metal-organic papers
m496
Du and Hu K+C7H3N2O6 doi:10.1107/S1600536806004417 Acta Cryst.(2006). E62, m496–m497 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Potassium 3,5-dinitrobenzoate
Yan-Ping Duaand Man-Cheng Hub*
a
School of Environment and Chemical Engineering, Xi’an University of Engineering Science and Technology, Xi’an 710068, People’s Republic of China, andbSchool of
Chemistry and Materials Science, ShaanXi Nomal University, Xi’an 710062, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 298 K
Mean(C–C) = 0.004 A˚
Rfactor = 0.036
wRfactor = 0.129
Data-to-parameter ratio = 10.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 2 December 2005 Accepted 6 February 2006
#2006 International Union of Crystallography All rights reserved
The title compound, K+C7H3N2O6
, crystallized from aqueous solution, is not isostructural with sodium 3,5-dinitro-benzoate. Both the anion and the cation lie on a twofold axis. The K+cation is surrounded by eight O atoms and the crystal structure is stabilized by–interactions.
Comment
During work on crystallization, potassium 3,5-dinitro-benzoate, (I), was obtained as single crystals by evaporation of an aqueous solution at room temperature, and its structure determined. Probably due to the difference in the radii of K+ and Na+cations, compound (I) is not isostructural with the Na
salt, sodium 3,5-dinitrobenzoate, whose structure has been reported in the space groupP3121 (Joneset al., 2005).
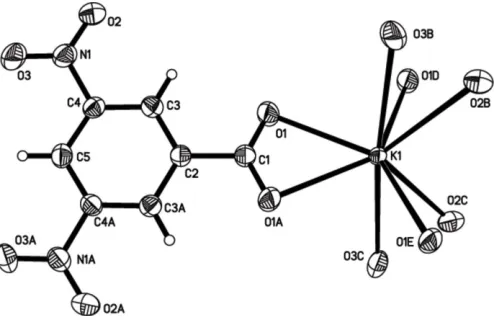
[image:1.610.281.383.358.445.2] [image:1.610.210.457.526.692.2]The structure of (I) is shown in Fig. 1. The K+cation and 3,5-dinitrobenzoate anion lie on a twofold axis. All atoms of the 3,5-dinitrobenzoate anion are almost coplanar, as observed in the corresponding Na+salt. The crystal structure is essentially
Figure 1
The ions and complete coordination of the K+ cation of the title compound. Displacement ellipsoids are drawn at the 30% probability level. Atoms with the suffixes A, B, C, D and E are at the symmetry positions (1x,y,1
2z), ( 1 2x,
1 2+y,
1 2z), (
1 2+x,
1
2+y,z+ 1), (x+ 1,
y+ 1,z) and (x,y+ 1,1
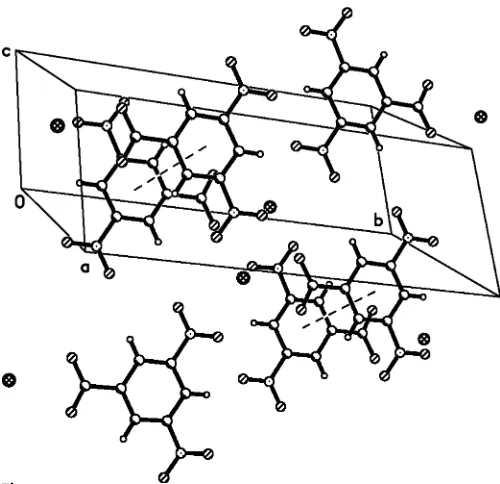
ionic, and each K+ cation interacts with eight O atoms belonging to nitro and carboxylate groups of four symmetry-related anions (Table 1). The crystal packing is stabilized by intermolecular – interactions (4.05 A˚ ) between aromatic rings (Fig. 2), forming a layer structure.
Experimental
3,5-Dinitrobenzoic acid (14.1 mmol) was added to a solution of KOH (14.1 mmol) in water (10 ml). The mixture was stirred for 2 h at 298 K. The solution was then filtered under reduced pressure and set aside for crystallization. After 7 d, pure colourless crystals of (I) were collected from the filtered solution.
Crystal data
K+C 7H3N2O6
Mr= 250.21
Monoclinic,C2=c a= 10.150 (3) A˚
b= 17.715 (4) A˚
c= 7.066 (2) A˚
= 133.075 (3)
V= 928.1 (4) A˚3
Z= 4
Dx= 1.791 Mg m
3
MoKradiation Cell parameters from 1239
reflections
= 3.0–27.6 = 0.59 mm1
T= 298 (2) K Block, colourless 0.450.320.25 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2002)
Tmin= 0.777,Tmax= 0.867
2333 measured reflections
809 independent reflections 667 reflections withI> 2(I)
Rint= 0.019 max= 25.0
h=12!11
k=20!16
l=8!8
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.036
wR(F2) = 0.129
S= 1.00 809 reflections 75 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0885P)2
+ 0.7104P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.21 e A˚ 3
min=0.22 e A˚ 3
Table 1
Selected bond lengths (A˚ ).
K1—O1i
2.636 (2)
K1—O1 2.801 (2)
K1—O3ii
3.021 (3) K1—O2ii
3.034 (3)
Symmetry codes: (i)xþ1;yþ1;z; (ii)xþ1 2;yþ
1 2;zþ1.
H atoms were placed in calculated positions and refined as riding, with C—H distances constrained to 0.93 A˚ and with Uiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 2001); cell refinement:SAINT (Bruker, 2001); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics: SHELXTL(Sheldrick, 2001); software used to prepare material for publication:SHELXTL.
Financial support from the National Natural Science Foundation of China (No. 20471035) is acknowledged.
References
Bruker (2001).SMARTandSAINT. Bruker AXS Inc., Madison, Wisconsin, USA.
Jones, H. P., Gillon, A. L. & Davey, R. J. (2005).Acta Cryst.E61, m1131– m1132.
Sheldrick, G. M. (1997). SHELXS97 and SHELXL97, University of Go¨ttingen, Germany.
Sheldrick, G. M. (2001).SHELXTL. Version 5. Bruker AXS Inc., Madison, Wisconsin, USA.
[image:2.610.314.564.71.313.2]Sheldrick, G. M. (2002).SADABS. University of Go¨ttingen, Germany.
Figure 2
supporting information
sup-1
Acta Cryst. (2006). E62, m496–m497
supporting information
Acta Cryst. (2006). E62, m496–m497 [https://doi.org/10.1107/S1600536806004417]
Potassium 3,5-dinitrobenzoate
Yan-Ping Du and Man-Cheng Hu
Potassium 3,5-dinitrobenzoate
Crystal data
K+·C7H3N2O6−
Mr = 250.21
Monoclinic, C2/c
Hall symbol: -C 2yc
a = 10.150 (3) Å
b = 17.715 (4) Å
c = 7.066 (2) Å
β = 133.075 (3)°
V = 928.1 (4) Å3
Z = 4
F(000) = 504
Dx = 1.791 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1239 reflections
θ = 3.0–27.6°
µ = 0.59 mm−1
T = 298 K Block, colourless 0.45 × 0.32 × 0.25 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 2002)
Tmin = 0.777, Tmax = 0.867
2333 measured reflections 809 independent reflections 667 reflections with I > 2σ(I)
Rint = 0.019
θmax = 25.0°, θmin = 2.3°
h = −12→11
k = −20→16
l = −8→8
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.036
wR(F2) = 0.129
S = 1.00 809 reflections 75 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0885P)2 + 0.7104P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.21 e Å−3 Δρmin = −0.22 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
K1 0.5000 0.54552 (4) 0.2500 0.0465 (4)
N1 0.3593 (4) 0.12417 (16) −0.2004 (5) 0.0606 (7)
O1 0.4719 (4) 0.40028 (12) 0.0686 (5) 0.0686 (7)
O2 0.2967 (4) 0.16050 (15) −0.3929 (5) 0.0737 (7)
O3 0.3584 (4) 0.05531 (14) −0.1966 (6) 0.0892 (9)
C1 0.5000 0.3686 (2) 0.2500 0.0465 (9)
C2 0.5000 0.2828 (2) 0.2500 0.0421 (8)
C3 0.4371 (4) 0.24349 (17) 0.0323 (5) 0.0455 (7)
H3 0.3964 0.2693 −0.1140 0.055*
C4 0.4355 (4) 0.16532 (16) 0.0351 (5) 0.0461 (7)
C5 0.5000 0.1243 (2) 0.2500 0.0506 (10)
H5 0.5000 0.0718 0.2500 0.061*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
K1 0.0830 (7) 0.0307 (5) 0.0366 (5) 0.000 0.0451 (5) 0.000
N1 0.0623 (16) 0.0551 (17) 0.0526 (16) 0.0056 (12) 0.0346 (13) −0.0065 (12)
O1 0.119 (2) 0.0469 (12) 0.0678 (14) 0.0015 (11) 0.0745 (15) 0.0050 (10)
O2 0.0925 (18) 0.0736 (16) 0.0507 (14) −0.0063 (12) 0.0472 (14) −0.0066 (11)
O3 0.107 (2) 0.0501 (15) 0.0717 (17) 0.0125 (13) 0.0463 (16) −0.0121 (11)
C1 0.058 (2) 0.043 (2) 0.049 (2) 0.000 0.041 (2) 0.000
C2 0.0463 (19) 0.041 (2) 0.045 (2) 0.000 0.0338 (17) 0.000
C3 0.0475 (14) 0.0479 (16) 0.0457 (16) 0.0039 (11) 0.0337 (13) 0.0025 (12)
C4 0.0449 (14) 0.0469 (16) 0.0434 (16) 0.0010 (11) 0.0288 (13) −0.0045 (11)
C5 0.048 (2) 0.042 (2) 0.054 (2) 0.000 0.032 (2) 0.000
Geometric parameters (Å, º)
K1—O1i 2.636 (2) N1—K1vi 3.313 (3)
K1—O1ii 2.636 (2) O1—C1 1.241 (3)
K1—O1 2.801 (2) O1—K1i 2.636 (2)
K1—O1iii 2.801 (2) O2—K1vi 3.034 (3)
K1—O3iv 3.021 (3) O3—K1vi 3.021 (3)
K1—O3v 3.021 (3) C1—O1iii 1.241 (3)
K1—O2iv 3.034 (3) C1—C2 1.521 (6)
K1—O2v 3.034 (3) C2—C3 1.385 (3)
K1—C1 3.134 (4) C2—C3iii 1.385 (3)
K1—N1iv 3.313 (3) C3—C4 1.385 (4)
supporting information
sup-3
Acta Cryst. (2006). E62, m496–m497
K1—K1i 3.8839 (11) C4—C5 1.380 (4)
N1—O2 1.220 (4) C5—C4iii 1.380 (4)
N1—O3 1.220 (4) C5—H5 0.9300
N1—C4 1.463 (4)
O1i—K1—O1ii 137.28 (10) O1iii—K1—N1v 127.54 (7)
O1i—K1—O1 88.87 (7) O3iv—K1—N1v 151.83 (7)
O1ii—K1—O1 133.53 (7) O3v—K1—N1v 21.58 (7)
O1i—K1—O1iii 133.53 (7) O2iv—K1—N1v 111.42 (8)
O1ii—K1—O1iii 88.87 (7) O2v—K1—N1v 21.58 (7)
O1—K1—O1iii 46.55 (9) C1—K1—N1v 114.87 (5)
O1i—K1—O3iv 110.34 (10) N1iv—K1—N1v 130.26 (10)
O1ii—K1—O3iv 67.08 (9) O1i—K1—K1i 46.14 (5)
O1—K1—O3iv 107.54 (8) O1ii—K1—K1i 174.50 (6)
O1iii—K1—O3iv 78.70 (8) O1—K1—K1i 42.74 (4)
O1i—K1—O3v 67.08 (9) O1iii—K1—K1i 88.37 (5)
O1ii—K1—O3v 110.34 (10) O3iv—K1—K1i 116.96 (6)
O1—K1—O3v 78.70 (8) O3v—K1—K1i 66.07 (7)
O1iii—K1—O3v 107.54 (8) O2iv—K1—K1i 114.75 (5)
O3iv—K1—O3v 173.42 (10) O2v—K1—K1i 97.98 (5)
O1i—K1—O2iv 80.84 (8) C1—K1—K1i 65.46 (2)
O1ii—K1—O2iv 70.74 (8) N1iv—K1—K1i 123.86 (5)
O1—K1—O2iv 136.92 (8) N1v—K1—K1i 78.01 (6)
O1iii—K1—O2iv 120.21 (7) O2—N1—O3 123.1 (3)
O3iv—K1—O2iv 41.50 (7) O2—N1—C4 118.3 (3)
O3v—K1—O2iv 132.21 (8) O3—N1—C4 118.6 (3)
O1i—K1—O2v 70.74 (8) O2—N1—K1vi 66.21 (16)
O1ii—K1—O2v 80.84 (8) O3—N1—K1vi 65.59 (17)
O1—K1—O2v 120.21 (7) C4—N1—K1vi 147.81 (18)
O1iii—K1—O2v 136.92 (8) C1—O1—K1i 164.64 (19)
O3iv—K1—O2v 132.21 (8) C1—O1—K1 93.6 (2)
O3v—K1—O2v 41.50 (7) K1i—O1—K1 91.13 (7)
O2iv—K1—O2v 95.66 (11) N1—O2—K1vi 92.21 (19)
O1i—K1—C1 111.36 (5) N1—O3—K1vi 92.83 (19)
O1ii—K1—C1 111.36 (5) O1—C1—O1iii 126.3 (4)
O1—K1—C1 23.27 (4) O1—C1—C2 116.87 (19)
O1iii—K1—C1 23.27 (4) O1iii—C1—C2 116.87 (19)
O3iv—K1—C1 93.29 (5) O1—C1—K1 63.13 (19)
O3v—K1—C1 93.29 (5) O1iii—C1—K1 63.13 (19)
O2iv—K1—C1 132.17 (6) C2—C1—K1 180.0
O2v—K1—C1 132.17 (6) C3—C2—C3iii 119.6 (4)
O1i—K1—N1iv 99.99 (8) C3—C2—C1 120.18 (18)
O1ii—K1—N1iv 61.33 (8) C3iii—C2—C1 120.18 (18)
O1—K1—N1iv 127.54 (7) C2—C3—C4 119.2 (3)
O1iii—K1—N1iv 99.40 (8) C2—C3—H3 120.4
O3iv—K1—N1iv 21.58 (7) C4—C3—H3 120.4
O3v—K1—N1iv 151.83 (7) C5—C4—C3 122.8 (3)
O2v—K1—N1iv 111.42 (8) C3—C4—N1 118.9 (3)
C1—K1—N1iv 114.87 (5) C4—C5—C4iii 116.4 (4)
O1i—K1—N1v 61.33 (8) C4—C5—H5 121.8
O1ii—K1—N1v 99.99 (8) C4iii—C5—H5 121.8
O1—K1—N1v 99.40 (8)