FIG. 5. Sequence stability and microheterogeneity of 16S-23S rDNA spacer sequences in conserved and more variable regions with relevance for the cleaving action ofHaeIII (GGCC) andCfoI (GCGC). Sequences not found in this study but published elsewhere are included (4, 8, 9). The respective sequevar designations are shown in brackets, and the number of strains sequenced for this study are shown in parentheses. Sequevars combined in one line exhibit base substitutions located in other regions of the spacer that are not displayed. Of Mav A to E and Min A to C, only Mav A or B and Min A were found. The sequevar Mgo B was not found among sevenM. gordonaeisolates examined.
on May 15, 2020 by guest
http://jcm.asm.org/
ously compromise the devised diagnostic algorithm or even make evaluation of additional enzymes necessary. Besides the genus specifity of the primers shown, the size variations of the PCR amplicons described are particularly useful because they provide a simple means for distinguishing rapidly growing from slowly growing species at first glance, since the latter produce amplicons larger than 250 bp. Furthermore, this valuable new feature of the method can prove helpful in unequivocally rec-ognizing mixed cultures. For example, a culture containing a slowly growing relevant pathogen likeM. malmoensecan easily be recognized when overgrowth by M. fortuitumorM. terrae
occurs because, irrespective of mixed patterns, the latter ex-hibit PCR products far larger than 220 bp.
Concerning intraspecies stability of RFLP patterns, we can state that the spacer-based method was successfully evaluated with respect to expanded groups of strains within slowly grow-ing species, such asM. tuberculosis,M. avium,M. xenopi, orM. gordonae. These are frequently found in a routine laboratory setting, and we were indeed satisfied to see that, with a few exceptions, all of these strains in a species were associated with only one RFLP pattern. This is in contrast to results obtained using the hsp65gene-based RFLP method, which exhibits a greater number of RFLP genotypes within one species (e.g., six patterns for M. gordonae) (5, 29). The finding of distinctM. kansasiisubgroups accurately defined by unique spacer RFLP genotypes is in perfect correlation with previous reports (1, 18, 20). Of note, the hsp65gene RFLP method is unable to dis-tinguish the clinically relevant subspecies M. kansasii II and Mka C from the nonpathogenic M. gastri (Table 2). By con-trast, the use of the spacer is flawed by the sequence identity of
M. marinumandM. ulcerans, but this represents a minor prob-lem from a clinical point of view, since these species appear under completely different epidemiological circumstances (7). The sequence variability of rapid growers was considerable. We can expect that additional spacer RFLP patterns will be found when more strains are analyzed. This may be particularly true for theM. terrae-M. nonchromogenicum complex or rap-idly growing species such as M. neoaurum, which exhibited species-specific results, although the small number of strains used probably underestimates their true genetic heterogeneity (32). Hence, we can state that data on most rapid growers are still insufficient and remain to be improved in further studies. Similar observations have been made concerning the hsp65
gene as a genetic target (21). It could be interesting to validate the biological significance of spacer RFLP genotypes in com-parison to type strains. Since 16S rDNA sequencing data for rapidly growing mycobacteria are still very incomplete (21, 26), it appears mandatory to look for the possibility that additional RFLP genotypes found may represent unknown infrasubspe-cific 16S rDNA genotypes. Evidence for this was shown here for M. flavescensand M. parafortuitum, but further investiga-tions of the exact phenotypes are certainly warranted because these reference strains were not reassessed by biochemical tests in this study. Besides phylogenetic or taxonomic consid-erations, such RFLP subgroups may reflect clinically, physio-logically, or epidemiologically significant subdivisions, as has been proposed forM. chelonae(19) or theM. aviumcomplex (4, 8, 9). Some additional RFLP genotypes in a species may not have recognizable phenotypic or genotypic correlates in either the physiological tests usually performed or in their complete 16S rDNAs, respectively, due to the higher phylogenetic res-olution of 16S-23S spacer sequences. This was nicely shown by 16S rDNA sequencing ofM. phleiandM. trivialeRFLP geno-types II.
The high similarity of M. aviumto M. bohemicumandM. kansasii IV represents an undesirable shortcoming of the
method, sinceM. aviumis the most frequently isolated mem-ber of the genus Mycobacterium. If gel electrophoresis was performed carefully, the above-mentioned similar patterns were not confused by technical staff in our laboratory (Fig. 3, compare lanes 6 and 7), and ultimately, application of a third enzyme resulted in a definite correct assignment in all cases. In addition, M. bohemicum and M. kansasiiRFLP genotype IV (sequevar Mka B) are very rare in clinical specimens (1, 20). A possible failing of the method in this case can be disregarded in most laboratories that use GenProbes for identification ofM. tuberculosisand theM. aviumcomplex. However, if RFLP is used as the sole procedure for identification, investigators should remain vigilant for this pattern by letting gels run longer in comparisons toM. aviumas the proposed internal standard. In this context, a conclusion that deserves mention is that judicious inclusion of closely related species is crucial for a complete assessment of the reliability and discriminatory power of these methods. In fact, the necessity to apply addi-tional enzymes in a few groups was only recognized because care was taken to study, if possible, closely related mycobac-teria as well (i.e.,M. simiaetogether withM. lentiflavumandM. triplex). Apparently, in contrast to thehsp65gene, the diversity of spacer sequences is not high enough at a species level in all phylogenetic groups to allow separation by only two digests. Nevertheless, we believe that this disadvantage is compensated for by the overall simplicity of the scheme for the majority of other species and the large amount of information yielded afterHaeIII digestion by itself. RFLP results for species such as M. lentiflavum-M. triplex, M. bohemicum (which is closely related to M. avium), Mycobacterium interjectum-Mycobacte-rium intermedium, M. farcinogenes, orMycobacterium obuense
have not yet been reported for thehsp65gene (5, 27–30). This issue must keep us alert to the fact that thehsp65data have yet to be perfected.
A major result that emerges from our study is the fact that the method presented reveals the potential to be used in my-cobacterial taxonomy. It is interesting that species closely re-lated to each other clustered in the same or similar HaeIII patterns. Examples are theM. aviumcomplex together withM. scrofulaceum andM. bohemicum,M. simiaeand relatives,M. fortuitum and M. senegalense, and similar HaeIII patterns found for M. terrae and M. nonchromogenicum. By contrast, one included taxon that probably represents a new species (termedMycobacteriumsp.xenopi), as indicated by a low 16S rDNA similarity of only 97% withM. xenopi, clearly possessed a distinctHaeIII pattern. Similarly,HaeIII patterns rather dif-ferent from the type strain pattern found in sixM. flavescens
reference strains suggested a genetic disintegrity of this group, a finding that was later confirmed by 16S sequencing. Organ-isms provisionally denoted subspecies, for example, Mycobac-teriumsp.malmoense, by contrast emerged as unique identifi-able entities but shared the sameHaeIII pattern with the most closely related species. Irrespective of the taxonomical validity of these observations, they are a reflection of a high degree of spacer sequence conservation and reinforce the previously dis-cussed view that 16S-23S rDNA spacer sequence analysis con-stitutes an adjunct to mycobacterial phylogeny (8, 9, 22). This study provides additional evidence that spacer sequence anal-ysis results are in good correlation with 16S rDNA data. Valid descriptions of new species or subspecies were not addressed in this study, but theM. kansasii-M. gastri and M. flavescens
cases illustrate the usefulness of our method in identifying novel taxons before more accurate but labor-intensive compar-ative sequencing investigations (or those involving numerical taxonomy) are initiated. The problem of pigmentedM. gastri
strains was addressed by Anz and Schro¨der as early as 1970 (2),
on May 15, 2020 by guest
http://jcm.asm.org/
and this debate was taken up again as the genetic heterogene-ity ofM. kansasiiwas recognized later (1, 20). The somewhat confusing nomenclature of different studies is gathered in Ta-ble 2, and it can be acknowledged that spacer-based methods are superior to all other genetic approaches, including the 16S rDNA method, which cannot differentiate deeply enough to account for the genotypes found, and the AccuProbe method, which misses oneM. kansasiisubgroup while the taxonomically indeterminate subgroup with the spacer genotype IV produces positive results. The latter finding has no impact on clinical reports because so far these strains have only been isolated in environmental samples (1).
In conclusion, 16S-23S rDNA PCR-RFLP is a promising new method with consistent advantages over the previously usedhsp65gene-based method for reliable and easy identifi-cation of mycobacteria. Compared to new but technically de-manding (and thus still cost prohibitive) techniques, such as the development of DNA probe arrays (31), this method has the advantage of being both simple and extensive in its diag-nostic spectrum and cost-effective at the same time. Despite allelic diversity, good intraspecies stabilities of recognizable RFLP genotypes were demonstrated. More significant poly-morphisms on a subspecies level do not preclude the use of this method; rather, finding correlations of specific genetic sub-types to medically relevant linkages represents an issue worthy of further investigation.
REFERENCES
1.Alcaide, F., I. Richter, C. Bernasconi, B. Springer, C. Hagenau, R. Schulze-Ro¨bbecke, E. Tortoli, R. Martin, E. Bo¨ttger, and A. Telenti.1997.
Hetero-geneity and clonality among isolates ofMycobacterium kansasii: implications
for epidemiological and pathogenicity studies. J. Clin. Microbiol.35:1959–1964.
2.Anz, W., and K.-H. Schro¨der.1970. Photochromogenic strains of Mycobac-terium gastri? Zentrabl. Bakteriol.214:553–554.
3.De Beenhouwer, H., Z. Liang, P. de Rijk, C. van Eekeren, and F. Portaels.
1995. Detection and identification of mycobacteria by DNA amplification and oligonucleotide-specific capture plate hybridization. J. Clin. Microbiol.
33:2994–2998.
4.De Smet, A. L., I. N. Brown, M. Yates, and J. Ivanyi.1995. Ribosomal
internal transcribed spacers are identical amongMycobacterium
avium-intra-cellularecomplex isolates from AIDS patients, but vary among isolates from
elderly pulmonary disease patients. Microbiology141:2739–2747.
5.Devallois, A., K. S. Goh, and N. Rastogi. 1997. Rapid identification of mycobacteria to species level by PCR-restriction fragment length
polymor-phism analysis of thehsp65gene and proposition of an algorithm to
differ-entiate 34 mycobacterial species. J. Clin. Microbiol.35:2969–2973.
6.Emler, S., B. Ninet, P. Rohner, R. Auckenthaler, D. Ja¨ger, and B. Hirschel.
1995. Molecular basis for cross-reactivity between a strain ofMycobacterium
terraeand DNA probes forMycobacterium tuberculosis complex. Eur. J.
Microbiol. Infect. Dis.14:627–629.
7.Falkinham, J. O.1996. Epidemiology of infection by nontuberculous
myco-bacteria. Clin. Microbiol. Rev.9:177–215.
8.Frothingham, R., and K. H. Wilson.1993. Sequence-based differentiation of
strains in theMycobacterium aviumcomplex. J. Bacteriol.175:2818–2825.
9.Frothingham, R., and K. H. Wilson. 1994. Molecular phylogeny of the
Mycobacterium aviumcomplex demonstrates clinically meaningful divisions.
J. Infect. Dis.169:305–312.
10. Good, R. C.1985. Opportunistic pathogens in the genusMycobacterium.
Annu. Rev. Microbiol.39:347–369.
11. Gu¨rtler, V., and V. A. Stanisch.1996. New approaches to typing and iden-tification of bacteria using the 16S-23S rDNA spacer region. Microbiology
142:3–16.
12. Kent, P. T., and G. P. Kubica.1985. Public health mycobacteriology—a guide for the level III laboratory. U.S. Department of Health and Human Services publication (CDC) 86-8230. Centers for Disease Control, Atlanta, Ga.
13. Kim, B.-J., S.-H. Lee, M.-A. Lyu, S.-J. Kim, G.-H. Bai, S.-J. Kim, G.-T. Chae, E.-C. Kim, C.-Y. Cha, and Y.-H. Kook.1999. Identification of mycobacterial species by comparative sequence analysis of the RNA polymerase gene
(rpoB). J. Clin. Microbiol.37:1714–1720.
14. Kirschner, P., B. Springer, U. Vogel, A. Meier, A. Wrede, M. Kiekenbeck,
F. C. Bange, and E. C. Bo¨ttger.1993. Genotypic identification of mycobac-teria by nucleic acid sequence determination: report of a 2-year experience
in a clinical laboratory. J. Clin. Microbiol.31:2882–2889.
15. Kox, L. F. F., J. van Leeuwen, S. Knijper, H. M. Jansen, and A. H. Kolk.
1995. PCR assay based on DNA coding for 16S rRNA for detection and
identification of mycobacteria in clinical samples. J. Clin. Microbiol.33:
3225–3233.
16. Lappayawichit, P., S. Rienthong, D. Rienthong, C. Chuchottaworn, A. Chaiprasert, W. Panbangred, H. Saringcarinkul, and P. Palittapongarnpim.
1996. Differentiation ofMycobacteriumspecies by restriction enzyme analysis
of amplified 16S-23S ribosomal DNA spacer sequences. Tubercle Lung Dis.
77:257–263.
17. Mu¨ller, K.-D., E. N. Schmid, and R. M. Kroppenstedt.1998. Improved identification of mycobacteria by using the Microbial Identification System in combination with additional trimethylsulfonium hydroxide pyrolysis. J. Clin.
Microbiol.36:2477–2480.
18. Picardeau, M., G. Prod’Hom, L. Raskine, M. P. LePennec, and V. Vincent.
1997. Genotypic characterization of five subspecies ofM. kansasii. J. Clin.
Microbiol.35:25–32.
19. Portaeles, F., P. de Rijk, G. Jannes, R. Lemans, W. Mijs, L. Riqouts, and R. Rossau.1996. The 16S-23S rRNA spacer, a useful tool for taxonomical and
epidemiological studies of theM. chelonaecomplex. Tubercle Lung Dis.
77(Suppl. 2):17–18.
20. Richter, E., S. Niemann, S. Ru¨sch-Gerdes, and S. Hoffner.1999.
Identifica-tion ofM. kansasii by using a DNA probe (AccuProbe) and molecular
techniques. J. Clin. Microbiol.37:964–970.
21. Ringuet, H., C. Akoua-Koffi, S. Honore, A. Varnerot, V. Vincent, P. Berche, J. L. Gaillard, and C. Pierre-Audigier.1999.hsp65sequencing for
identifi-cation of rapidly growing mycobacteria. J. Clin. Microbiol.37:852–857.
22. Roth, A., M. Fischer, H. E. Hamid, W. Ludwig, S. Michalke, and H. Mauch.
1998. Differentiation of phylogenetically related slowly growing mycobacte-ria based on 16S-23S rRNA gene internal transcribed spacer sequences.
J. Clin. Microbiol.36:139–147.
23. Sanguinetti, M., B. Pasteraro, F. Ardito, S. Zanetti, A. Cingolani, L. Sechi, A. De Luca, L. Ortona, and G. Fadda.1998. Routine use of PCR-reverse
cross-blot hybridization assay for rapid identification ofMycobacterium
spe-cies growing in liquid media. J. Clin. Microbiol.36:1530–1533.
24. Schro¨der, K.-H., L. Naumann, R. M. Kroppenstedt, and U. Reischl.1997.
Mycobacterium hassiacumsp. nov., a new rapidly growing thermophilic
my-cobacterium. Int. J. Syst. Bacteriol.47:86–91.
25. Shinnick, T. M., and R. C. Good.1994. Mycobacterial taxonomy. Eur. J. Clin.
Microbiol. Infect. Dis.13:884–901.
26. Springer, B., L. Stockman, K. Teschner, G. D. Roberts, and E. C. Bo¨ttger.
1996. Two-laboratory collaborative study on identification of mycobacteria:
molecular versus phenotypic methods. J. Clin. Microbiol.34:296–303.
27. Steingrube, A. V., J. L. Gibson, B. A. Brown, Y. Zhang, R. W. Wilson, M. Rajagopalan, and R. J. Wallace.1995. PCR amplification and restriction endonuclease analysis of a 65-kilodalton heat shock protein gene sequence for taxonomic separation of rapidly growing mycobacteria. J. Clin. Microbiol.
33:149–153.
28. Steingrube, V. A., R. W. Wilson, B. A. Brown, K. C. Jost, Z. Blacklock, J. L. Gibson, and R. J. Wallace.1997. Rapid identification of clinically significant
species and taxa of aerobic actinomycetes, includingActinomadura,
Gor-donia,Nocardia,Rhodococcus,Streptomyces, andTsukamurellaisolates, by DNA amplification and restriction endonuclease analysis. J. Clin. Microbiol.
35:817–822.
29. Taylor, T. B., C. Patterson, Y. Hlae, and W. W. Safranek.1997. Routine use of PCR-restriction fragment length polymorphism analysis for identification
of mycobacteria growing in liquid media. J. Clin. Microbiol.35:79–85.
30. Telenti, A., F. Marchesi, M. Balz, F. Bally, E. C. Bo¨ttger, and T. Bodmer.
1993. Rapid identification of mycobacteria to the species level by polymerase
chain reaction and restriction enzyme analysis. J. Clin. Microbiol.31:175–
178.
31. Troesch, A., H. Nguyen, C. G. Miyada, S. Desvarenne, T. R. Gingeras, P. M. Kaplan, P. Cros, and C. Mabilat.1999. Mycobacterium species identification and rifampin resistance testing with high-density DNA probe arrays. J. Clin.
Microbiol.37:49–55.
32. Torkko, P., M. Suutari, S. Suomalainen, L. Paulin, L. Larsson, and M.-L. Katila.1998. Separation among species ofMycobacterium terraecomplex by lipid analysis: comparison with biochemical tests and 16S rRNA sequencing.
J. Clin. Microbiol.36:499–505.
33. Wallace, R. J.1994. Recent changes in taxonomy and disease manifestations of the rapidly growing mycobacteria. Eur. J. Clin. Microbiol. Infect. Dis.
13:953–960.
34. Wilson, R. W., V. A. Steingrube, B. A. Brown, and R. J. Wallace.1998. Clinical application of PCR-restriction enzyme pattern analysis for rapid
identification of aerobic actinomycete isolates. J. Clin. Microbiol.36:148–
152.