organic papers
Acta Cryst.(2007). E63, o15–o16 doi:10.1107/S1600536806050690 Buttet al. C
9H5I2NO
o15
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
5,7-Diiodoquinolin-8-ol
M. Saeed Butt,aZareen Akhter,a* Michael Bolteband Humaira M. Siddiqia
aDepartment of Chemistry, Quaid-I-Azam
University, Islamabad 45320, Pakistan, and
b
Institut fu¨r Anorganische Chemie, J.-W.-Goethe-Universita¨t Frankfurt, Max-von-Laue-Str. 7, 60438 Frankfurt/Main, Germany
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 173 K
Mean(C–C) = 0.006 A˚
Rfactor = 0.031
wRfactor = 0.077
Data-to-parameter ratio = 18.7
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 31 August 2006 Accepted 24 November 2006
#2007 International Union of Crystallography All rights reserved
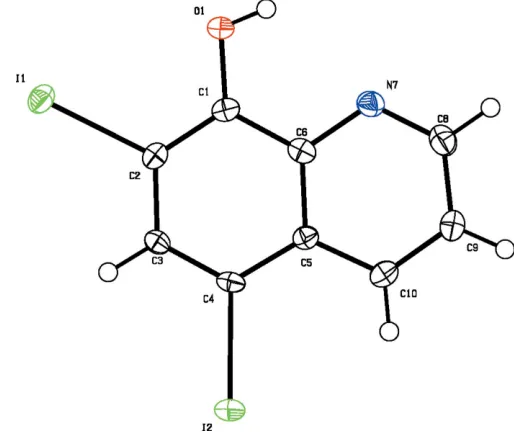
The title compound, C9H5I2NO, features an almost planar
molecule. Geometric parameters are in the usual ranges. The crystal packing shows that two hydrogen-bonded molecules are related by a twofold rotation axis.
Comment
5,7-Diiodoquinolin-8-ol, (I), is of therapeutic interest.
Quinolin-8-ol and its derivatives have antibacterial activity and form chelate complexes with divalent metal ions (Rohde
et al., 1976). The importance of metallic oxinates in analytical chemistry is also well known. Oxine and its derivatives have found extensive application as analytical reagents (Guerreiro
et al., 2002) in absorption spectroscopy, fluorimetry, extraction with solvents and chromatography. In an attempt to prepare the manganese complex of 5,7-diiodo-8-hydroxyquinoline we obtained crystals of the ligand, (I). A perspective view of the title compound is shown in Fig. 1. Bond lengths and angles can be regarded as normal (Cambridge Structural Database; Version 5.27, November 2005, updated August 2006; Allen, 2002). The molecule is essentially planar (r.m.s. deviation for all non-H atoms is 0.058 A˚ ). The packing diagram (Fig. 2) reveals that two molecules, which are related by a twofold rotation axis, are connected by an O—H N hydrogen bonds (Table 1).
Experimental
An attempt was made to complex 5,7-diiodoquinolin-8-ol with manganese. Unfortunately, ligand (I) crystallized out in an attempt to recrystallize the complex (m.p. 471–473 K) from toluene.
Crystal data
C9H5I2NO
Mr= 396.94
Monoclinic,P2=c a= 14.1699 (13) A˚
b= 4.2915 (4) A˚
c= 16.1565 (13) A˚
= 96.801 (7) V= 975.57 (15) A˚3
Z= 4
Dx= 2.703 Mg m
3
MoKradiation
= 6.40 mm1
T= 173 (2) K Rod, yellow
Data collection
Stoe IPDS-II two-circle diffractometer
!scans
Absorption correction: multi-scan (MULABS; Spek, 2003; Blessing, 1995)
Tmin= 0.318,Tmax= 0.527
7396 measured reflections 2244 independent reflections 2163 reflections withI> 2(I)
Rint= 0.046
max= 27.5
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.031
wR(F2) = 0.077
S= 1.17 2244 reflections 120 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0385P)2
+ 2.4632P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 1.25 e A˚
3
min=1.23 e A˚
3
Extinction correction:SHELXL97
[image:2.610.307.564.73.289.2]Extinction coefficient: 0.0083 (5)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O1—H1 N7i
0.84 1.98 2.757 (5) 154
Symmetry code: (i)xþ1;y;zþ3 2.
H atoms were found in a difference Fourier map but they were constrained, with C—H = 0.95 A˚ and O—H = 0.84 A˚, and were refined using a riding model;Uiso(H) = 1.2Ueq(C,O). The hydroxyl
group was allowed to rotate but not to tip. The highest peak in the final difference Fourier map is located at 0.77 A˚ from I1 and the deepest hole is at 0.98 A˚ from I2.
Data collection: X-AREA (Stoe & Cie, 2001); cell refinement:
X-AREA; data reduction: X-AREA; program(s) used to solve structure: SHELXS86(Sheldrick, 1990); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
PLATON(Spek, 2003) andXPinSHELXTL-Plus(Sheldrick, 1991); software used to prepare material for publication:SHELXL97and
PLATON.
The authors are grateful to the Department of Chemistry, Quaid-I-Azam University, Islamabad, Pakistan.
References
Allen, F. H. (2002).Acta Cryst.B58, 380–388. Blessing, R. H. (1995).Acta Cryst.A51, 33–38.
Guerreiro, C. T. R., Ribeiro, C. A. & Crespi, M. S. (2002).J. Therm. Anal. Calorim.70, 437–445.
Rohde, W., Mikelens, P., Jackson, J., Blackman, J., Whitcher, J. & Levinson, W. (1976).Antimicrob. Agents Chemother.10, 234–240.
Sheldrick, G. M. (1990).Acta Cryst.A46, 467–473.
Sheldrick, G. M. (1991).SHELXTL-Plus. Release 4.1. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
[image:2.610.315.563.338.476.2]Stoe & Cie (2001).X-AREA. Stoe & Cie, Darmstadt, Germany.
Figure 1
Molecular structure of (I). Displacement ellipsoids are drawn at the 50% probability level.
Figure 2
supporting information
sup-1
Acta Cryst. (2007). E63, o15–o16
supporting information
Acta Cryst. (2007). E63, o15–o16 [https://doi.org/10.1107/S1600536806050690]
5,7-Diiodoquinolin-8-ol
M. Saeed Butt, Zareen Akhter, Michael Bolte and Humaira M. Siddiqi
5,7-Diiodoquinolin-8-ol
Crystal data
C9H5I2NO Mr = 396.94
Monoclinic, P2/c
Hall symbol: -P 2yc
a = 14.1699 (13) Å
b = 4.2915 (4) Å
c = 16.1565 (13) Å
β = 96.801 (7)°
V = 975.57 (15) Å3 Z = 4
F(000) = 720
Dx = 2.703 Mg m−3 Melting point: 472(1) K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 7152 reflections
θ = 3.6–27.2°
µ = 6.40 mm−1 T = 173 K Rod, yellow
0.31 × 0.11 × 0.10 mm
Data collection
Stoe IPDS-II two-circle diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: multi-scan
(MULABS; Spek, 2003; Blessing, 1995)
Tmin = 0.318, Tmax = 0.527
7396 measured reflections 2244 independent reflections 2163 reflections with I > 2σ(I)
Rint = 0.046
θmax = 27.5°, θmin = 3.6°
h = −18→18
k = −5→5
l = −20→21
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.031 wR(F2) = 0.077 S = 1.17 2244 reflections 120 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0385P)2 + 2.4632P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 1.25 e Å−3 Δρmin = −1.23 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
I1 0.199166 (19) 1.16860 (6) 0.595651 (15) 0.02198 (12)
I2 0.062028 (18) 0.50962 (6) 0.884895 (17) 0.02272 (12)
O1 0.3950 (2) 0.8966 (8) 0.68130 (19) 0.0244 (6)
H1 0.4428 0.7834 0.6942 0.029*
C1 0.3270 (3) 0.8246 (9) 0.7300 (2) 0.0179 (7)
C2 0.2343 (3) 0.9234 (9) 0.7074 (2) 0.0168 (7)
C3 0.1599 (3) 0.8430 (9) 0.7548 (2) 0.0167 (7)
H3 0.0975 0.9198 0.7390 0.020*
C4 0.1780 (3) 0.6540 (9) 0.8236 (2) 0.0168 (7)
C5 0.2723 (3) 0.5472 (9) 0.8502 (2) 0.0159 (7)
C6 0.3471 (3) 0.6446 (9) 0.8039 (2) 0.0178 (7)
N7 0.4399 (3) 0.5647 (9) 0.8275 (2) 0.0211 (7)
C8 0.4603 (3) 0.3821 (11) 0.8938 (3) 0.0244 (8)
H8 0.5248 0.3261 0.9097 0.029*
C9 0.3907 (3) 0.2676 (11) 0.9418 (3) 0.0228 (8)
H9 0.4081 0.1343 0.9880 0.027*
C10 0.2973 (3) 0.3523 (10) 0.9205 (3) 0.0206 (8)
H10 0.2497 0.2806 0.9526 0.025*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
I1 0.02860 (18) 0.02110 (17) 0.01693 (16) 0.00530 (10) 0.00559 (10) 0.00259 (9)
I2 0.01802 (17) 0.02671 (18) 0.02479 (17) −0.00249 (9) 0.00822 (11) 0.00300 (10)
O1 0.0171 (13) 0.0334 (16) 0.0241 (14) 0.0032 (13) 0.0079 (11) 0.0093 (14)
C1 0.0193 (18) 0.0184 (18) 0.0166 (17) −0.0029 (14) 0.0044 (14) −0.0008 (15)
C2 0.0219 (19) 0.0141 (16) 0.0146 (16) −0.0002 (14) 0.0023 (14) −0.0009 (14)
C3 0.0138 (16) 0.0178 (17) 0.0185 (17) −0.0010 (13) 0.0016 (13) −0.0024 (14)
C4 0.0131 (17) 0.0203 (18) 0.0176 (17) −0.0038 (13) 0.0048 (13) −0.0040 (15)
C5 0.0169 (17) 0.0154 (16) 0.0154 (16) −0.0031 (14) 0.0019 (13) −0.0011 (14)
C6 0.0161 (17) 0.0189 (18) 0.0187 (17) −0.0014 (14) 0.0026 (13) −0.0052 (15)
N7 0.0188 (16) 0.0257 (17) 0.0191 (16) 0.0013 (14) 0.0039 (13) −0.0005 (14)
C8 0.0211 (19) 0.028 (2) 0.0237 (19) 0.0050 (17) 0.0008 (15) −0.0021 (18)
C9 0.024 (2) 0.0229 (19) 0.0203 (19) 0.0025 (16) −0.0005 (15) 0.0028 (16)
supporting information
sup-3
Acta Cryst. (2007). E63, o15–o16
Geometric parameters (Å, º)
I1—C2 2.098 (4) C5—C10 1.420 (6)
I2—C4 2.110 (4) C5—C6 1.430 (5)
O1—C1 1.350 (5) C6—N7 1.370 (5)
O1—H1 0.8400 N7—C8 1.332 (6)
C1—C2 1.388 (6) C8—C9 1.413 (6)
C1—C6 1.421 (6) C8—H8 0.9500
C2—C3 1.417 (5) C9—C10 1.376 (6)
C3—C4 1.375 (6) C9—H9 0.9500
C3—H3 0.9500 C10—H10 0.9500
C4—C5 1.430 (5)
C1—O1—H1 109.5 C4—C5—C6 118.2 (4)
O1—C1—C2 119.7 (4) N7—C6—C1 117.4 (4)
O1—C1—C6 121.9 (4) N7—C6—C5 121.8 (4)
C2—C1—C6 118.4 (4) C1—C6—C5 120.8 (4)
C1—C2—C3 121.6 (4) C8—N7—C6 118.8 (4)
C1—C2—I1 120.1 (3) N7—C8—C9 123.2 (4)
C3—C2—I1 118.1 (3) N7—C8—H8 118.4
C4—C3—C2 120.1 (3) C9—C8—H8 118.4
C4—C3—H3 120.0 C10—C9—C8 119.0 (4)
C2—C3—H3 120.0 C10—C9—H9 120.5
C3—C4—C5 120.7 (3) C8—C9—H9 120.5
C3—C4—I2 118.2 (3) C9—C10—C5 119.8 (4)
C5—C4—I2 121.1 (3) C9—C10—H10 120.1
C10—C5—C4 124.4 (4) C5—C10—H10 120.1
C10—C5—C6 117.4 (4)
O1—C1—C2—C3 177.5 (4) O1—C1—C6—C5 −174.2 (4)
C6—C1—C2—C3 −1.5 (6) C2—C1—C6—C5 4.7 (6)
O1—C1—C2—I1 2.3 (5) C10—C5—C6—N7 −2.5 (6)
C6—C1—C2—I1 −176.6 (3) C4—C5—C6—N7 176.9 (4)
C1—C2—C3—C4 −2.2 (6) C10—C5—C6—C1 176.3 (4)
I1—C2—C3—C4 173.0 (3) C4—C5—C6—C1 −4.3 (6)
C2—C3—C4—C5 2.7 (6) C1—C6—N7—C8 −176.4 (4)
C2—C3—C4—I2 −174.5 (3) C5—C6—N7—C8 2.4 (6)
C3—C4—C5—C10 179.9 (4) C6—N7—C8—C9 −0.5 (7)
I2—C4—C5—C10 −3.0 (6) N7—C8—C9—C10 −1.3 (7)
C3—C4—C5—C6 0.5 (6) C8—C9—C10—C5 1.2 (7)
I2—C4—C5—C6 177.6 (3) C4—C5—C10—C9 −178.7 (4)
O1—C1—C6—N7 4.7 (6) C6—C5—C10—C9 0.6 (6)
C2—C1—C6—N7 −176.4 (4)
Hydrogen-bond geometry (Å, º)
O1—H1···N7i 0.84 1.98 2.757 (5) 154