organic papers
Acta Cryst.(2005). E61, o2479–o2481 doi:10.1107/S1600536805020192 Awalehet al. C
14H14S2
o2479
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
1,2-Bis(phenylsulfanyl)ethane
Mohamed Osman Awaleh,* Antonnella Badia and Franc¸ois Brisse
De´partement de Chimie, Universite´ de Montre´al, CP 6128, Succ. Centre-ville, Montre´al, Que´bec, Canada H3C 3J7
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 220 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.036
wRfactor = 0.107
Data-to-parameter ratio = 16.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
In the title crystal structure, C14H14S2,L 2
, the molecules lie on crystallographic inversion centers. The phenyl rings are nearly perpendicular to the planar S—C—C—S group of atoms.
Comment
One of the interests of metal–organic polymer coordination is the design of predictable networks with useful properties (Leininger et al., 2000; Holliday & Mirkin, 2001). Linear bifunctional ligands are often used as building blocks for the construction of metal–organic framework materials (MOF) (Carlucciet al., 2002). The S atom is a soft base and has a good ability to coordinate to silver(I), which is a soft acid. The dithiol group affords two coordination sites to metal centers and can be used to construct supramolecular architectures (Blacket al., 1995; Buet al., 2002). We report here the struc-ture of 1,2-bis(phenylsulfanyl)ethane,L2, (I).
The molecular structure of (I) is shown in Fig. 1. There is a crystallographic center of symmetry at the midpoint of the
central C—C bond. Hence, the torsion angle S1—C7—C7i—
S1iis 180[symmetry code: (i) 2x,y, 1z]. The C1—S1—
C7—C7i torsion angle is 83.9 (2), and thus the aliphatic
segment (S—CH2—CH2—S) is nearly perpendicular to the
phenyl rings. In contrast, the longer chain molecule
1,10-bis(phenylsulfanyl)decane, L10, is nearly wholly planar
(Awalehet al., 2005). The bond distances and angles inL2are in the normal range (Table 1) (International Tables for Crys-tallography, 1995, Vol. C).
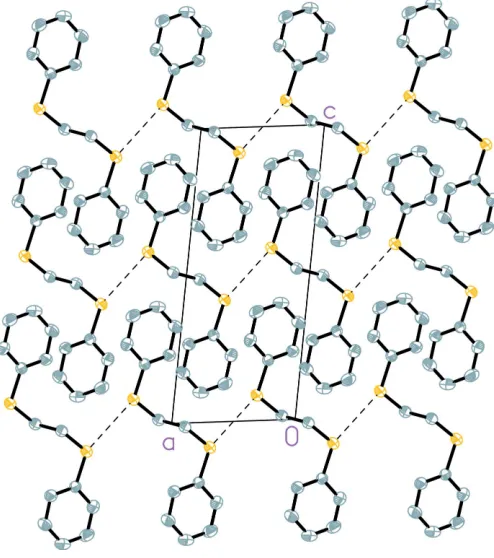
In the crystal packing of (I), illustrated in Fig. 2, there are no
significant – stacking interactions. The S S distance
between adjacent molecules, with a value of 3.5136 (8) A˚ , is slightly less than the sum of the van der Waals radii (3.60 A˚ ; Porterfield, 1994). These short contacts are shown in Fig. 3.
Another determination of the title compound is reported in the following paper (Houet al., 2005).
Experimental
The title compound was synthesized according to the published procedure of Hartleyet al.(1979). The1H NMR spectrum, recorded
in acetone-d6, confirms the purity of the compound. Compound (I)
was obtained as a crystalline powder from which a needle-like crystal suitable for X-ray analysis was isolated (yield 43%). Analysis, found: C 68.25, H 6.09%; calculated for C14H14S2: C 68.24, H 5.73%.
1
H NMR (acetone-d6 , p.p.m.): 3.14 [s, 4H, –S-(CH2)2-S–], 7.32 (m, 10H,
C6H5–).
Crystal data
C14H14S2
Mr= 246.37
Monoclinic, P21=c a= 5.8654 (1) A˚
b= 7.5638 (1) A˚
c= 14.0472 (2) A˚ = 97.600 (1)
V= 617.73 (2) A˚3
Z= 2
Dx= 1.325 Mg m
3
CuKradiation Cell parameters from 4189
reflections = 3.2–72.7 = 3.63 mm1
T= 220 (2) K Needle, colorless 0.320.060.05 mm
Data collection
Bruker SMART 2000 area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Sheldrick,1996)
Tmin= 0.375,Tmax= 0.825 5018 measured reflections
1184 independent reflections 1099 reflections withI> 2(I)
Rint= 0.026
max= 72.7
h=5!6
k=9!9
l=17!16
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.036
wR(F2) = 0.107
S= 1.03 1184 reflections 74 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0794P)2 + 0.1086P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.23 e A˚ 3
min=0.23 e A˚ 3
Extinction correction:SHELXL97
(Sheldrick, 1997)
Extinction coefficient: 0.018 (2)
organic papers
o2480
Awalehet al. C [image:2.610.111.244.71.301.2] [image:2.610.316.563.73.352.2]14H14S2 Acta Cryst.(2005). E61, o2479–o2481
Figure 1
The molecular structure and atomic numbering of (I). Displacement ellipsoids are drawn at the 50% probability level. H atoms have been omitted. The unlabeled part of the molecule is related by the symmetry transformation (2x,y, 1z).
Figure 2
[image:2.610.118.229.362.639.2]A crystal packing diagram of (I), viewed along theaaxis. H atoms have been omitted.
Figure 3
Table 1
Selected geometric parameters (A˚ ,).
S1—C1 1.7667 (15)
S1—C7 1.8123 (16)
C7—C7i
1.515 (3)
C1—S1—C7 105.08 (7) C7i
—C7—S1 112.48 (14)
C1—S1—C7—C7i
83.90 (17)
Symmetry code: (i) 2x;y;1z.
H atoms were placed in calculated positions (C—H = 0.93–0.98 A˚ ) and refined as riding atoms, withUiso(H) = 1.2Ueq(C). A final
veri-fication of possible voids was performed using the VOID routine of thePLATONprogram (Spek, 2003).
Data collection:SMART(Bruker, 1999); cell refinement:SAINT
(Bruker, 1999); data reduction: SAINT; program(s) used to solve structure: SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL (Bruker, 1997); software used to prepare material for publication:UdMX(Maris, 2004).
This work was supported by the Natural Sciences and Engineering Research Council of Canada (FB). MOA thanks
the Programme Canadien de Bourse de la Francophonie and the Organization Internationale de la Francophonie for a graduate scholarship.
References
Awaleh, M. O., Badia, A. & Brisse, F. (2005).Acta Cryst.E61, o2473–o2475. Black, J. R., Champness, N. R., Levason, W. & Reid, G. (1995).J. Chem. Soc.
Dalton Trans.pp. 3439–3445.
Bruker (1997). SHELXTL. Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999).SAINT(Version 6.06) andSMART(Version 5.059). Bruker AXS Inc., Madison, Wisconsin, USA.
Bu, X. H., Chen, W., Hou, W. F., Du, M., Zhang, R. H. & Brisse, F. (2002).
Inorg. Chem.41, 3477–3482.
Carlucci, L., Ciani, G., Proserpio, D. M. & Rizzato, S. (2002).CrystEngComm, 4, 413–425.
Hartley, F. R., Murray, S. G., Levason, W., Soutter, H. E. & McAuliffe, C. A. (1979).Inorg. Chim. Acta,35, 265–277.
Holliday, B. J. & Mirkin, C. A. (2001).Angew. Chem. Int. Ed.40, 2022–2043. Hou, B.-H., Zhou, L.-N., Yin, Q.-X., Wang, J.-K. & Chen, W. (2005).Acta
Cryst.E61, o2482–o2483.
Leininger, S., Olenyuk, B. & Stang, P. J. (2000).Chem. Rev.100, 853–908. Maris, T. (2004).UdMX.Universite´ de Montre´al, Canada.
Porterfield, W. W. (1994).Inorganic Chemistry: A Unified Approach, p. 168. London: Addison–Wesley.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
organic papers
Acta Cryst.(2005). E61, o2479–o2481 Awalehet al. C
supporting information
sup-1
Acta Cryst. (2005). E61, o2479–o2481
supporting information
Acta Cryst. (2005). E61, o2479–o2481 [https://doi.org/10.1107/S1600536805020192]
1,2-Bis(phenylsulfanyl)ethane
Mohamed Osman Awaleh, Antonnella Badia and Fran
ç
ois Brisse
1,2-Bis(phenylthio)ethane
Crystal data
C14H14S2
Mr = 246.37 Monoclinic, P21/c Hall symbol: -P 2ybc
a = 5.8654 (1) Å
b = 7.5638 (1) Å
c = 14.0472 (2) Å
β = 97.600 (1)°
V = 617.73 (2) Å3
Z = 2
F(000) = 260
Dx = 1.325 Mg m−3
Cu Kα radiation, λ = 1.54178 Å Cell parameters from 4189 reflections
θ = 3.2–72.7°
µ = 3.63 mm−1
T = 220 K
Needle-like, colorless 0.32 × 0.06 × 0.05 mm
Data collection
Bruker SMART 2000 area-detector diffractometer
Radiation source: X-ray sealed tube Graphite monochromator
Detector resolution: 5.5 pixels mm-1
ω scans
Absorption correction: multi-scan (SADABS; Sheldrick,1996)
Tmin = 0.375, Tmax = 0.825
5018 measured reflections 1184 independent reflections 1099 reflections with I > 2σ(I)
Rint = 0.026
θmax = 72.7°, θmin = 6.4°
h = −5→6
k = −9→9
l = −17→16
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.036
wR(F2) = 0.107
S = 1.03 1184 reflections 74 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0794P)2 + 0.1086P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.23 e Å−3 Δρmin = −0.23 e Å−3
supporting information
sup-2
Acta Cryst. (2005). E61, o2479–o2481
Special details
Experimental. X-ray crystallographic data for I were collected from a single-crystal sample, which was mounted on a loop fiber. Data were collected using a Bruker Platform diffractometer, equipped with a Bruker SMART 2 K Charged-Coupled Device (CCD) Area Detector using the program SMART and normal focus sealed tube source graphite monochromated Cu—Kα radiation. The crystal-to-detector distance was 4.908 cm, and the data collection was carried out in 512 x 512 pixel mode, utilizing 4 x 4 pixel binning. The initial unit-cell parameters were determined by a least-squares fit of the angular setting of strong reflections, collected by a 9.0 degree scan in 30 frames over four different parts of the reciprocal space (120 frames total). One complete sphere of data was collected, to better than 0.8 Å resolution. Upon completion of the data collection, the first 101 frames were recollected in order to improve the decay correction analysis.
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
S1 0.66983 (6) 0.07504 (6) 0.41416 (3) 0.0399 (2) C1 0.7328 (3) 0.05351 (17) 0.29514 (11) 0.0307 (4) C4 0.7900 (3) 0.0336 (2) 0.10185 (12) 0.0453 (4)
H4 0.8092 0.0265 0.0366 0.054*
C6 0.9244 (3) −0.0306 (2) 0.26748 (11) 0.0374 (4)
H6 1.0358 −0.0806 0.3140 0.045*
C5 0.9501 (3) −0.0404 (2) 0.17069 (12) 0.0417 (4)
H5 1.0788 −0.0985 0.1520 0.050*
C2 0.5714 (3) 0.1287 (2) 0.22523 (12) 0.0403 (4)
H2 0.4421 0.1866 0.2433 0.048*
C3 0.6005 (3) 0.1185 (2) 0.12942 (13) 0.0477 (5)
H3 0.4908 0.1697 0.0826 0.057*
C7 0.8901 (3) −0.0522 (2) 0.48666 (12) 0.0375 (4)
H7A 0.8315 −0.0905 0.5455 0.045*
H7B 0.9244 −0.1582 0.4511 0.045*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3
Acta Cryst. (2005). E61, o2479–o2481
Geometric parameters (Å, º)
S1—C1 1.7667 (15) C6—H6 0.94
S1—C7 1.8123 (16) C5—H5 0.94
C1—C6 1.391 (2) C2—C3 1.381 (2)
C1—C2 1.392 (2) C2—H2 0.94
C4—C5 1.375 (2) C3—H3 0.94
C4—C3 1.383 (3) C7—C7i 1.515 (3)
C4—H4 0.94 C7—H7A 0.98
C6—C5 1.390 (2) C7—H7B 0.98
C1—S1—C7 105.08 (7) C3—C2—C1 120.32 (16)
C6—C1—C2 119.27 (15) C3—C2—H2 119.8
C6—C1—S1 125.75 (12) C1—C2—H2 119.8
C2—C1—S1 114.98 (12) C2—C3—C4 120.41 (16)
C5—C4—C3 119.46 (16) C2—C3—H3 119.8
C5—C4—H4 120.3 C4—C3—H3 119.8
C3—C4—H4 120.3 C7i—C7—S1 112.48 (14)
C5—C6—C1 119.60 (14) C7i—C7—H7A 109.1
C5—C6—H6 120.2 S1—C7—H7A 109.1
C1—C6—H6 120.2 C7i—C7—H7B 109.1
C4—C5—C6 120.94 (16) S1—C7—H7B 109.1
C4—C5—H5 119.5 H7A—C7—H7B 107.8
C6—C5—H5 119.5
C7—S1—C1—C6 −5.28 (15) C6—C1—C2—C3 0.5 (2) C7—S1—C1—C2 174.47 (12) S1—C1—C2—C3 −179.25 (13)
C2—C1—C6—C5 −0.9 (2) C1—C2—C3—C4 0.0 (3)
S1—C1—C6—C5 178.87 (12) C5—C4—C3—C2 −0.2 (3) C3—C4—C5—C6 −0.2 (3) C1—S1—C7—C7i 83.90 (17) C1—C6—C5—C4 0.7 (3)