Methyl β N (3 nitrophenylmethylene)dithiocarbazate
Full text
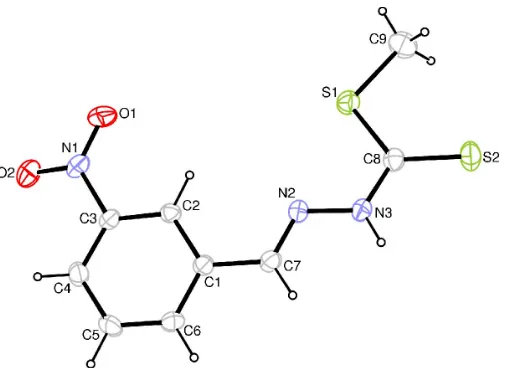
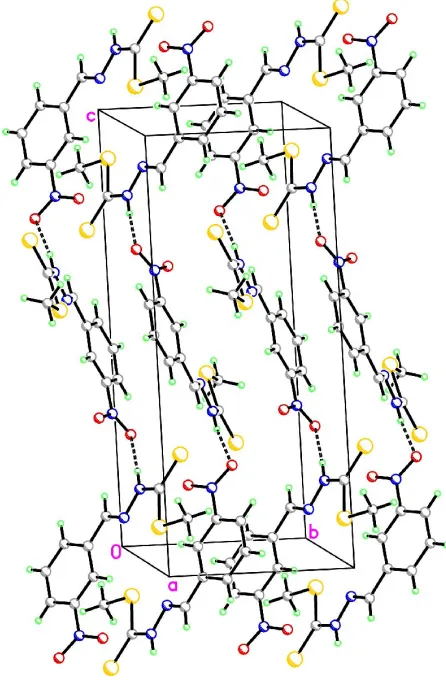
Figure
Related documents
limitation de l’égalité d’accès aux positions des autorités publiques rémunératrices et stratégiquement importantes, favorisant ainsi le népotisme et
Fur samtliche genannten Veroffentlichungen sind in der Cro nos-Datenbank van Eurostat Zeitreihen verfugbar, und sie konnen auf Wunsch in Form van Ustenausdrucken oder
PRODOTTI TRANSFORMATI DI CERE.ALI E Dl RISO SCI AOPPI F PROOOTT I DEL SETT ORE DEL LO /UCCHERO. TUTTO
The Uruguay Round - General Agreement on Tariffs and Trade (GATT).. The Community and the United States cooperated to launch the Uruguay Round of
Los aportes del Análisis Crítico del Discurso (ACD), son útiles para este estudio por cuanto que se trata de un campo interdisciplinario que se ocupa de las relaciones entre
In the second stage, we evaluate the preference for linear hedging by measuring the extent of total notional hedged through linear contracts over the total notional amount of
Now, if we look at the migration pattern according to the duration of residence, we find that for majority of the states, as of 2001, a large volume of migrants came to their
We introduce ALTO , an interactive framework that combines both active learning selections with topic model overviews to both help users induce a label set and assign labels
