Role of Copy Number Variants in Structural
Birth Defects
abstract
BACKGROUND AND OBJECTIVE:Human genomes include copy number variants (CNVs), defined as regions with DNA gains or losses. Patho-logic CNVs, which are larger and often occur de novo, are increasingly associated with disease. Given advances in genetic testing, namely microarray-based comparative genomic hybridization and single nucleotide polymorphism arrays, previously unidentified genotypic aberrations can now be correlated with phenotypic anomalies. The objective of this study was to conduct a nonsystematic literature review to document the role of CNVs as they relate to isolated structural anomalies of the craniofacial, respiratory, renal, and cardiac systems.
METHODS:All full-length articles in the PubMed database through May 2011 that discussed CNVs and isolated structural defects of the craniofacial, respiratory, renal, and cardiac systems were considered. Search terms queried include CNV, copy number variation, array comparative genomic hybridization, birth defects, craniofacial defects, respiratory defects, renal defects, and congenital heart disease. Reports published in languages other than English and articles regarding CNVs and neurocognitive deficits were not considered.
RESULTS:Evidence supports that putatively pathogenic CNVs occur at an increased frequency in patients with isolated structural birth defects and implicate specific regions of the genome. Through CNV de-tection, advances have been made in identifying genes and specific loci that underlie isolated birth defects.
CONCLUSIONS: Although limited studies have been published, the promising evidence reviewed here warrants the continued investiga-tion of CNVs in children with isolated structural birth defects. Patient care and genetic counseling stand to improve through a better under-standing of CNVs and their effect on disease phenotype. Pediatrics 2012;129:755–763
AUTHORS:Abigail E. Southard, BA,aLisa J. Edelmann, PhD,b
and Bruce D. Gelb, MDb,c
aMount Sinai School of Medicine, New York, New York; bDepartment of Genetics & Genomic Sciences; andcDepartment
of Pediatrics, Child Health and Development Institute, Mount Sinai School of Medicine, New York, New York
KEY WORDS
DNA copy number variations, comparative genomic hybridization, congenital abnormalities, craniofacial abnormalities, congenital heart disease
ABBREVIATIONS
aCGH—array comparative genomic hybridization CDH—congenital diaphragmatic hernia CHD—congenital heart disease CLP—cleft lip and/or palate CNVs—copy number variants
FGFR—fibroblast growth factor receptor HLHS—hypoplastic left heart syndrome MDS—Miller-Dieker syndrome
NMLFS—nablus masklike facial syndrome SNP—single nucleotide polymorphism TOF—tetralogy of Fallot
UT—urinary tract
All authors made substantial intellectual contributions to the submitted manuscript; Ms Southard and Dr Gelb were responsible for the intellectual conception of the review; Ms Southard was primarily responsible for acquisition and interpretation of the articles viewed and for drafting the manuscript; and Drs Edelman and Gelb were primarily responsible for revising the manuscript, providing specific feedback, and forfinal approval of the submitted text. www.pediatrics.org/cgi/doi/10.1542/peds.2011-2337
doi:10.1542/peds.2011-2337
Accepted for publication Dec 7, 2011
Address correspondence to Bruce D. Gelb, MD, Child Health and Development Institute, Mount Sinai School of Medicine, One Gustave Levy Place, Box 1040, New York, NY 10029. E-mail: bruce. [email protected]
PEDIATRICS (ISSN Numbers: Print, 0031-4005; Online, 1098-4275).
Copyright © 2012 by the American Academy of Pediatrics
FINANCIAL DISCLOSURE:Dr Gelb received an honorarium for Signature Genomics for speaking at a conference that they sponsored; Ms Southard and Dr Edelmann have indicated they have nofinancial relationships relevant to this article to disclose.
A remarkable discovery enabled by the Human Genome Project is that our ge-nome contains multiple areas of DNA gains and losses, now formally termed copy number variants (CNVs)(See Sup-plemental Information). These regions are DNA larger than 1000 bp that are present with variable copy numbers in comparison with the reference ge-nome.1Many CNVs are inherited
poly-morphisms and account for a significant proportion of the healthy human ge-nome.1There is an interplay between
CNV detection and array platform reso-lution. As platforms with higher resolu-tion are applied to clinical and research settings, additional copy number vari-able regions are detected.2,3 Although
many CNVs do not produce pathologic phenotypes but enable normal pop-ulation variance, larger CNVs, often de novo, are increasingly associated with human disease. To date, numerous pub-lications implicate specific pathogenic CNVs in intellectual disability, autism, and schizophrenia.4,5
One explanation for the seemingly un-predictable effect of CNVs on phenotype is that disease states commonly involve CNVs containing dosage-dependent genes or regulatory sequences.6 Further
in-vestigation into the existence and effect of CNVs is vital to better understand the etiology of microdeletion and micro-duplication syndromes. This is under-scored by the variable expressivity exhibited for specific phenotypes, as well the incomplete penetrance of many genomic disorders.
Although a clear relationship exists between CNVs and disease, and genetic causes have been implicated in both syndromic and nonsyndromic congenital defects, few reports address the role of pathogenic CNVs in structural birth de-fects. This is surprising, considering that many defects are part of the spectrum of features associated with particular microdeletion or microduplication syn-dromes or may occur in a subset of
patients with those syndromes; more-over, a birth defect may not be recog-nized as syndromic until developmental delays manifest.
Birth defects are the leading cause of mortality among infants (aged 1–12 months) in the United States and, in up to 70% of cases, an underlying cause cannot be determined.7To that end, the
purpose of this review is to document the role of CNVs as they relate to isolated structural abnormalities of the cranio-facial, renal, respiratory, and cardio-vascular systems (Table 1). Although this list encompasses most structural birth defects, it is important to note that this review does not discuss neural tube defects or orthopedic abnormalities (including polydactyly and syndactyly), which are also relatively common struc-tural malformations, but have not yet been studied as isolated defects.
CNVs AND ISOLATED CRANIOFACIAL DEFECTS
Isolated Cleft Lip and Palate
Cleft lip and/or palate (CLP) occur in isolation or as features of a broader syndrome. The etiologies of these con-genital craniofacial malformations are complex and most likely result from major and minor genetic factors.8 To
identify candidate genes implicated in CLP, Shi and coworkers9analyzed 725
affected families from 2 Scandinavian populations of shared ancestry for CNVs at 333 candidate genes by using single nucleotide polymorphism (SNP) arrays. They confirmed 7 novel microdeletions, including de novo and familial changes, andSUMO1,TBX1,andTFAP2Aloss CNVs were deemed pathogenic. Interestingly, SUMO1had previously been implicated in CLP10based on a chromosomal
trans-location involvingSUMO1that resulted in haploinsufficiency in a patient and a murineSumo1hypomorphic allele that caused CLP.
To further explore the utility of array comparative genomic hybridization (aCGH) in identifying CNVs implicated in orofacial clefting, 3 separate patient populations were studied: those with Van der Woude syndrome or lower lip pits, those with syndromic CLP, and those with nonsyndromic CLP.11 The
array resolution enabled detection of CNVs∼300 kb or larger. Of 83 syndromic cases, 1 patient harbored a previously unidentified large deletion of the 22q11.21 region, overlapping the DiGeorge syn-drome region. The authors also note that 18 of the syndromic cases had phenotypic features of Van der Woude syndrome, with deletions encompass-ing the interferon regulatory factor 6 gene in 5 of these patients. Of the 104 nonsyndromic cases scanned, a large de novo deletion at chromosome
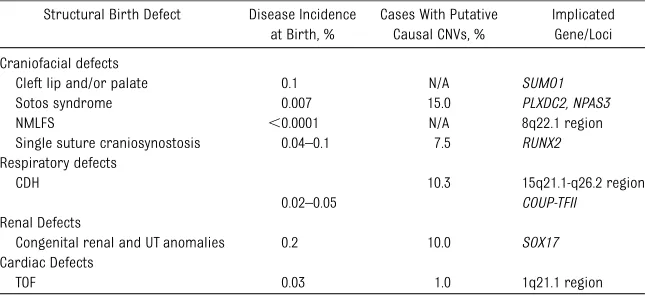
TABLE 1 Clinically Significant Genes and Loci Implicated in the Development of Isolated Structural Birth Defects as Determined by CNV Identification
Structural Birth Defect Disease Incidence at Birth, %
Cases With Putative Causal CNVs, %
Implicated Gene/Loci
Craniofacial defects
Cleft lip and/or palate 0.1 N/A SUMO1
Sotos syndrome 0.007 15.0 PLXDC2, NPAS3
NMLFS ,0.0001 N/A 8q22.1 region
Single suture craniosynostosis 0.04–0.1 7.5 RUNX2
Respiratory defects
CDH 10.3 15q21.1-q26.2 region
0.02–0.05 COUP-TFII
Renal Defects
Congenital renal and UT anomalies 0.2 10.0 SOX17
Cardiac Defects
TOF 0.03 1.0 1q21.1 region
6p25.1-25.2 was identified in 1 indivi-dual. In addition, a deletion at 10q26.11-26.13 was also discovered, which was inherited from a parent who also had CLP. By using gene prioritization soft-ware, estrogen receptor 1 and fi bro-blast growth factor receptor 2 (FGFR2) were identified as likely causative genes from the 6p23.1-25.2 and 10q26.11-26.3 regions, respectively. Given the discovery of such presumed pathogenic aberra-tions, the authors concluded that aCGH is an effective method for isolating can-didate loci in both syndromic and non-syndromic forms of CLP.
Micro/Macrocephaly
Although microcephaly and macro-cephaly most commonly occur in the context of a syndrome, individuals with only head size abnormalities were identified as harboring microdeletions (n= 21) or microduplications (n= 15) of the 1q21.1 region.12 The 1q21.1
re-gion is known to contain multiple CNVs and was previously associated with congenital heart disease,13
develop-mental delay,14,15schizophrenia, and
psychosis.16,17 By using aCGH,
duplica-tions and deleduplica-tions of this region were shown to produce macrocephaly and microcephaly, respectively. Thus, the genes located at 1q21.1 appear to be dosage sensitive and important in de-termining head size.
Embryonic CNS development occurs concurrently with cardiac morpho-genesis, particularly valvular forma-tion, which, in turn, affects systemic blood flow. Hinton et al18 examined
head circumference in infants with hypoplastic left heart syndrome (HLHS) and found a significant reduction in circumferential growth when compar-ing neonates with HLHS with fetuses with HLHS. The authors posit that the restricted head growth may depend on the genetic abnormality and the timing of the alterations in blood flow distri-bution. Hopefully, further work to isolate
the genetic basis of HLHS will elucidate these phenotypic observations.
Single-Suture Craniosynostosis
The genetic aberrations underlying iso-lated craniosynostosis remain largely unknown. To further identify novel can-didate genes responsible for this mal-formation, Mefford and colleagues19
conducted aCGH on 186 patients with single-suture craniosynostosis who had screened negative for mutations in the hot spots of genes associated with craniosynostosis:FDFR1,FGFR2, FGFR3,TWIST1,MSX2, and EFNB1.Based on aCGH analysis, 7.5% of participants possessed a rare deletion or duplica-tion of candidate genes. A pair of cous-ins with craniosynostosis shared a 1-Mb duplication at 6p21 involving RUNX2, which was deemed pathogenic. RUNX2 encodes a pro-osteogenic protein that is necessary for osteoblast differentiation. RUNX2 is expressed in fusing cranial sutures and is repressed by TWIST1. This study revealed that novel CNVs can cause craniosynostosis, suggesting that further evaluation is warranted to de-tect additional candidate loci.
IDENTIFICATION OF CNVs TO ELUCIDATE GENES IMPLICATED IN SYNDROMES CHARACTERIZED BY SPECIFIC CRANIOFACIAL
PHENOTYPES
Sotos Syndrome
Sotos syndrome, characterized by ad-vanced bone age, macrocephaly, sca-phocephaly, language and motor delays, advanced osseous maturation, and en-largement of the central ventricles and extracerebralfluid spaces, results from haploinsufficiency of NSD1 in 60% to 90% of cases but is unexplained in the remainder. Visser and colleagues20
ana-lyzed 26 patients with phenotypic changes suggestive of Sotos syndrome via high-resolution whole-genome SNP arrays. Four potential pathogenic CNVs were detected: deletions at chromosomes
10p12.32-p12.31, 14q13.1, Xq21.1-q21.31, and a duplication at chromosome 15q11.2-q13.1. A potential candidate gene for bone overgrowth (PLXDC2) and intellectual deficits (NPAS3) were con-tained within these regions.
Interestingly, Franco and colleagues21
described 2 patients of different back-ground and ethnicity who had pheno-typic and genopheno-typic changes related to Sotos syndrome. The area surround-ing NSD1, known as the Sotos critical region, contains ∼21 genes and is
flanked by 2 low copy number repeats: Sos-PREP and Sos-DREP. Both patients were shown via aCGH to have duplica-tion CNVs corresponding to the Sotos critical region. These 2 patients, how-ever, possessed phenotypic features opposite from those commonly observed in Sotos syndrome: microcephaly, short stature, delayed bone maturation, and no osseous dysmorphism. Given that oppo-site phenotypic results originated from a duplication compared with a deletion, the authors attributed the microcephaly and short stature to gain-of-function effects.
Nablus Masklike Facial Syndrome
Nablus masklike facial syndrome (NMLFS) is characterized by tight, glis-tening facial skin, upswept frontal hair pattern, ocular hypertelorism, sparse eyebrows, abnormal ear configuration, and blepharophimosis. Given the limited number of patients with NMLFS, the etiology has not been discovered. Two unrelated children with NMLFS were found via aCGH to have 4-Mb micro-deletions in the 8q21.3-8q22.1 region.22
In 2009, aCGH results for 2 additional patients with NMFLS were reported.23In
conjunction with the previously
identi-fied CNVs, the authors narrowed the deleted region in common to 2.78 Mb on chromosome 8q22.1. Refinement of the deletion size is crucial for identi-fying future patients and interpreting the significance of overlapping regions.
Interpreting deletions at 8q22.1 be-comes more complex when considering a case report of a boy with speech delay and autisticlike features who was found via aCGH to have a 1.6-Mb deletion at 8q22.1, but lacked the facial charac-teristics associated with NMFLS.24
No-tably, this smaller deletion only partially overlapped with those previously asso-ciated with NMFLS. Thus, the authors posited that the NMFLS critical region was not deleted in their patient. Addi-tional patient samples must be analyzed to better understand this microdeletion syndrome and the effects of aberrations at 8q22.1 on phenotype.
Isolated Lissencephaly
Miller-Dieker syndrome (MDS), caused by a continuous gene deletion that includes 17p13.3, manifests as severe lissencephaly with dysmorphic facies and additional organ involvement. Al-though isolated lissencephaly results from the PAFAH1B1 deletion, and de-letion ofPAFAH1B1andYWHAEtogether produces MDS, limited research exists regarding phenotypic outcomes of iso-lated YWHAE deletions. To that end, Nagamani et al25 conducted aCGH, by
using custom 44K oligonucleotide arrays, on 8 patients, 5 of whom had known isolatedYWHAE deletions. Their results demonstrated that those with deletions including YWHAE, but not PAFAH1B1, present with growth restriction, cranio-facial abnormalities and structural brain malformations, but with milder neuro-cognitive deficits. Interestingly, 1 subject possessed mosaicism for both YWHAE andPAFAH1B1deletions. This patient
demonstrated a simplified gyral pat-tern, less severe than what is typically observed with classic lissencephaly and, as the authors noted, provided an important opportunity for studying gene dosage effects. Schiff et al26
fur-ther explored the role of the YWHAE deletion on the phenotypic manifes-tations of MDS, examining 4 patients with 17p13.3 deletions distal to the PAFAH1B1region but involvingYWHAE. They confirmed that the distinct phe-notype of MDS is caused by the deletion distal toPAFAH2B1.
Considering these results together, the phenotypic changes associated with MDS appear to stem from aberrations involving YWHAE,and mutations invol-ving PAFAH1B1may be solely respon-sible for the agyria.
CNVs AND CONGENITAL RESPIRATORY DEFECTS
Congenital Diaphragmatic Hernias
Although limited research has exam-ined the relationship between patho-genic CNVs and congenital respiratory defects, work published has focused on congenital diaphragmatic hernias (CDH). Although commonly observed in associ-ation with the autosomal recessive dis-order, Fryns syndrome (Online Mendelian Inheritance in Man #229850), CDH occurs as an isolated defect nearly 50% of the time.27
To identify candidate regions for syndromic and nonsyndromic CDH, Klaassens and colleagues28studied 200
subject with karyotyping, finding 24 (12%) were abnormal. Of these, 14 patients had numeric abnormalities (trisomy 18 or 21) and the remaining 10 had structural defects. Three patients harbored a deletion of part of chromo-some 15q, which was then defined with aCGH. Combining this information with 4 published cases of congenital heart disease (CHD) with 15q defects,29–32the
authors identified a CDH critical region at 15q26.1-q26.2 harboring 4 genes, of
which 2 (NR2F2andCHD2) were deemed interesting candidates for CDH. None of these 4 genes had previously been im-plicated in diaphragm development or CDH.
Slavotinek et al33 confirmed the
im-portance of the 15q26.1-26.2 region in their study of 29 patients with CDH. By using aCGH, the authors ultimately found that 3 of 29 patients with CDH possessed 15q26.1-26.2 deletions. In addition, the authors examined 3 known hotspot regions for CDH: a 8p23.1 de-letion associated with a Fryns syn-drome–like phenotype that includes CDH and deletions at 4p16.3-4pter and 1q41-1q42 in patients with CDH plus ad-ditional cardiac and musculoskeletal anomalies.34The authors concluded that
detection of additional patients with deletions in key CHD regions strength-ened the idea that haploinsufficiency of gene(s) in these areas led to CDH phenotype.
Scott and coworkers35examined CNVs
via aCGH to identify candidate genes for syndromic CDH. Of the 150 CNVs oc-curring in the 26 patients with CDH scanned, 20 changes exclusively exis-ted in patients with CDH and were confirmed via a secondary method. Of the 5 de novo CNVs identified, 1 altered the long arm of chromosome 15. Build-ing on Klaassens et al’s work,28coupled
with the fact that this de novo aberration alteredCOUP-TFII, which had been pre-viously implicated in CDH, the authors refined their area of interest on 15q26 to COUP-TFII and 8 additional genes.36–38
This refinement was further supported by the observation that targeted abla-tion of COUP-TFII in mice resulted in CDH.39Importantly, CDH is not seen in all
humans with a 15q26 deletion involving COUP-TFII. This suggests that although deletion of the gene may play a role in CHD development, additional factors must also underlie the defect.35
Srisupundit and colleagues40designed
oligonucleotide probes targeted to specific genes and loci previously as-sociated with CDH. The authors found genomic imbalances existed in 3 (4%) of 79 patients with CDH with their cus-tomized platform. A deletion at 8p22-p23.2, a duplication of Xq13.1 involving EFNB1, and mosaic trisomy 2 were de-tected in the 3 separate patients. Al-though the authors recognized that the rarity of trisomy 2 makes it an un-likely cause of CDH, their duplication involvingEFNB1(a previously suspected CDH gene) further implicated it as a CHD candidate gene.
CNVs AND CONGENITAL KIDNEY DEFECTS
Congenital Renal and Urinary Tract Anomalies
Attention has turned to mapping novel candidate loci implicated in renal and urinary tract (UT) anomalies, which underlie 50% of end-stage renal disease in children. Although previous studies identified autosomal dominant and recessive loci for renal and UT anom-alies, the etiology remains unknown in most cases.41,42 To that end, patients
with abnormal renal and UT systems have been studied to identify patho-genic CNVs. Weber and colleagues43
performed aCGH on 30 children with various forms of renal and UT malfor-mations plus at least 1 extrarenal problem. They identified 3 patients harboring putatively causal CNVs, cor-responding to an overall prevalence of 10%. Their work implicated 4 chro-mosomal regions in the pathogenesis of renal and UT anomalies: 1q21.1, 2q37. 1-q37.3, 3q23-q25.1, and 7q36.2-q36.3.
Following the identification of a young girl with renal and UT abnormalities, chronic constipation, and mild mental retardation who harbored a de novo pseudodicentric duplication of the long arm of chromosome 8 via aCGH, Gimelli and colleagues44 identified SOX17 as
the most promising candidate among
the 38 genes affected by this duplica-tion because of its known expression throughout the urogenital tract in de-veloping mouse embryos. The authors isolated a point mutation inSOX17 in the initial proband and 7 subsequent patients with nonsyndromic renal and UT malformations. In addition, of the 58 familial cases of vesico-ureteral reflux analyzed, 2 of the families and several sporadic cases harbored the same p. Y259N mutation of the SOX17 gene. Given the significant occurrence of SOX17mutations in patients with renal and UT defects, the authors concluded thatSOX17is involved in controlling the development of the urinary tract and advocated for further exploration into this region and gene mutation.
CNVs AND CONGENITAL HEART DISEASE
Despite recent medical and surgical advances, congenital heart disease (CHD) remains the most deadly birth defect, affecting nearly 1% of all new-borns.45 Although ∼20% of CHD is
attributable to Mendelian disorders (high-penetrance monogenic diseases) or chromosomal aneuploidies, and these etiologies are relatively well understood, the remaining 80% of CHD is attributable to non-Mendelian causes that are poorly understood.46 Non-Mendelian causes of
genetic diseases suggest polygenic in-heritance with a strong environmental component. Specific environmental fac-tors linked to CHD include viral infections (such as maternal rubella), toxic expo-sures (including retinoic acid, dilantin, ethanol, and halogenated hydro-carbons), and maternal diseases (such as diabetes mellitus). Although the rela-tive contribution of genetic and envi-ronmental factors to CHD pathogenesis remains unclear, the best available epi-demiologic data suggest that the genetic factors predominate.47To date, a limited
number of studies have established the relevance of CNVs in the etiology of CHD.
In 2007, Thienpont and colleagues48
es-tablished the ability of aCGH to identify chromosomal aberrations in patients with CHD plus additional extracardiac abnormalities (so-called CHD+). The group scanned 60 patients with CHD+ by using a customized 1-Mb Bacterial Ar-tificial Chromosome array. Novel chro-mosomal aberrations were detected in 18 of 60 patients, corresponding to a prevalence of 30%. Ten of the imbal-ances detected were deemed patho-genic. In addition, 3-deletion CNVs involved genes previously implicated in CHD (NKX2.5, NOTCH1, NSD1, EHMT). Of the 3 deletions identified, 1 altered both NOTCH1 and EHMT. Thus, the authors demonstrated the applicability of aCGH technology in detecting large numbers of clinically significant CNVs. By adding additional subjects to the 60 patients already screened by Thienpont et al,48
Breckpot and colleagues49interrogated
genomic structural variants in 150 patients with syndromic CHD of un-known cause via aCGH at 1-Mb resolu-tion. Forty-three aberrations were detected via this method, of which 26 were deemed to be causative. CNVs were considered causative if they met the following criteria: de novo, segre-gating with phenotype, not described in public databases of healthy individuals, a deletion, and occur in a gene-rich area. Twenty-nine patients who had normal results following 1-Mb array analysis underwent subsequent screening via a 244-K oligo-microarray to increase di-agnostic yield. From this analysis, 75 additional aberrations were detected, but only 2 were considered causative. Ultimately, the authors concluded that higher resolution arrays do not neces-sarily increase diagnostic yield.
Erdogan et al45scanned 105 patients with
occurring de novo. The authors stated that their study was thefirst to dem-onstrate that CNVs occur in syndromic and nonsyndromic CHD. They advo-cated for the routine application of aCGH to screen patients with isolated CHD, citing the capability of revealing syndromic disease before additional symptoms manifest and argued that screening via aCGH is useful for ad-vancing our understanding of patho-genic and benign CNVs.
Richards and colleagues50scanned 20
patients with diverse forms of CHD and additional birth defects who had normal karyotypes with aCGH. Interestingly, the authors selected individuals with CHD but without additional birth defects as their matched control population. The authors identified 5 children with CNVs in their study population, but did not observe any chromosomal changes among the controls. Importantly, the chromosomal abnormalities occurred predominately in children with CHD plus neurologic defects. Given these results, coupled with similarfindings reported by Breckpot et al,49 the authors
sup-ported screening children with CHD and additional birth defects, specifically neurologic defects, via aCGH when a chromosomal anomaly cannot be iden-tified by standard karyotyping.50 In
contrast to Erdogan and colleagues,45
Richards and colleagues50 did not
ad-vocate routinely screening children with isolated CHD via aCGH based on their results. They also cited the number of CNVs of uncertain significance that arise from routine screening via aCGH.
More recently, Greenway et al in-vestigated the relevance of de novo CNVs in isolated tetralogy of Fallot (TOF).51By conducting a genome-wide
survey of 114 patients with isolated TOF and their parents, the group identified 11 de novo CNVs that rarely existed in reference controls. The group then ex-plored a second cohort of patients with TOF for CNVs particularly occurring at
these loci, and discovered CNVs at 1q21.1 in 1% of nonsyndromic, spo-radic cases of TOF. Additional areas of interest were located at 3p25.1, 7p21.3, and 22q11.2. Based on their findings, the authors concluded that at least 10% of sporadic, nonsyndromic cases of TOF can be attributed to novel CNVs and that further investigation of these loci is warranted to elucidate the eti-ology of TOF.
Most recently, Fakhro et al52examined
rare CNVs to assess genes implicated in sporadic heterotaxy. Heterotaxy re-sults from asymmetric development along the left-right body axis, resulting in thoracic and abdominal organ de-fects. By using SNP arrays, the authors genotyped 262 patients with heterotaxy and 991 controls. They observed a sta-tistically significant increase in rare CNVs among the cohort with heterotaxy compared with the controls. By exam-ining conserved genes located within these unique regions functionally by using gene knockdown in the frog Xenopus tropicalis, the authors dem-onstrated 5 genes contained within CNV areas to be significant. Specifically, knockdown of NEK2, ROCK2, TCGBR2, GALNT11, and NUP188 disrupted left-right development; none had been pre-viously implicated in left-right patterning. Such findings further support investi-gating isolated congenital anomalies by examining rare CNVs to uncover un-derlying genetic etiologies.
LIMITATIONS OF aCGH FOR CLINICAL USE IN STRUCTURAL BIRTH DEFECTS
Although aCGH is a powerful technology for isolating genetic aberrations that underlie structural birth defects, there are important limitations to this testing modality. At present, it remains difficult to consistently determine whether a particular variant is, in fact, pathologic. Although public databases that catalog genetic abnormalities with phenotypic
data are available and continue to evolve, they are not comprehensive, particularly for smaller genomic lesions. Thus, it is possible that presumably pathologic CNVs actually represent polymorphisms and do not underlie phenotypic defects. This current pit-fall further underscores the need to account for parental genotype (although incomplete penetrance complicates even this information) and phenotype when interpreting aberration signifi -cance. The remedy for this problem is to continue to catalog genetic information on both healthy and diseased individu-als systematically. By using a standard-ized nomenclature and classification system, our understanding of genotype-phenotype relationships will undoubt-edly improve.
In addition, it is important to recognize the growing belief that although “one hit” may potentiate certain diseases (particularly neuropsychiatric diseases,) a “second hit,” either in the form of a single nucleotide point mutation or CNV, is required for some disease phenotypes to manifest.53This
obser-vation is particularly important when considering the effect of inherited polymorphisms and parental genotype on offspring phenotype. Although it is important to highlight the potentially benign nature of“one-hit”aberrations, most CNVs discussed in this review were found to be de novo and specific for particular birth defects.
CONCLUSIONS
birth defects, including those with isolated ones.
Through the detection of CNVs, signifi -cant advances have been made in iden-tifying genes that contribute to isolated birth defects. Mutations alteringSUMO1, PAFAH1B1,NSD1,andRUNX2appear to contribute directly to the development of isolated craniofacial defects, deletions affectingCOUP-TFIIare associated with CDH, and aberrations altering SOX17 have been linked to congenital renal and UT malformations. In addition, CNV in-terrogation has elucidated how muta-tions at specific loci translate into clinically significant phenotypes. For in-stance, the Seidman group was able to pinpoint 4 genomic regions as areas of particular interest in nonsyndromic, sporadic TOF in this manner.
Genome-wide scanning has enabled researchers and clinicians to detect potentially pathogenic aberrations in children with normal karyotypes. Al-though the optimal approach for detecting CNVs is influx, and the
con-fidence with which particular ones can be declared pathogenic continues to improve, it is already clear that certain CNVs are clinically significant and critical to understanding birth defects. To that end, a multicenter National Institutes of Health study led by Ronald Wapner at Columbia University is cur-rently conducting a blinded prospec-tive comparison of prenatal detection rates of cytogenetic abnormalities by using microarray versus metaphase
molecular karyotyping. Such work will not only examine the accuracy of micro-array analysis in determining cytogenetic diagnosis, but will also provide important information regarding the de novo rate of pathogenic CNVs as well as the eco-nomic feasibility of conducting prenatal screening via microarray on a large scale.54
In addition, the Ledbetter group is cur-rently attempting to create a compre-hensive, standardized CNV atlas by leveraging existing clinical CNV data-bases into a large, streamlined CNV re-search database.55The group also aims
to standardize array design, genotype analysis, and clinical phenotypic data. If successful, this project will provide a standardized CNV catalog and become a useful companion to current, publi-cally available CNV databases, such as the Database of Genomic Variants.55
When managing a patient with struc-tural birth defects from a clinical perspective, the guidelines regarding cytogenetic testing remain nebulous. Although there is a clear diagnostic benefit to conducting microarray testing over G-banding karyotypes for patients with autism spectrum disorder, non-syndromic developmental delay, and multiple congenital anomalies, the utility of completing microarray-based cy-togenetic analysis on patients with isolated structural defects remains un-certain.56Additional studies are needed
to better understand the potential of screening infants with isolated
malformations via microarray tech-nology. If cytogenetic testing via microarray is to be conducted, how-ever, it is vital that clinicians have a firm understanding of testing plat-forms, their technological limitations, and optimal testing conditions. For example, parental studies should be conducted after abnormal results to rule out the presence of inherited chromosomal rearrangement. It is only through proper clinical imple-mentation that CNV detection will function as a successful diagnostic tool.
Although several questions remain unanswered regarding the role of CNVs in birth defects, it is clear from the literature reviewed here that CNVs occur, often at an increased prevalence, in individuals with isolated birth de-fects and that they alter genes that lead to disease phenotypes. Therefore, as cytogenetic diagnosis has currently evolved into the cytogenomic diagnosis of pathogenic CNVs, the clinical indi-cations for which chromosome ab-normalities are considered has also progressed to include specific isolated birth defects. As more specific infor-mation is developed about genotype-phenotype associations, including the breadth of abnormalities associated with specific CNVs and their pene-trance, this class of genetic lesions will factor even more largely in the care of patients with birth defects as well as for counseling their families.
REFERENCES
1. Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome.Nature. 2006;444(7118):444–454 2. Conrad DF, Pinto D, Redon R, et al;
Well-come Trust Case Control Consortium. Ori-gins and functional impact of copy number variation in the human genome. Nature. 2010;464(7289):704–712
3. Park H, Kim JI, Ju YS, et al. Discovery of common Asian copy number variants using
integrated high-resolution array CGH and massively parallel DNA sequencing. Nat Genet. 2010;42(5):400–405
4. Weiss LA, Shen Y, Korn JM, et al; Autism Consortium. Association between micro-deletion and microduplication at 16p11.2 and autism.N Engl J Med. 2008;358(7):667– 675
5. Walsh T, McClellan JM, McCarthy SE, et al. Rare structural variants disrupt multiple genes in
neurodevelopmental pathways in schizophre-nia.Science. 2008;320(5875):539–543 6. Stankiewicz P, Lupski JR. Structural
varia-tion in the human genome and its role in disease.Annu Rev Med. 2010;61:437–455 7. Lu XY, Phung MT, Shaw CA, et al. Genomic
8. Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts.Hum Mol Genet. 2004;13(spec no 1):R73–R81 9. Shi M, Mostowska A, Jugessur A, et al.
Identification of microdeletions in candi-date genes for cleft lip and/or palate.Birth Defects Res A Clin Mol Teratol. 2009;85(1): 42–51
10. Alkuraya FS, Saadi I, Lund JJ, Turbe-Doan A, Morton CC, Maas RL. SUMO1
haploinsuf-ficiency leads to cleft lip and palate. Sci-ence. 2006;313(5794):1751
11. Osoegawa K, Vessere GM, Utami KH, et al. Identification of novel candidate genes as-sociated with cleft lip and palate using array comparative genomic hybridisation.
J Med Genet. 2008;45(2):81–86
12. Brunetti-Pierri N, Berg JS, Scaglia F, et al. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat Genet. 2008; 40(12):1466–1471
13. Christiansen J, Dyck JD, Elyas BG, et al. Chromosome 1q21.1 contiguous gene de-letion is associated with congenital heart disease.Circ Res. 2004;94(11):1429–1435 14. Sharp AJ, Hansen S, Selzer RR, et al.
Dis-covery of previously unidentified genomic disorders from the duplication architec-ture of the human genome.Nat Genet. 2006; 38(9):1038–1042
15. Shaffer LG, Kashork CD, Saleki R, et al. Targeted genomic microarray analysis for identification of chromosome abnormali-ties in 1500 consecutive clinical cases.
J Pediatr. 2006;149(1):98–102
16. Stefansson H, Rujescu D, Cichon S, et al; GROUP. Large recurrent microdeletions as-sociated with schizophrenia.Nature. 2008; 455(7210):232–236
17. Stone JL; International Schizophrenia Con-sortium. Rare chromosomal deletions and duplications increase risk of schizophrenia.
Nature. 2008;455(7210):237–241
18. Hinton RB, Andelfinger G, Sekar P, et al. Prenatal head growth and white matter injury in hypoplastic left heart syndrome.
Pediatr Res. 2008;64(4):364–369
19. Mefford HC, Shafer N, Antonacci F, et al. Copy number variation analysis in single-suture craniosynostosis: multiple rare variants including RUNX2 duplication in two cousins with metopic craniosynos-tosis. Am J Med Genet A. 2010;152A(9): 2203–2210
20. Visser R, Gijsbers A, Ruivenkamp C, et al. Genome-wide SNP array analysis in patients with features of Sotos syndrome.Horm Res Paediatr. 2010;73(4):265–274
21. Franco LM, de Ravel T, Graham BH, et al. A syndrome of short stature, microcephaly and speech delay is associated with duplications reciprocal to the common Sotos syndrome deletion.Eur J Hum Genet. 2010;18(2):258–261
22. Shieh JT, Aradhya S, Novelli A, et al. Nablus mask-like facial syndrome is caused by a microdeletion of 8q detected by array-based comparative genomic hybridization.
Am J Med Genet A. 2006;140(12):1267–1273 23. Raas-Rothschild A, Dijkhuizen T, Sikkema-Raddatz B, et al. The 8q22.1 microdeletion syndrome or Nablus mask-like facial syn-drome: report on two patients and review of the literature.Eur J Med Genet. 2009;52 (2-3):140–144
24. Jain S, Yang P, Farrell SA. A case of 8q22.1 microdeletion without the Nablus mask-like facial syndrome phenotype. Eur J Med Genet. 2010;53(2):108–110
25. Nagamani SC, Zhang F, Shchelochkov OA, et al. Microdeletions including YWHAE in the Miller-Dieker syndrome region on chromosome 17p13.3 result in facial dys-morphisms, growth restriction, and cogni-tive impairment.J Med Genet. 2009;46(12): 825–833
26. Schiff M, Delahaye A, Andrieux J, et al. Further delineation of the 17p13.3 micro-deletion involving YWHAE but distal to PAFAH1B1: four additional patients.Eur J Med Genet. 2010;53(5):303–308
27. Tibboel D, Gaag AV. Etiologic and genetic factors in congenital diaphragmatic hernia.
Clin Perinatol. 1996;23(4):689–699 28. Klaassens M, van Dooren M, Eussen HJ,
et al. Congenital diaphragmatic hernia and chromosome 15q26: determination of a candidate region by use offluorescent in situ hybridization and array-based com-parative genomic hybridization.Am J Hum Genet. 2005;76(5):877–882
29. de Jong G, Rossouw RA, Retief AE. Ring chromosome 15 in a patient with features of Fryns’syndrome.J Med Genet. 1989;26 (7):469–470
30. Rosenberg C, Blakemore KJ, Kearns WG, et al. Analysis of reciprocal translocations by chromosome painting: applications and limitations of the technique. Am J Hum Genet. 1992;50(4):700–705
31. Chen CP, Lee CC, Pan CW, Kir TY, Chen BF. Partial trisomy 8q and partial monosomy 15q associated with congenital hydro-cephalus, diaphragmatic hernia, urinary tract anomalies, congenital heart defect and kyphoscoliosis. Prenat Diagn. 1998;18 (12):1289–1293
32. Schlembach D, Zenker M, Trautmann U, Ulmer R, Beinder E. Deletion 15q24-26 in
prenatally detected diaphragmatic hernia: increasing evidence of a candidate region for diaphragmatic development. Prenat Diagn. 2001;21(4):289–292
33. Slavotinek AM, MoshrefiA, Davis R, et al. Array comparative genomic hybridization in patients with congenital diaphragmatic hernia: mapping of four CDH-critical re-gions and sequencing of candidate genes at 15q26.1-15q26.2.Eur J Hum Genet. 2006; 14(9):999–1008
34. Kantarci S, Casavant D, Prada C, et al. Findings from aCGH in patients with con-genital diaphragmatic hernia (CDH): a pos-sible locus for Fryns syndrome.Am J Med Genet A. 2006;140(1):17–23
35. Scott DA, Klaassens M, Holder AM, et al. Genome-wide oligonucleotide-based array comparative genome hybridization analysis of non-isolated congenital diaphragmatic hernia.Hum Mol Genet. 2007;16(4):424–430 36. Castiglia L, Fichera M, Romano C, et al. Narrowing the candidate region for con-genital diaphragmatic hernia in chromo-some 15q26: contradictory results. Am J Hum Genet. 2005;77(5):892–894, author re-ply 894–895
37. Major D, Cadenas M, Fournier L, Leclerc S, Lefebvre M, Cloutier R. Retinol status of newborn infants with congenital diaphrag-matic hernia. Pediatr Surg Int. 1998;13(8): 547–549
38. Greer JJ, Babiuk RP, Thebaud B. Etiology of congenital diaphragmatic hernia: the reti-noid hypothesis. Pediatr Res. 2003;53(5): 726–730
39. You LR, Takamoto N, Yu CT, et al. Mouse lacking COUP-TFII as an animal model of Bochdalek-type congenital diaphragmatic hernia.Proc Natl Acad Sci U S A. 2005;102 (45):16351–16356
40. Srisupundit K, Brady PD, Devriendt K, et al. Targeted array comparative genomic hybridisation (array CGH) identifies ge-nomic imbalances associated with iso-lated congenital diaphragmatic hernia (CDH). Prenat Diagn. 2010;30(12-13):1198– 1206
41. Ashraf S, Hoskins BE, Chaib H, et al. Map-ping of a new locus for congenital anom-alies of the kidney and urinary tract on chromosome 8q24. Nephrol Dial Trans-plant. 2010;25(5):1496–1501
42. Nakayama M, Nozu K, Goto Y, et al. HNF1B alterations associated with congenital anomalies of the kidney and urinary tract.
Pediatr Nephrol. 2010;25(6):1073–1079 43. Weber S, Landwehr C, Renkert M, et al.
comparative genomic hybridization. Neph-rol Dial Transplant. 2011;26(1):136–143 44. Gimelli S, Caridi G, Beri S, et al. Mutations
in SOX17 are associated with congenital anomalies of the kidney and the urinary tract.Hum Mutat. 2010;31(12):1352–1359 45. Erdogan F, Larsen LA, Zhang L, et al. High
frequency of submicroscopic genomic aber-rations detected by tiling path array com-parative genome hybridisation in patients with isolated congenital heart disease.J Med Genet. 2008;45(11):704–709
46. Bentham J, Bhattacharya S. Genetic mecha-nisms controlling cardiovascular develop-ment.Ann N Y Acad Sci. 2008;1123:10–19 47. Oyen N, Poulsen G, Wohlfahrt J, Boyd HA,
Jensen PK, Melbye M. Recurrence of discordant congenital heart defects in families.Circ Cardiovasc Genet. 2010;3(2): 122–128
48. Thienpont B, Mertens L, de Ravel T, et al. Submicroscopic chromosomal imbalances
detected by array-CGH are a frequent cause of congenital heart defects in selected patients. Eur Heart J. 2007;28(22):2778– 2784
49. Breckpot J, Thienpont B, Peeters H, et al. Array comparative genomic hybridization as a diagnostic tool for syndromic heart defects. J Pediatr. 2010;156(5):810–817, 817.e1–817.e4
50. Richards AA, Santos LJ, Nichols HA, et al. Cryptic chromosomal abnormalities iden-tified in children with congenital heart disease.Pediatr Res. 2008;64(4):358–363 51. Greenway SC, Pereira AC, Lin JC, et al. De
novo copy number variants identify new genes and loci in isolated sporadic tetral-ogy of Fallot.Nat Genet. 2009;41(8):931–935 52. Fakhro KA, Choi M, Ware SM, et al. Rare copy number variations in congenital heart disease patients identify unique genes in left-right patterning.Proc Natl Acad Sci U S A. 2011;108(7):2915–2920
53. Girirajan S, Rosenfeld JA, Cooper GM, et al. A recurrent 16p12.1 microdeletion sup-ports a two-hit model for severe de-velopmental delay. Nat Genet. 2010;42(3): 203–209
54. Wapner R. Prenatal cytogenetic diagnosis by array-based copy number analysis (microarray). Available at: http://clinicaltrials. gov/ct2/show/NCT01279733. Accessed April 1, 2011
55. Riggs E, Church D, Hanson K, et al. Towards an evidence-based process for the clinical interpretation of copy number variation [published online ahead of print November 19, 2011].Clin Genet. doi: 10.1111/j.1399-0004.2011.01818.x
56. Manning M, Hudgins L; Professional Prac-tice and Guidelines Committee. Array-based technology and recommendations for uti-lization in medical genetics practice for detection of chromosomal abnormalities.
Genet Med. 2010;12(11):742–745
THE VIEW FROM THE INSIDE:For a number of years, our kids have had petfish. Thefish have been housed in aquariums of various sizes; from those that hold a gallon or so to those that hold 40 or more. We had never really paid too much attention to the size of the aquarium but did notice that many of ourfish seemed more aggressive than expected. Moreover, the life expectancy of ourfish pop-ulation has been less than anticipated despite paying close attention to the water temperature, pH, and feeding practices. According to an article inThe New York Times(Science: December 26, 2011), part of the problem may be the size of the fish tanks. Evidently, freshwaterfish kept in small tanks are substantially more aggressive than those kept in larger ones. Researchers studied young common freshwaterfish in two experiments. In one,fish were added to a tank while in the other, threefish were placed in larger tanks with more complex ecosystems. The behaviors were recorded for two hours after feeding. Thefish subject to over-crowding became aggressive while thefish placed in the larger tanks demon-strated far less aggressive behavior. Researchers suspect that in their natural environment,fish have places to hide and are not in constant contact with each other. While other studies have confirmed that overcrowding changes animal behavior,fish owners are unlikely to purchase 100 gallon tanks to keep theirfish content. According to the article, even the Humane Society opted not to wade into the issue. On a more practical aspect, overcrowding can affect the health of afish population. This is most important for commercialfish farms. While some species can tolerate dense living conditions, most do not. Overcrowding can lead to poor fish health and the rapid spread of disease. In Scotland, the farmed salmon are so riddled with sea lice that the government may ban coastal farms. We are not planning to eat any of our petfish. Still, we avoid buying too many and are content with only twofish for every 20 gallons of water.
DOI: 10.1542/peds.2011-2337 originally published online March 19, 2012;
2012;129;755
Pediatrics
Abigail E. Southard, Lisa J. Edelmann and Bruce D. Gelb
Role of Copy Number Variants in Structural Birth Defects
Services
Updated Information &
http://pediatrics.aappublications.org/content/129/4/755 including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/129/4/755#BIBL This article cites 52 articles, 11 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/genetics_sub Genetics
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
DOI: 10.1542/peds.2011-2337 originally published online March 19, 2012;
http://pediatrics.aappublications.org/content/129/4/755
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
http://pediatrics.aappublications.org/content/suppl/2012/03/14/peds.2011-2337.DCSupplemental Data Supplement at:
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.