Molecular Dynamic Simulation of Coalescence
between Silver and Palladium Clusters
Hyun You Kim, Sung Hoon Lee
*1, Hyoung Gyu Kim, Ji Hoon Ryu and Hyuck Mo Lee
*2Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology, Kusung-Dong 373-1, Yusung-Gu, Taejon, 305-701 Korea
The coalescence between 135-Ag and 16-Pd clusters was studied through constant temperature molecular dynamic (MD) simulations at 300 K. Initially, the surface energy reduction was dominant. Later, some of the Pd atoms at the surface penetrated into the cluster and induced a further energy decrease. Surface atoms were rearranged to a local five-folded icosahedron structure (Ihp). Pd atoms gradually penetrated the cluster but did not segregate into the cluster core. As a result, a core-shell cluster structure was not observed, which could be explained by the strong mixing nature between Ag and Pd atoms and low kinetic energy. [doi:10.2320/matertrans.48.455]
(Received October 25, 2006; Accepted December 12, 2006; Published February 25, 2007)
Keywords: molecular dynamic simulation, silver-palladium nano cluster
1. Introduction
Bimetallic clusters composed of two different metal elements have recently attracted considerable attention, because their catalytic properties can be tuned with compo-sition or surface atomic distribution.1–3)Previous studies of bimetallic clusters such as Ag-Pd, Au-Pd, and Ag-Cu systems revealed that the different atomic size and surface energy between the two metal components cause preferential surface segregation of the component having relatively large size and low surface energy.3–6) In general, the other element segregates into the cluster core, forming an overall core-shell cluster structure.4–6)
In spite of recent remarkable development in chemical synthesis methods7–12) and the technical importance of bimetallic clusters, there have been few studies on the formation mechanism and structure of bimetallic clusters. Current computational research has been mainly oriented to the physical properties of single component metal clusters13) or surface segregation and reaction of bimetallic sur-faces.1–3,14)
The most notable computational study on the bimetallic cluster is the molecular simulation work of the Ferrando group.4–6)They have studied the overall structures of some bimetallic core-shell clusters using a genetic algorithm and a many-body potential.4–6)They produced a core-shell bimet-allic cluster structure by colliding metal atoms with relative large atomic radii and low surface energy one by one into a large metal cluster composed of single metal elements with small atomic radii and high surface energy.4–6)Even though the genetic algorithm is suitable for producing the final global minimum cluster morphology, at present, the solid solution type nanoparticle alloys appearing in the experimental synthesis of bimetallic clusters cannot be explained.11,12)
In this work, we study coalescence at 300 K between an Ag cluster composed of 135 atoms and a Pd cluster of 16 atoms using a molecular dynamic (MD) simulation method in order
to investigate the structural evolution and the related energy change of the Ag-Pd bimetallic cluster. Because Ag atoms are known to have a strong tendency to segregate to the cluster surface,10–12)we set the Pd/Ag atomic ratio at 1/9 to analyze the relationship between energy and cluster structure regarding the formation of a core-shell or a solid solution type.
2. Computational Details
2.1 Quantum sutton-chen potential
All MD simulations were performed using a quantum Sutton-Chen (Q-SC) potential.15,16) Based on the Sutton-Chen (SC) type potential,17)the total energy of the system can be predicted as follows:
Utot¼X i
Ui¼X i
" X i6¼j
1
2VðrijÞ c 1=2
i
" #
ð1Þ
where rij is the distance between atoms i and j, c is a dimensionless parameter scaling the attractive terms, and"
sets the overall energy scale. Here,VðrijÞis a pair potential to account for the repulsive force between atoms i and j resulting from Pauli repulsion between the core electrons:
VðrijÞ ¼ a
rij n
ð2Þ
The contribution of the local electron density associated with an atom i is given by
i¼X j6¼i
ðrijÞ ¼X j6¼i
a
rij m
ð3Þ
Here,is a functional that specifies how the electron density of an atom depends on the distances from the neighboring atoms and a is a length parameter scaling all spacings (leading to dimensionless V and). The Q-SC potential, an advanced form of the SC potential proposed by the Goddard group,15)has the same form as the SC potential.17)It includes quantum corrections and takes into account the zero-point energy (quantum) effect, thus allowing better prediction of temperature dependent properties.15,16,18)As such, it can be *1Present address: Department of Materials Science and Engineering, The
Pennsylvania State University, University Park, PA 16802, USA *2Corresponding author: E-mail: [email protected]
successfully applied to predict properties involving surface energies, vacancy energies, and stacking-fault energies, which cannot be precisely predicted with the SC poten-tial.16,18,19)The Q-SC potential parameters used for this study are listed in Table 1. The parameter values have been optimized by fitting to density, cohesive energy, and phonon frequencies.18–20)We employed combination rules to predict inter-atomic potentials between different metal elements. The geometric mean was used to obtain the energy scaling parameter,", and an arithmetic mean was used for n, m, and a.
2.2 MD simulation details
Classical MD simulations were carried out under a NVT ensemble. A constant temperature Nose-Hoover thermo-stat21) with a Nose-Hoover coupling constant Q¼0:1 was used. The temperature was set to 300 K to minimize the effect of temperature and precisely identify the structure changing mechanism. To solve the Newton’s dynamic equation, we applied a leap-frog Verlet algorithm.22)The time step for all simulations was set to 0.0002 ps.
Initially, the Ag cluster was composed of 135 atoms and the Pd cluster 16 atoms (the atomic fraction of Pd equal to 0.109), which corresponds to a fcc structure. Later, they were independently equilibrated at 300 K for 200 ps followed by a production time of 500 ps for structure relaxation.
In order to create the necessary physical collision conditions, the structure-relaxed Ag and Pd clusters were placed in the center of a large simulation box (50nm
50nm50nm) at a distance of 0.35 nm from each other. MD simulations of collisions were carried out for 1000 ps (five million time steps). The effect of the cluster surface orientation was neglected. The total energy vs. simulation time diagram was analyzed in conjunction with the cluster structure and various structure factors listed in Table 2. Structure factors such as total number of inter atomic bonds (Pd-Pd and Ag-Pd), aspect ratio of collided clusters, number of Pd atoms beneath the surface layer, and number of Pd atoms contacting Ag atoms were considered in this study. In addition, the molar Gibbs free energy diagram of the Ag-Pd
bulk alloy system was calculated using a Thermo-Calc program23)with the general alloy solution database V4.824) and was applied to explain the Ag-Pd mixing.
3. Results and Discussion
3.1 Total energy decreasing mechanism and cluster surface morphology
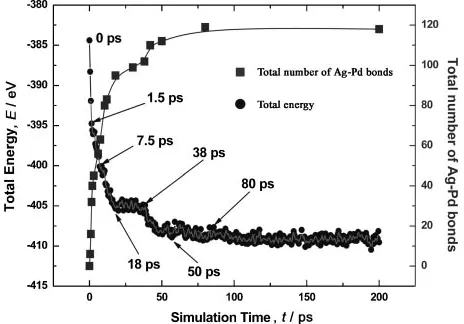
Figure 1 shows a diagram plotting the variation of total energy with simulation time. The total energy readily converged during an early simulation stage (before 150 ps). Because it is important to find the exact energy decreasing mechanism that accompanies the cluster structure change after collision, the energy values were plotted only up to 200 ps. Numerical energy values and relevant structure factors corresponding to the selected simulation time are presented in Table 2. The total energy diagram can be divided into the following three sections according to structure changes in relation to the energy decreasing mechanism.
3.1.1 Initial stage (until 18 ps)
[image:2.595.311.543.74.236.2]In this early stage, the total energy decreased rapidly (about 80% of the decreased energy) and 40% of the energy decrease took place before 1.5 ps, as shown in Fig. 1 and Table 2. The total number of Pd-Pd bonds and the aspect ratio gradually decreased. On the contrary, the total number Table 1 Q-SC potential parameters used in this study. Parameter values for
Ag and Pd atoms are from Ref. 19) and Ref. 18), respectively.
element n m "(meV) C a (nm)
Ag 11 6 4.0072 94.948 0.40691
[image:2.595.45.291.94.134.2]Pd 12 6 3.2864 148.205 0.38813
Table 2 Total energy and structure factors vs. simulation time.
time (ps)
total energy (eV)
number of Pd-Pd bond
number of
Ag-Pd bond aspect ratio
number of penetrated Pd atoms
number of Pd atoms in contact with Ag
0 384:399 51 0 — 0 0
1.5 394:759 28 32 1.44 1 9
7.5 400:093 24 63 1.24 2 16
18 405:324 15 95 1.12 6 16
38 405:282 19 102 1.05 6 16
50 408:286 16 112 1.07 9 16
200 409:535 12 118 1.02 9 16
[image:2.595.48.549.663.783.2]of Ag-Pd bonds rapidly increased (see Fig. 1). Both the number of Pd atoms in contact with Ag and penetrated Pd atoms increased. Notably, most structure factors in Table 2 decreased or increased monotonically until 18 ps except for the total number of penetrated Pd atoms, which started to increase remarkably after 7.5 ps, and the number of Pd-Pd bonds, which was stagnant between 1.5 ps and 7.5 ps.
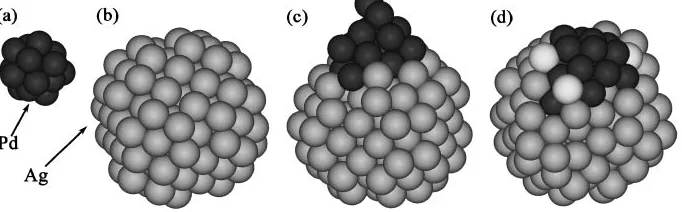
Figures 2(a) to 2(c) show that there is no remarkable change in the surface or overall structure of the Ag cluster until 1.5 ps. Immediately after collision, nine Pd atoms contacted surface Ag atoms at 1.5 ps. Only one Pd atom penetrated the surface layer. Therefore, the total number of Pd-Pd bonds decreased to about half that before collision and the number of Ag-Pd bonds increased rapidly (see Table 2). Because there was little change in the morphology of the Ag cluster, the energy change must have been induced by the decreased surface energy resulting from the reduced surface area of both clusters. This indicates that a strong driving force that decreases the surface energy and reduces the total number of low coordinated surface atoms is dominant during an early stage of coalescence between Ag and Pd clusters.
Table 2 shows that from 1.5 ps to 7.5 ps, there was no significant change in the number of Pd-Pd bonds. However, the number of Ag-Pd bonds increased continuously because the Pd cluster fanned out and all the Pd atoms established bonds with the surface Ag atoms (see Fig. 2(d)). At 7.5 ps, two or three Ag atoms, colored white in Fig. 2(d), started to climb up the surface Pd atoms, making additional Ag-Pd bonds. This suggests that the surface arrangement of a large Ag cluster undergoes a change after 7.5 ps. As noted in previous studies dealing with the formation of a core-shell structure in a bimetallic cluster system with a global minimization method,5,6) this can occur spontaneously due to the relatively low surface energy of Ag.
After 7.5 ps, the Pd atoms in contact with Ag started to penetrate the cluster. As shown in Fig. 3, the penetrated Pd atoms induced rearrangement of the surrounding Ag atoms at the surface. Table 3 shows that the penetrated Pd atoms moved closer to the cluster center by an average of 0.3 nm between 7.5 ps and 18 ps. The main energy reduction resulted from the increase in Ag-Pd bonds by penetration of Pd atoms due to the surface energy difference between Ag and Pd atoms.
According to a previous study on coalescence between two
565-Pb clusters,25)the aspect ratio rapidly decreased down to one during coalescence. However, in this work, the aspect ratio was still larger than one even after 18 ps. The large aspect ratio results from Ag atoms that climbed over the surface or squeezed Ag atoms induced by Pd penetration (see Fig. 2(d)).
3.1.2 Stationary stage (from 18 ps to 38 ps)
According to Table 2, only the number of Ag-Pd and Pd-Pd bonds increased slightly during this stage while other structure factors remained more or less the same. However, there was a remarkable change in surface arrangement. Between 7.5 ps and 18 ps, penetrating Pd atoms induced disordering of surrounding surface Ag atoms. From 18 ps, disordered Ag atoms started to form a local Ihp structure around the surface Pd atoms (see Ag atoms colored white in Fig. 2 Overall structure of the clusters before and after coalescence. (a) 16-Pd cluster before coalescence, (b) 135-Ag cluster before
coalescence, (c) Ag-Pd cluster simulated at 1.5 ps after collision and (d) Ag-Pd cluster simulated at 7.5 ps after collision. Ag atoms are colored light blue and Pd atoms colored dark blue. Ag atoms colored white are those that climbed over Pd atoms during collision (the same coloring is used throughout the figures).
[image:3.595.129.470.79.185.2]Fig. 3 A schematic diagram that shows penetration of Pd atoms and scattering of Ag atoms at the surface. The left snapshot was taken at 7.5 ps and the right at 18 ps. The number of penetrated Pd atoms is 2 at the left (7.5 ps) and 6 at the right (18 ps).
Table 3 Distance of Pd atoms from the cluster center and total number of atoms contributing to formation of the Ihp structure. Distance from cluster center represents the averaged distance between the cluster center and the six selected Pd atoms that penetrated the cluster.
time (ps)
distance from cluster center
(nm)
total number of atoms forming Ihps
7.5 0.7353 —
18 0.4371 12
38 0.4331 22
50 0.3829 24
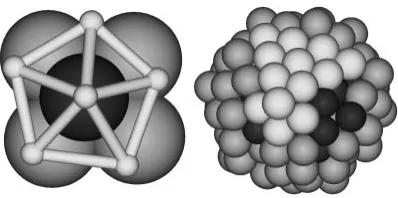
[image:3.595.306.549.491.587.2]Fig. 4). The Ihp structure mainly consisted of Ag atoms, with Pd atoms sometimes involved. As shown in Table 3, the number of atoms that belong to local Ihp increased in this stage.
The stationary stage implies that there is no remarkable change in the structure factors, the relative location of Pd atoms, and the total energy. The average distance of Pd atoms from the cluster center did not change, as seen in Table 3. The little change in energy is explained as follows. There was no Pd penetration, which resulted in Ag disordering at the surface and the number of Ag atoms that contributed to formation of Ihp increased. Thus, the formation of the Ihp structure itself is expected to decrease the total energy of the system,26) but instead it is compensated by extra energy incurred during surface diffusion of Ag atoms.
3.1.3 Final stage (from 38 ps to 200 ps)
The total energy decreased again in this stage. Pd atoms started again to penetrate into the interior of the cluster after 38 ps (Table 3) and the total number of atoms forming the Ihp increased substantially after 50 ps (Table 3). At 50 ps, the Ihp also formed at the opposite side of the cluster. Interestingly, throughout the stationary and final stages, all the Ihp structures formed just above the penetrated Pd atoms (see the embedded Pd atom beneath the Ihp in Fig. 5). This again proves that a strong relationship exists between surface disordering caused by Pd penetration and Ihp formation. Nam
et al.26)reported that the formation of an icosahedral structure in the Au cluster is induced by surface atoms. Further, the energy barrier for formation of Ihp is lower than that of the close packed surface arrangement.27)In this study, the Ihp phase also started to form at the cluster surface. The morphology of the cluster became closer to a sphere (aspect ratio = 1.02). Still, the penetrated Pd atoms moved more deeply inside the cluster. Figure 5 shows that some of the atoms of the Ihp structure form (111) planes.
A time step of about 150 ps was needed to stabilize the overall cluster structure after collision. However, approx-imately 80% of the total energy reduction was concentrated in the initial stage. An additional energy reduction is followed by penetration of Pd atoms and formation of Ihp in coalescence between Ad and Pd clusters. Even after 1000 ps of the MD simulation, the structure of the bimetallic Ag-Pd cluster was far from an exact core-shell type.
3.2 Overall cluster structure
In this work, the core-shell structure, which was reported to have been achieved by simulations using a genetic algorithm,4–6) was not attained at 200 ps (Fig. 5) nor even after 1000 ps (five million time step). Although the Pd atoms preferred the inside of the cluster to the cluster surface, the penetrated Pd atoms did not come together into the cluster center to form a typical bimetallic core-shell structure. They instead appeared to make a solid solution with Ag atoms. One of the reasons for this behavior will be the low temperature of simulation. The room temperature was not so high to supply sufficient energy for the Pd atoms to penetrate into the cluster interior. Therefore, a higher temperature study is under way now.
Another possibility is as follows. It is well known that the Ag-Pd alloy forms a complete solid solution at room temperature,28) and the Ferrando group6) reported that the degree of core-shell formation in the bimetallic cluster may be related with the mixing nature of the bulk alloy.6)Leeet al.29)reported that the binary nano phase diagram is shifted downward in terms of temperature because of the melting temperature drop in the nano system. However, the overall shape of the phase diagram did not change. Lianget al.30)also reported that there is no change in the phase diagram shape of a complete solid solution system even when the system size is reduced to the nano scale. The phase diagram is only shifted rather than taking on a completely different form.30)
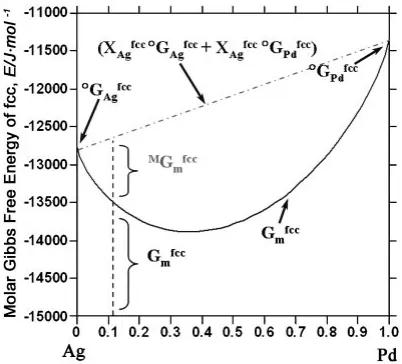
Thus, in order to analyze the Ag-Pd intermixing nature of the nano sized system observed in this study, the Gibbs free energy of the Ag-Pd bulk alloy system was calculated using the Thermo-Calc program.23)As shown in Fig. 6, the molar Gibbs free energy of mixing has a negative value at 0.109 atomic fraction of Pd, which corresponds to the exact composition of this study. This implies that not only the Ag-Pd bulk alloy but also the nano-sized Ag-Ag-Pd system shows a strong tendency of intermixing between Ag and Pd. This verifies that Pd atoms prefer to make a solid solution with Ag rather than to segregate into the cluster center and thereby form a core-shell structure.
Six Pd atoms remained at the cluster surface even after 1000 ps of simulation time. Because this work was performed at low temperature in order to investigate the exact energy-Fig. 4 Structure of the Ag-Pd cluster obtained at 18 ps and viewed at a
different angle. Ag atoms colored white locally formed a five-folded Ihp structure.
[image:4.595.85.253.75.235.2] [image:4.595.69.268.298.397.2]structure relationship, Ag and Pd atoms were not supplied with sufficient thermal energy. This might have prevented individual atoms to move more dynamically and overcome the potential barrier for further movement. The exact temperature-dependent nature of the cluster structure and the surface distribution of atoms requires further study.
4. Summary
The energy decreasing mechanism during coalescence of Ag and Pd clusters was investigated through constant temperature MD simulations at 300 K. Initially, the energy decrease was mainly caused by surface energy reduction. The increased total number of Ag-Pd bonds and the decreased number of Pd-Pd bonds reflect that a smaller Pd cluster was dismantled and Pd atoms came into contact with surface Ag atoms, thereby reducing the total number of low coordinated surface atoms. Afterwards, some of the surface Pd atoms started to penetrate into the cluster, causing an additional energy decrease due to the difference in surface energy of Ag and Pd. Approximately 80% of the total energy reduction was concentrated in the initial energy decrease stage.
In the stationary stage, the surface atoms were rearranged to a local five-folded Ihp structure, which resulted from surface atomic disordering caused by penetrated Pd atoms. The formation of the Ihp structure decreased the total energy of the system but was compensated by extra energy incurred during surface diffusion of Ag atoms. Throughout the stationary and final stages, all the Ihp structures formed just above the penetrated Pd atoms, which were embedded beneath the Ihp.
The Pd atoms preferred the inside of the cluster to the cluster surface but the penetrated Pd atoms did not come together into the cluster center to form the typical bimetallic core-shell structure. They instead appeared to make a solid solution with Ag atoms. Even the nano-sized Ag-Pd system
showed a strong tendency of intermixing between Ag and Pd at room temperature.
Acknowledgments
Many discussions with Dr. Sang Soo Han at the Materials and Process Simulation Center of CALTECH are greatly appreciated.
REFERENCES
1) T. Jacob and W. A. Goddard III: J. Phys. Chem. B108(2004) 8311– 8323.
2) T. Jacob, B. V. Merinov and W. A. Goddard III: Chem. Phys. Lett.385 (2004) 374–377.
3) J. Greeley and M. Mavrikakis: Nature Mater.3(2004) 810–815. 4) F. Baletto, C. Mottet, A. Rapallo, G. Rossi and R. Ferrando: Surf. Sci.
566–568(2004) 192–196.
5) F. Baletto, C. Mottet and R. Ferrando: Phys. Rev. B66(2002) 155420. 6) G. Rossi, R. Ferrando, A. Rapallo, A. Fortunelli, B. C. Curley, L. D.
Lloyd and R. L. Johnston: J. Chem. Phys.122(2005) 194309. 7) S. W. Kim, J. N. Park, Y. J. Jang, Y. H. Chung, S. J. Hwang, T. H.
Hyeon and Y. W. Kim: Nano Lett.9(2003) 1289–1291.
8) J. N. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. H. Hyeon: Nature Mater.3(2004) 891–895. 9) T. H. Hyeon, Y. H. Chung, J. N. Park, S. S. Lee, Y.-W. Kim and B. H.
Park: J. Phys. Chem. B106(2002) 6831–6833.
10) S. J. Park, S. S. Kim, S. Y. Lee, Z. G. Khim, K. Char and T. H. Hyeon: J. Am. Chem. Soc.122(2000) 8581–8582.
11) J. I. Park and J. W. Cheon: J. Am. Chem. Soc.123(2001) 57443–5746. 12) X. Teng, D. Black, N. J. Watkins, Y. Gao and H. Yang: Nano Lett.3
(2003) 261–264.
13) J. H. Shim, B. J. Lee and Y. W. Cho: Surf. Sci.512(2002) 262–268. 14) T. Jacob, R. P. Muller and W. A. Goddard III: J. Phys. Chem. B107
(2003) 9465–9476.
15) T. Cagin, Y. Kimura, Y. Qi, H. Li, H. Ikeda, W. L. Johnson and W. A. Goddard III: Bulk Metallic Glasses, MRS Symposia Proceedings No. 554, ed. by W. L. Johnson, C. T. Liu and A. Inoue, (Materials Research Society, Pittsburgh, 1999) pp. 43–48.
16) Y. Qi, T. Cagin, Y. Kimura and W. A. Goddard III: J. Comput-Aided Mater. Des.8(2001) 233–243.
17) A. P. Sutton and J. Chen: Phil. Mag. Lett.61(1990) 139–146. 18) Y. Qi, T. Cagin, Y. Kimura and W. A. Goddard III: Phys. Rev. B59
(1999) 3527–3533.
19) S. K. R. S. Sankaranarayanan, V. R. Bhethanaotla and B. Joseph: Phys. Rev. B71(2005) 195415.
20) S. K. R. S. Sankaranarayanan, V. R. Bhethanaotla and B. Joseph: Phys. Rev. B72(2005) 195405.
21) W. G. Hoover: Phys. Rev. A34(1986) 2499–2500. 22) R. W. Hockey: Methods Comput. Phys.9(1970) 136–211.
23) B. Sundman, B. Jansson and J. O. Andersson: CALPHAD9(1985) 153–190.
24) P. Shi: SGTE Solutions Database, SGTE, Scientific Group Thermodata Europe, (Version 4.8, 2003/2004).
25) S. Hendy, S. A. Brown and M. Hyslop: Phys. Rev. B 68 (2003) 241403(R).
26) H. S. Nam, N. M. Hwnag, B. D. Yu and J. K. Yoon: Phys. Rev. Lett.89 (2002) 275502.
27) H. S. Nam, N. M. Hwnag, B. D. Yu, D. Y. Kim and J. K. Yoon: Phys. Rev. B71(2005) 233401.
28) I. Karakaya and W. T. Thompson: Bull. Alloy Phase Diagrams9(1988) 237–243.
29) J. G. Lee, J. H. Lee, T. Tanaka, H. Mori and K. Pentila¨: JOM57(2005) 56–59.
30) L. H. Liang, D. Liu and G. Jiang: Nanotechnology14(2003) 438–442. Fig. 6 Molar Gibbs free energy of the solid solution fcc phase of the Ag-Pd
[image:5.595.68.269.72.254.2]