1535-9778/05/$08.00⫹0 doi:10.1128/EC.4.6.1018–1028.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
ACE2
,
CBK1
, and
BUD4
in Budding and Cell Separation
Warren P. Voth, Aileen E. Olsen, Mohammed Sbia,† Karen H. Freedman,‡
and David J. Stillman*
Department of Pathology, University of Utah Health Sciences Center, Salt Lake City, Utah 84132
Received 14 January 2005/Accepted 6 April 2005
Mutations in the RAM network genes, includingCBK1,MOB2,KIC1,HYM1, andTAO3, cause defects in bud site selection, asymmetric apical growth, and mating projections. Additionally, these mutants show altered colony morphology, cell separation defects, and reducedCTS1expression, phenotypes also seen by mutating the Ace2 transcription factor. We show that anACE2multicopy plasmid suppresses the latter three defects of RAM network mutations, demonstrating that Ace2 is downstream of the RAM network and suggesting that these phenotypes are caused by reduced expression of Ace2 target genes. We show that wild-type W303 strains have abud4mutation and that combiningbud4with eitherace2orcbk1in haploids results in altered colony morphology. We describe a timed sedimentation assay that allows quantitation of cytokinesis defects and subtle changes in budding pattern and cell shape. Experiments examining budding patterns and sedimentation rates both show that Ace2 and Cbk1 have independent functions in addition to their common pathway in transcrip-tion of genes such asCTS1.SWI5encodes a transcription factor paralogous toACE2. Additive effects are seen
incbk1 swi5strains, and we show that activation of some target genes, such asEGT2, requires either Swi5 or
Ace2 with Cbk1. The relative roles and interactions of Ace2, Cbk1, and Bud4 in bud site selection, polarized growth, and cell separation are discussed.
Cells are innately asymmetric, and even the most basic in-ternal polarization can be exploited to program division pat-terns which lead to organized structures. The morphology of multicellular organisms or any multicellular assemblage is based chiefly on the shapes which cells adopt, the patterns of cell division, and the retention or loss of cellular contacts after division. Changes in these processes correlate with morphoge-netic transitions in organismal development, as well as in re-source exploitation and pathogenesis in so-called unicellular
organisms.Saccharomyces cerevisiaehas been shown to
regu-late these phenomena, in part by controlling intrinsic cell po-larity as well as reorganization of popo-larity in response to ex-ternal cues, the selection of sites at which budding initiates depending on cell type and environment, and by controlling separation of divided cells after mitosis (7, 8, 23, 27).
The yeast Cbk1 protein is a member of the AGC
serine-threonine protein kinase family, which also includesS.
cerevi-siaeDbf2,Schizosaccharomyces pombeOrb6 and Sid2, Ndr1,
WARTS/LATS, and others (15, 28, 35). Cells lacking Cbk1 show defects in a number of processes, including bud site selection, asymmetric apical growth of the emerging bud, re-sponse to mating pheromone, morphogenesis of mating
pro-jections, and cell separation after mitosis (3, 28).CBK1 is a
component of the RAM (regulation ofAce2 activity and
cel-lularmorphogenesis) network, which includes the Cbk1
bind-ing partnerMOB2andKIC1,HYM1, andTAO3, all strains of
which have several phenotypes similar tocbk1 mutants when
mutated (25). cbk1 mutant cells lack the normal ability to
select budding locations near the polar sites of previous bud-ding and instead form buds at random locations across the cell surface. Random budding is symptomatic of a more general
loss of axis incbk1 cells, which also fail to display the usual
ellipsoid cell shape which arises from apically oriented growth
in the emerging bud. Instead,cbk1mutants grow isotropically
to form more spherical cells. This lack of polarity is also
evi-dent as reduced development of mating projections incbk1
and other RAM mutant cells, a consequence of loss of orga-nization in the underlying actin cytoskeleton. Strains mutant in
CBK1also possess defects in colony formation, with a loss of
the normal hemispherical shape and smooth opalescent
sur-face, instead appearing irregular and rough. Mutations incbk1
or any other RAM network gene are not tolerated in some genetic backgrounds, including those used for global knockout and expression studies. This strain-dependent difference in
lethality is due to polymorphism at theSSD1locus; thus lack of
a RAM network gene is tolerated only ifSSD1is also mutated,
and most studies of RAM network genes have been carried out
inssd1strain backgrounds, such as W303 (20).
The zinc finger transcription factor Ace2 activates a set of
genes in G1, many of which code for enzymes responsible for
locally specific cell wall degradation (11, 12, 14). Loss or re-duction in Ace2 activity causes daughter cells to remain at-tached by the cell wall to mothers at the bud site septum, but with no delay in subsequent nuclear or cytoplasmic cell cycles. This results in the accumulation of multicellular clusters or clumps. In diploids, or in haploids in some strain backgrounds,
ace2mutant strains also show the rough colony morphological
defects similar to those incbk1strains (22). Ace2 activity and
concomitant breakage of the mother-daughter connection is tightly controlled with respect to the cell cycle and to cell type. Several transcriptional and posttranscriptional mechanisms act through Ace2 to ensure that septum separation occurs only
* Corresponding author. Mailing address: Department of Pathology, University of Utah, 30 North 1900 East, Salt Lake City, UT 84132-2501. Phone: (801) 581-5429. Fax: (801) 581-4517. E-mail: david.stillman @path.utah.edu.
† Present address: Department of Oncological Sciences, University of Utah, Salt Lake City, UT 84132.
‡ Present address: University of Wisconsin, Madison, WI 53706.
1018
on September 8, 2020 by guest
http://ec.asm.org/
after the completion of cytokinesis and only on the daughter-proximal side of the chitinous budding ring. The ability of Ace2 to activate transcription is regulated by Cbk1 and other mem-bers of the RAM network in ways that are not fully under-stood, but deletion of any member of the network results in cell separation and target gene activation phenotypes similar to
ace2mutations without changes in Ace2 protein levels.
More than 70 genes have been identified which are neces-sary for yeast’s normal budding pattern programs (26). These genes have been classified into several groups based on the type of altered budding pattern seen in the mutant. Vegeta-tively growing haploids follow an axial budding program, in which subsequent buds form in close apposition to the initial bud site, itself adjacent to the birth scar, resulting in a chain of bud scars visible around the long axis of the cell. Mutation of
theBUD4gene gives rise to haploids which fail in axial
bud-ding but instead execute an otherwise normal bipolar program typical of diploids, in which subsequent rounds of budding are directed to alternating poles of the mother cell (9). There are several other genes in which mutation causes this same
axial-to-bipolar change in haploids, includingAXL1(18). Bud4
pos-sesses pleckstrin homology and GTP-binding domains and fa-cilitates axial budding by functioning as a molecular tag marking the position of the bud neck from previous budding
event. Bud4 is modeled as an activator of Axl1, which is itself a repressor of the bipolar programming proteins Rax1 and Rax2 and the bipolar tags Bud8 and Bud9 (17). In diploids,
whileBUD4is still expressed, its action on Axl1 is obviated by
thea1/␣2-dependent repression ofAXL1transcription, which
allows the bipolar landmarks Bud8 and Bud9 to supercede the axial pattern (17).
Here we report that the widely used W303 strain
back-ground has abud4 mutation, and we investigate the relative
roles ofBUD4,ACE2, andCBK1in controlling cell separation.
A bud4 mutation shows additive effects with either ace2 or
cbk1, resulting in an altered colony morphology. We find that
the random budding pattern associated withcbk1haploids (28)
is only seen in combination with abud4mutation but that a
cbk1 diploid buds randomly irrespective ofBUD4 genotype.
Thus, the random budding seen in acbk1 mutant is only
ob-served in a strain that would otherwise exhibit bipolar budding, but not in an axial budding strain. We describe a sedimentation assay that allows us to quantify defects in cell separation and also detects modest changes in budding pattern and cell shape.
MATERIALS AND METHODS
All yeast strains used are listed in Table 1 and are isogenic in the W303 background (37). Standard genetic methods were used for strain construction
TABLE 1. Strains used in this study
Straina Relevant characteristics
DY150MATa...ade2 can1 his3 leu2 trp1 ura3
DY151MAT␣...ade2 can1 his3 leu2 trp1 ura3
DY1640MATa/MAT␣...ade2 can1 his3 leu2 trp1 ura3
DY1654MATa...CTS1-lacZ::LEU2 ace2::HIS3 ade2 can1 his3 leu2 trp1 ura3
DY2396MATa...ade2 can1 his3 leu2 lys2 trp1 ura3
DY3923MATa...ace2::HIS3 ade2 can1 his3 leu2 lys2 trp1 ura3
DY3925MATa...swi5::TRP1 ade2 can1 his3 leu2 lys2 trp1 ura3
DY4653MATa...ace2::HIS3 swi5::TRP1 ade2 can1 his3 leu2 lys2 trp1 ura3
DY5786MATa...CTS1-lacZ::LEU2 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY5794MATa...cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY5795MAT␣...cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY5796MATa...ace2::HIS3 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY5798MATa...swi5::TRP1 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY5800MATa...ace2::HIS3 swi5::TRP1 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY6604MATa...BUD4 ade2 can1 his3 leu2 lys2 trp1 ura3
DY6605MAT␣...BUD4 ade2 can1 his3 leu2 lys2 trp1 ura3
DY6984MATa...CTS1-lacZ::LEU2 hym1::ADE2 ade2 can1 his3 leu2 lys2 trp1 ura3
DY6986MAT␣...CTS1-lacZ::LEU2 mob2::TRP1 ade2 can1 his3 leu2 trp1 ura3
DY7294MATa...CTS1-lacZ::LEU2 kic1::KanMX ade2 can1 his3 leu2 trp1 ura3
DY8497MATa/MAT␣...BUD4 ade2 can1 his3 leu2 lys2/⫹trp1 ura3
DY8566MATa/MAT␣...ace2::HIS3/ace2::KanMX ade2 can1 his3 leu2 trp1 ura3
DY8567MATa/MAT␣...cbk1::KanMX/cbk1::HphMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9184MATa...bud4::URA3::BUD4 ace2::HIS3 swi5::TRP1 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9186MATa...bud4::URA3::BUD4 ace2::HIS3 swi5::TRP1 ade2 can1 his3 leu2 lys2 trp1 ura3
DY9188MATa...bud4::URA3::BUD4 ace2::HIS3 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9190MATa...bud4::URA3::BUD4 ace2::HIS3 ade2 can1 his3 leu2 lys2 trp1 ura3
DY9192MATa...bud4::URA3::BUD4 swi5::TRP1 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9194MATa...bud4::URA3::BUD4 swi5::TRP1 ade2 can1 his3 leu2 lys2 trp1 ura3
DY9195MATa...bud4::URA3::BUD4 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9197MATa...bud4::URA3::BUD4 ade2 can1 his3 leu2 lys2 trp1 ura3
DY9216MATa/MAT␣...bud4::URA3::BUD4/bud4 cbk1::KanMX/cbk1::HphMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9259MATa...bud4::URA3::BUD4 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9260MAT␣...bud4::URA3::BUD4 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
DY9399MATa/MAT␣...bud4::URA3::BUD4/BUD4 ace2::HIS3 ade2 can1 his3 leu2 lys2 trp1 ura3
DY9400MATa/MAT␣...ace2::HIS3 cbk1::HphMX/cbk1::KanMX ade2 can1 his3 leu2 lys2/⫹trp1 ura3
DY9401MATa/MAT␣...bud4::URA3::BUD4/BUD4 ace2::HIS3 cbk1::KanMX ade2 can1 his3 leu2 lys2 trp1 ura3
aAll strains are in the W303 background and have abud4mutation unless otherwise noted.
on September 8, 2020 by guest
http://ec.asm.org/
(29, 32). Thecbk1::KanMXdisruption and theCTS1-lacZ::LEU2reporter have been previously described (13, 15). Marker swap plasmids (39) were used to convert the cbk1::KanMX allele to cbk1::HphMX and hym1::TRP1 (15) to
hym1::ADE2. Thekic1::KanMXandmob2::TRP1gene deletions were made by PCR (1) and verified by PCR or by Southern blot analysis. The bud4::
URA3::BUD4allele was integrated at thebud4locus by transforming abud4 ace2
strain with SalI-cleaved M4213. Passage of this strain over 5-fluoroorotic acid to select for the loss of the integratedURA3YIp plasmid resulted in bothbud4 ace2
andBUD4 ace2progeny, which could be distinguished by colony morphology and budding pattern. Cells were grown in yeast extract-peptone-dextrose (YEPD) medium (32) at 30°C or in synthetic complete medium with 2% glucose supple-mented with adenine and appropriate amino acids but lacking uracil to select for plasmids.
Plasmids used in this study are listed in Table 2. Plasmids with theBUD4gene were isolated from a YCp50 library as complementing the rough colony mor-phology ofkic1andmob2mutants. A 6-kb BamHI-MluI fragment with theBUD4
gene was first subcloned into pUC21 (38), and then from this plasmid a 6-kb BamHI-KpnI fragment withBUD4was cloned into pRS406 and pRS416 (4), constructing M4213 and M4214, respectively. M4104 contains a 4.4-kb SacII-BamHI fragment with theCBK1gene in pRS426. M4105 contains a 1.9-kb AflII-KpnI fragment with theHYM1gene in pRS426. M4298 contains a 2.9-kb SpeI-Sau3AI fragment with theMOB2 gene in pRS426. M4102 contains a 4.9-kb HindIII-KpnI fragment with theKIC1gene in pRS426. Details of plasmid con-struction are available on request.
RNA levels were determined with S1 nuclease protection assays as described previously (2), using oligonucleotides specific forEGT2(CCGCTACTCAATG ATGCAGTAACCGCAACTTCAGGGATAGCGGATGATTGCGAAGTAT AAGAAGAAAGCGGGTCCG),SCW11(TGCTTGGCTGCAAAGTAGAGG TTGGCTGCGAAGATGTACTCATTTCAGCCAAAAGG),DSE2(ATGTAC CTGACGATTCGATACTTTGAGATGATTCGATATCGTATGTAGAGCT AGAGGTACAAC),CTS1(CGCAAAATCAATGTCTGCATTTTCCAACAA GTCACCAACAGAAGCATCCGGGTATGGACATTGTGGTGCGATGA GG), andCMD1(GGGCAAAGGCTTCTTTGAATTCAGCAATTTGTTCTT CGGTGGAGCC).
To measure sedimentation rates, late-log cultures were vortexed vigorously, an aliquot was placed in a cuvette, and the optical density at 660 nm was measured over time. Because each culture had different starting and endpoint values, the data were normalized. The sedimentation assays were conducted in triplicate, and the graphs show the average optical density of the three cultures any indi-vidual time point. The sedimentation half-time was defined as the time required for the initial density to decrease to one-half of the difference between the initial and the final lower plateau values. The sedimentation half-time was calculated independently for each of the three replicates, and is given as the average⫾the standard error.
Budding patterns on Calcofluor-stained cells were detected as described pre-viously (22). Activity of theCTS1-lacZreporter was assayed by X-Gal (5-bromo-4-chloro-3-indolyl--D-galactopyranoside) filter assays (5).
RESULTS
Multicopy ACE2 suppresses RAM pathway mutations.We
identifiedCBK1andHYM1by mutations causing a mild defect
in transcriptional repression by a LexA-Sin3 fusion protein (15). This screen identified other mutants with similar pheno-types that had not been cloned by complementation at the time
of that report. These mutants aremob2,kic1, andace2. All of
these mutants have two phenotypes in common: an altered colony morphology (see below) and reduced expression of a
CTS1-lacZreporter. We performed multicopy suppression ex-periments to investigate the relationships between these
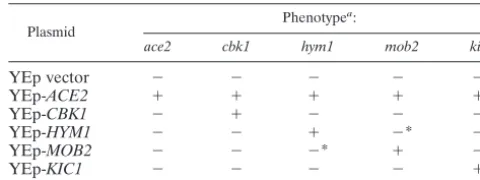
fac-tors. As shown in Table 3, a multicopy plasmid with theACE2
gene can suppress the colony morphology andCTS1-lacZ
ex-pression phenotypes of thecbk1, hym2, mob2, and kic1
mu-tants. These results suggest that CBK1, MOB2, HYM1, and
KIC1all act upstream ofACE2, consistent with the report that
multicopy ACE2 suppresses the cell separation defects in
RAM mutants (25, 28). However, we do not have a clear
understanding of why mutating eitherACE2or upstream
fac-tors should affect repression by LexA-Sin3.
Genetic interactions between Ace2 and Bud4. An altered
colony morphology is seen in an ace2 orcbk1mutant strain
(Fig. 1), and a similar phenotype is seen inmob2, hym1, and
kic1mutants. Colonies of these mutants lose the typical
hemi-spherical shape and smooth opalescent surface and instead exhibit scalloped colony edges and a roughened surface with many branched channels and invaginations. Interestingly, these
rough colonies are seen inace2W303 but not in ace2S288c
strains (18; W. P. Voth and D. J. Stillman, unpublished obser-vations). When we used a visual screen for complementation of
the rough morphology to cloneCBK1, MOB2, HYM1, KIC1,
andACE2, we identified several plasmids that complemented
the colony morphology of all of the mutants. These plasmids
containedBUD4and several adjacent genes, and subcloning
demonstrated that theBUD4gene was sufficient for
comple-mentation. These results suggested that the parent W303 strain
has abud4mutation and that two mutations are required for
the colony phenotype. To test this hypothesis, we constructed
a YIp-URA3-BUD4plasmid which was integrated at thebud4
locus, creating abud4::URA3::BUD4allele. Growth on
5-fluo-roorotic acid allowed us to recover W303BUD4⫹strains,
with-out theURA3marker. In crosses betweenace2 bud4andace2
bud4::URA3::BUD4strains, the smooth colony phenotype
al-ways segregated withURA3. Additionally, we used the genetic
linkage of theIME1andBUD4genes to demonstrate that the
bud4::URA3::BUD4allele is at thebud4locus. A strain with
thisbud4::URA3::BUD4allele was crossed to a strain with an
ime1::TRP1allele, andbud4::URA3::BUD4showed tight
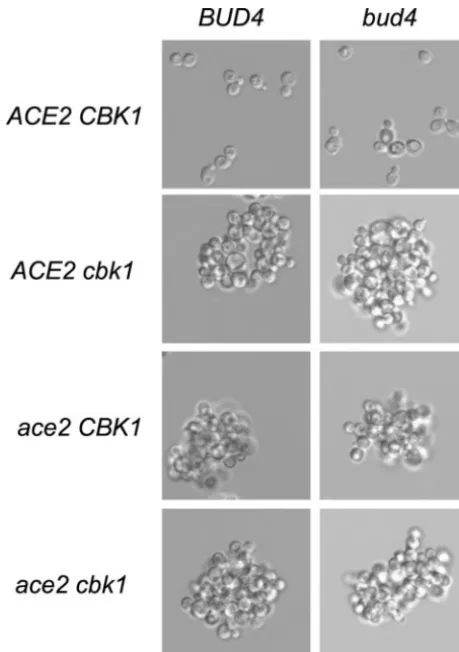
link-age to IME1⫹ in this cross. As shown in Fig. 1A, a smooth
colony phenotype is seen in strains with a single mutation in
either CBK1or BUD4. Importantly, roughened colonies are
TABLE 2. Plasmids used in this study
Plasmid Description Source or reference
M4213 BUD4in YIp-URA3plasmid This work M4214 BUD4in YCp-URA3plasmid This work
pRS426 YEp-URA3vector 4
pGB1-1 ACE2in YEp-URA3plasmid 6 M4104 CBK1in YEp-URA3plasmid This work M4105 HYM1in YEp-URA3plasmid This work M4298 MOB2in YEp-URA3plasmid This work M4102 KIC1in YEp-URA3plasmid This work
TABLE 3. MulticopyACE2suppresses RAM network mutants
Plasmid
Phenotypea:
ace2 cbk1 hym1 mob2 kic1
YEp vector ⫺ ⫺ ⫺ ⫺ ⫺
YEp-ACE2 ⫹ ⫹ ⫹ ⫹ ⫹
YEp-CBK1 ⫺ ⫹ ⫺ ⫺ ⫺
YEp-HYM1 ⫺ ⫺ ⫹ ⫺* ⫺
YEp-MOB2 ⫺ ⫺ ⫺* ⫹ ⫺*
YEp-KIC1 ⫺ ⫺ ⫺ ⫺ ⫹
a⫺
, mutant phenotype, altered colony morphology, and lowCTS1-lacZ ex-pression;⫹, normal phenotype, normal colony morphology, and normal CTS1-lacZexpression (CTS1-lacZexpression based on blue/white filter assays in the presence of X-Gal); *, no suppression ofCTS1-lacZexpression defect, but mild suppression of colony morphology defect.
on September 8, 2020 by guest
http://ec.asm.org/
seen only when bothCBK1andBUD4are mutant. Similarly, theace2 bud4double mutant has the altered colony morphol-ogy (Fig. 1A).
To investigate the roles of BUD4 and CBK1 on budding
patterns, isogenic strains were stained with Calcofluor, which stains bud scars. As shown in Table 4 (top 12 strains), wild-type W303 haploid strains display a mixture of axial and bipolar budding patterns (strains DY150 and DY151), due to their
naturalbud4defect, while theBUD4derivatives are exclusively
axial (strains DY6604 and DY6605). Acbk1mutation has no
effect on budding pattern in BUD4 haploid cells (compare
strains DY6604 and DY6605 to strains DY9259 and DY9260).
Interestingly, combining thebud4andcbk1mutations in
hap-loid cells (strains DY5794 and DY5795) causes essentially all cells to bud randomly, but this is not seen in either single
mutant. Abud4 mutation does not alter budding pattern in
diploids (compare strains DY8497 and DY1640), as reported
previously (9). We also note that in diploids acbk1mutation
results in exclusively random budding, even when the wild-type
BUD4 gene is present (strain DY9216). We conclude that
Cbk1 is required for normal budding in both haploid and
diploid cells and that the combination ofcbk1with loss of Bud4
activity, either through mutation or cell-type-specific repres-sion, results in random budding.
It has been previously noted thatbud4 cbk1haploid cells are
very round (28). We find that BUD4 cbk1 haploid cells are
equivalently round and that acbk1mutation affects cell shape
in both haploids and diploids. Budding defects are often
asso-ciated with a loss of cell polarity (3, 26), andbud4 cbk1haploid
FIG. 1.ace2,cbk1, andbud4affect colony morphology in haploids and diploids. (Top) Cells were grown on a YEPD plate for 3 days at 30°C. Haploid strains DY2396 (bud4), DY3923 (bud4 ace2), DY5794 (bud4 cbk1), DY5796 (bud4 ace2 cbk1), DY9197 (BUD4), DY9190 (BUD4 ace2), DY9195 (BUD4 cbk1), and DY9188 (BUD4 ace2 cbk1) were used. (Bottom) Cells were grown on a YEPD plate for 2 days at 30°C. Diploid strains DY1640 (bud4), DY8566 (bud4 ace2), DY8567 (bud4 cbk1), DY9400 (bud4 ace2 cbk1), DY8497 (BUD4), DY9399 (BUD4 ace2), DY9216 (BUD4 cbk1), and DY9401 (BUD4 ace2 cbk1) were used.
TABLE 4. cbk1causes a random budding phenotypea
Strain Genotype
% with budding:
Axial Bipolar Random
DY6604 MATaBUD4 CBK1 99.5⫾0.9 0.3⫾0.6 0.2⫾0.3
DY6605 MAT␣BUD4 CBK1 99.8⫾0.3 0.2⫾0.3 0.0⫾0.0
DY8497 MATa/MAT␣BUD4 CBK1 2.3⫾1.9 94.9⫾5.9 2.8⫾4.0
DY150 MATabud4 CBK1 32.1⫾2.9 64.5⫾3.7 3.3⫾0.8
DY151 MAT␣bud4 CBK1 41.7⫾5.6 51.5⫾7.1 6.6⫾1.8
DY1640 MATa/MAT␣bud4 CBK1 1.8⫾0.8 89.7⫾0.3 8.5⫾1.0
DY9259 MATaBUD4 cbk1 99.3⫾0.3 0.2⫾0.3 0.5⫾0.5
DY9260 MAT␣BUD4 cbk1 100⫾0.0 0.0⫾0.0 0.0⫾0.0
DY9216 MATa/MAT␣BUD4 cbk1 0.3⫾0.6 0.0⫾0.0 99.7⫾0.6
DY5794 MATabud4 cbk1 1.5⫾0.5 0.0⫾0.0 98.5⫾0.5
DY5795 MAT␣bud4 cbk1 1.0⫾0.5 0.0⫾0.0 99.0⫾0.5
DY8567 MATa/MAT␣bud4 cbk1 0.0⫾0.0 0.0⫾0.0 100⫾0.0
DY9197 MATaBUD4 ACE2 96.0⫾1.7 3.7⫾1.2 0.3⫾0.6
DY8497 MATa/MAT␣BUD4 ACE2 2.3⫾1.5 85.0⫾2.0 12.7⫾0.6
DY2396 MATabud4 ACE2 22.3⫾5.5 62.0⫾4.6 15.7⫾2.3
DY1640 MATa/MAT␣bud4 ACE2 1.7⫾0.6 98.0⫾0.6 0.7⫾0.6
DY9190 MATaBUD4 ace2 99.3⫾0.6 0.3⫾0.6 0.3⫾0.6
DY9399 MATa/MAT␣BUD4 ace2 7.1⫾2.6 86.0⫾4.2 6.7⫾2.9
DY3923 MATabud4 ace2 1.7⫾0.6 96.0⫾1.0 2.3⫾0.6
DY8566 MATa/MAT␣bud4 ace2 1.0⫾0.0 93.0⫾1.2 5.7⫾1.2
aLog-phase cultures were sonicated, fixed with 3.7% formaldehyde, and stained with Calcofluor to visualize bud scars. For each strain, at least 200 cells were examined.
on September 8, 2020 by guest
http://ec.asm.org/
cells both bud randomly and have lost cell polarity (3, 28).
However, the fact thatBUD4 cbk1haploids bud normally but
retain the round phenotype associated with a defect in polar-ized cell growth suggests that cell polarity defects may not always be revealed by a random budding pattern.
Strains with either anace2or acbk1mutation have a defect
in cell separation, resulting in grape-like clusters of cells visible
under the microscope (12, 28). To investigate the role ofBUD4
in this phenotype, we examined isogenic strains under the microscope. The results show no difference in the size of the
clusters betweencbk1 BUD4andcbk1 bud4strains or between
ace2 BUD4andace2 bud4strains (Fig. 2).
To quantitate this cell separation defect, we developed a timed sedimentation assay. We have observed that when liquid
cultures oface2orcbk1mutants are placed on the bench, the
turbidity of the culture rapidly clears as the cells fall to the bottom of the tube. We have ascribed this rapid sedimentation
at 1⫻gto the large clusters of cells, and we thought we could
use this sedimentation in a quantitative assay. Cells growing in liquid culture were vortexed vigorously and transferred to a cuvette, and the optical density at 660 nm was measured over time. We defined the sedimentation half-time as how long it takes the optical density to fall to one-half of the difference
between the initial and the final lower plateau values. The rapid sedimentation in these mutants is not reversed by
addi-tion of EDTA, and thus it is not due to the Ca2⫹-dependent
flocculation described in some mutants (36).
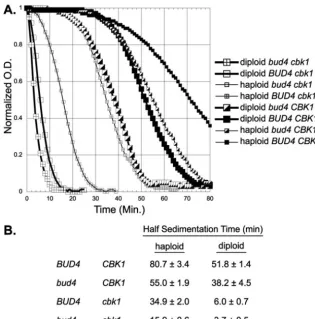
As shown in Fig. 3, thebud4 cbk1cultures clear rapidly, with
sedimentation half-times of 16 min for haploids and 4 min for diploids. In contrast, there is little change in the optical density
for the haploid BUD4 CBK1 strain until after 50 min. The
results from this assay allow us to make several conclusions.
First, thebud4and cbk1mutations each separately cause an
increase in sedimentation. Second, thebud4 cbk1double
mu-tant shows an additive increase in sedimentation, compared to the single mutants. Moreover, the effect of other RAM
muta-tions in combination withbud4is identical to that ofcbk1 in
the sedimentation assay (data not shown). Finally, we note that diploid cells show a faster sedimentation rate than haploids for all four genotypes tested. This could be because of the
incom-plete penetrance of the bipolar phenotype inbud4 haploids
relative to diploids and also because of the larger overall size of diploids.
Correlation between budding, sedimentation, and colony morphology. In the sedimentation assay, we noted that the
cbk1diploid cells sedimented rapidly whilecbk1haploids
sedi-mented slowly. Based on this difference, we examined the colony morphology of these strains and observed an altered
morphology in theBUD4 cbk1diploid that is different from
that in the BUD4 cbk1 haploid (Fig. 1B). Examining these
three phenotypes, colony morphology, sedimentation rate, and budding pattern, we noted an important correlation. All of the strains listed in Table 5 showing axial or bipolar budding are smooth and sediment slowly, while the strains with a random budding pattern display the altered colony morphology and sediment rapidly. The correlation in these results suggests that the random budding pattern is responsible for the changes in colony appearance and sedimentation rate.
To investigate this idea further, we examined sedimentation
rates and budding patterns in ace2 mutants, since an ace2
mutation has a similar effect on colony morphology ascbk1.
Figure 4 shows the results of sedimentation assays for isogenic
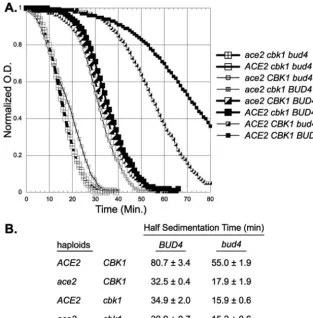
haploid strains differing at theACE2,CBK1, andBUD4loci. In
BUD4 strains, either an ace2 or a cbk1 mutation causes a
considerable increase in sedimentation. Importantly, theace2
bud4andcbk1 bud4strains show a significantly increased
sed-imentation rate compared to theace2 BUD4andcbk1 BUD4
strains. Thus, ace2 and cbk1 are similar in that both show
additive effects when combined with abud4mutation. There is
a very modest increase in sedimentation in theace2 cbk1
dou-ble mutant compared to the rate in each single mutant. This
increased sedimentation in theace2 cbk1strain is seen in both
the BUD4 or bud4 genotypes but is more significant in the
BUD4strains. In diploid cells, a similar additive increase in
sedimentation rate is also seen inace2 bud4double mutants
compared to that in the single mutants (data not shown). We
conclude that while ace2 and cbk1 mutations are similar in
both showing strong additive effects when combined with a
bud4mutation, the additive effect of combiningace2withcbk1
suggests they have independent as well as overlapping func-tions. As described in the Discussion, we believe the indepen-dent functions of Ace2 and Cbk1 are distinct from their over-lapping functions in cell separation.
FIG. 2. Clumpy morphology oface2andcbk1mutants. Cells were grown in liquid YEPD medium and examined microscopically using Nomarski optics. Strains DY9197 (BUD4), DY9195 (BUD4 cbk1), DY9190 (BUD4 ace2), DY9188 (BUD4 ace2 cbk1), DY2396 (wild type), DY5794 (bud4 cbk1), DY3923 (bud4 ace2), and DY5796 (bud4 ace2 cbk1) were used.
on September 8, 2020 by guest
http://ec.asm.org/
The measurements of budding patterns show a marked
dif-ference between ace2 and cbk1 mutants (Table 4). Haploid
bud4 ace2 cells show bipolar budding, while haploid bud4
cbk1 cells bud randomly (compare DY3923 to DY5794 and
DY5795). Additionally, while acbk1mutation causes random
budding in diploids (DY9216 and DY8567), diploidace2and
bud4 ace2 strains show the typical bipolar pattern (DY9399
and DY8566). We conclude that an ace2 mutation does not
affect budding pattern. This marked difference betweenace2
andcbk1phenotypes suggests independent functions.
Table 6 summarizes the effects oface2andbud4mutations
on three phenotypes: budding, sedimentation, and colony mor-phology. Importantly, the rough colony morphology does cor-relate with a rapid sedimentation rate. However, unlike the
situation withcbk1mutants (Table 5), none of theace2
mu-tants shows random budding morphology. We conclude that a random budding pattern is not sufficient for the rough colony
appearance and the rapid sedimentation, as the ace2 bud4
strain with bipolar budding also shows these phenotypes.
Additive effects betweencbk1andswi5mutations.We used the sedimentation assay to investigate the genetic interactions
between ACE2, SWI5, and CBK1. We have previously
ob-served qualitatively increasing severity of cell separation
de-fects inswi5,ace2, andace2 swi5double-mutant strains (12),
and here we see a corresponding decrease in sedimentation
half-time, withswi5at 33 min,ace2at 18 min, andace2 swi5at
6 min (Table 7). Interestingly, the 16-min sedimentation
half-time in thecbk1single mutant is similar to that seen in theace2
FIG. 3.bud4andcbk1affect sedimentation rates. (A) Cells were grown in liquid YEPD medium, and the sedimentation rate at 1⫻gwas determined as described in Materials and Methods. Strains DY8567 (diploidbud4 cbk1), DY9216 (diploidBUD4 cbk1), DY5794 (haploidMATa bud4 cbk1), DY9195 (haploidMATaBUD4 cbk1), DY1640 (diploidbud4), DY8497 (diploidBUD4), DY2396 (haploidMATabud4), and DY9197 (haploidMATaBUD4) were used. Similar results were seen with haploidMAT␣strains. (B) From the data in panel A, the sedimentation half-time is the time for the optical density (O.D.) to fall to one-half of the difference between the initial and the final lower plateau values.
TABLE 5. Budding, sedimentation, and colony morphology incbk1mutants
Mutant
Haploid Diploid
Colony
appearance Sedimentation rate Budding pattern
Colony
appearance Sedimentation rate Budding pattern
BUD4 CBK1 Smooth Slow Axial Smooth Slow Bipolar
bud4 CBK1 Smooth Slow Bipolar Smooth Intermediate Bipolar
BUD4 cbk1 Smooth Intermediate Axial Rough Rapid Random
bud4 cbk1 Rough Rapid Random Rough Rapid Random
on September 8, 2020 by guest
http://ec.asm.org/
single mutant, consistent with thecbk1mutation affecting Ace2 activity. We see a strong additive effect on sedimentation in the
ace2 swi5double mutant, compared to that in the single mu-tants, consistent with the fact that transcriptional activation of some cell separation and other genes requires either Ace2 or Swi5.
To investigate the additive effect ofcbk1andswi5mutations
on sedimentation, we examined the effect of these mutations,
along with ace2 and bud4, on transcription of known Ace2
target genes. RNA was prepared from 16 isogenic strains
dif-fering at the ACE2, BUD4, CBK1, and SWI5 loci, and S1
protection assays were performed to measure mRNA levels for
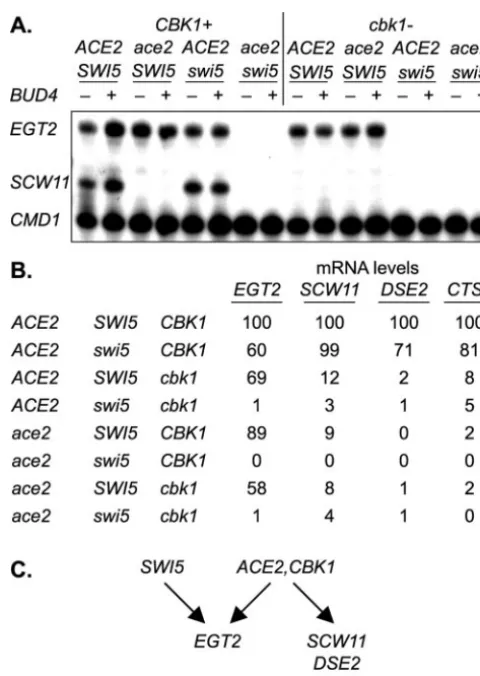
EGT2, SCW11, DSE2, andCTS1 (Fig. 5) (data not shown).
First, we can conclude that abud4mutation has no effect on
expression of these genes. Additionally, Western blots probed with antibody against Ace2 show the same protein level in
BUD4orbud4extracts, indicating that there is also no
post-transcriptional effect of Bud4 on Ace2 (data not shown).
Sec-ond, the pattern of regulation is different forEGT2from those
forSCW11,DSE2, andCTS1. EGT2expression is only
mod-estly reduced when there is a single mutation inACE2,SWI5,
orCBK1, but expression is reduced in theace2 swi5or thecbk1
swi5double mutant. Importantly,EGT2is still expressed in the
ace2 cbk1double mutant, consistent with Ace2 and Cbk1 func-tioning in the same pathway, separate from the Swi5 pathway.
We conclude thatEGT2expression can be activated by either
of the two related DNA-binding proteins, Ace2 or Swi5. In
contrast, expression ofSCW11,DSE2, andCTS1is unaffected
FIG. 4.bud4andace2affect sedimentation rates. (A) Cells were grown in liquid YEPD medium, and the sedimentation rate at 1⫻gwas determined as described in Materials and Methods. Strains DY5796 (bud4 ace2 cbk1), DY5794 (bud4 cbk1), DY3923 (bud4 ace2), DY9188 (BUD4 ace2 cbk1), DY9190 (BUD4 ace2), DY9195 (BUD4 cbk1), DY2396 (bud4), and DY9197 (BUD4) were used. (B) From the data in panel A, the sedimentation half-time is the time for the optical density (O.D.) to fall to one-half of the difference between the initial and the final lower plateau values.
TABLE 6. Budding, sedimentation, and colony morphology inace2mutants
Mutant
Haploid Diploid
Colony
appearance Sedimentation rate Budding pattern
Colony
appearance Sedimentation rate Budding pattern
BUD4 ACE2 Smooth Slow Axial Smooth Slow Bipolar
bud4 ACE2 Smooth Slow Bipolar Smooth Intermediate Bipolar
BUD4 ace2 Smooth Intermediate Axial Rough Rapid Bipolar
bud4 ace2 Rough Rapid Bipolar Rough Rapid Bipolar
on September 8, 2020 by guest
http://ec.asm.org/
by answi5mutation and these genes require bothACE2and
CBK1for full expression.
DISCUSSION
A cbk1mutation sharply reduces expression of genes
acti-vated by the Ace2 transcription factor, and bothcbk1andace2
mutants display an altered colony phenotype. A multicopy
ACE2plasmid suppresses both the transcriptional defects and
the altered colony morphology of thecbk1mutant and other
mutants in the RAM pathway, demonstrating that Ace2 is downstream of Cbk1. Many of the Ace2 target genes, such as
CTS1,DSE2, andSCW11, facilitate cytokinesis, andace2and
cbk1mutants both display defects in cell separation. In
addi-tion to their common role in transcripaddi-tional activaaddi-tion, we show that Ace2 and Cbk1 have distinct functions in cellular morphology and cell division. The W303 strain background has
a mutation in theBUD4gene, required for axial budding in
haploid cells. Combining abud4mutation with eitherace2or
cbk1has an additive effect, seen as a rough colony morphology.
The random budding pattern resulting from overall loss of
polarity incbk1 mutants is evident only due to genetic
inter-action between cbk1 and bud4. In addition, we also used a
sedimentation assay to show that thebud4 cbk1double mutant
has quantitatively additive effects on multicellular morphology.
A mutation in SWI5, an ACE2 paralog, has additive effects
when combined with cbk1 on both sedimentation rate and
EGT2expression, suggesting that Cbk1 has functions distinct
from that of Ace2. In summary, we conclude that Ace2 and Cbk1 have independent as well as overlapping functions.
The RAM network comprisesCBK1,HYM1,KIC1,MOB2,
andTAO3. Mutations in any of these genes have similar
phe-notypes, including decreasedCTS1expression, failure in cell
separation, altered colony morphology, and defects in polar-ized cell growth (3, 25, 28). Additionally, RAM gene products show many mutual physical and functional interactions (16, 19,
25, 28). Similar effects onCTS1expression, cell separation, and
colony morphology are seen in anace2mutant. We show that
multicopy plasmids withACE2can suppress the effects ofcbk1,
hym1,kic1, andmob2RAM network mutations with respect to
the rough colony morphology and failure of cell separation after mitosis, as well as the defect in transcriptional activation of genes necessary for that cell separation. This multicopy suppression shows that Ace2 is functionally downstream of the RAM pathway components, consistent with other studies (3, 25, 28, 40). Strains lacking Cbk1 show a loss of expression in
CTS1andSCW11to an extent that is similar to, but slightly less
severe than, that inace2mutants. This suggests that Ace2 may
have some residual function in RAM network-mutated strains, and elevating levels of Ace2 with a multicopy plasmid may provide enough active Ace2 to restore some function. Finally,
polarized cell growth is normal inace2mutants, demonstrating
that the RAM network has functions independent of Ace2.
W303 strains have a bud4 mutation.Although ace2 gene disruptions cause a failure of cell separation in all genetic backgrounds tested, there is a difference in colony morphology
between strain backgrounds. S288cace2strains have a smooth,
wild-type colony surface, while W303 ace2 strains show the
same altered colony morphology seen in W303 strains with RAM network mutations. This difference can be explained by
the fact that W303 strains contain a bud4 mutation. While
FIG. 5.cbk1andswi5mutations have an additive effect onEGT2
expression. (A) S1 nuclease protection assays were performed using probes specific for EGT2, SCW11, and CMD1 (internal control). RNAs were prepared from the following strains: DY2396 (bud4), DY9197 (BUD4), DY3923 (bud4 ace2), DY9190 (BUD4 ace2), DY3925 (bud4 swi5), DY9194 (BUD4 swi5), DY4653 (bud4 ace2 swi5), DY9186 (BUD4 ace2 swi5), DY5794 (bud4 cbk1), DY9195 (BUD4 cbk1), DY5796 (bud4 ace2 cbk1), DY9188 (BUD4 ace2 cbk1), DY5798 (bud4 swi5 cbk1), DY9192 (BUD4 swi5 cbk1), DY5800 (bud4 ace2 swi5 cbk1), and DY9184 (BUD4 ace2 swi5 cbk1). (B) mRNA levels were determined by PhosphorImager forEGT2andSCW11(from the data in panel A) and forDSE2andCTS1(data not shown). mRNA values are normalized to the wild-type strain. For this experiment, results are only shown forbud4strains, but similar results were seen withBUD4
strains. (C)EGT2activation requires either Swi5 or both Ace2 and Cbk1, whileSCW11,DSE2, andCTS1expression requires both Ace2 and Cbk1 but not Swi5.
TABLE 7. cbk1andswi5mutations have an additive effect on sedimentation
Mutant
ACE2 ace2
Strain
Sedimentation half-time
(min)
Strain
Sedimentation half-time
(min)
SWI5 CBK1 DY2396 55.0⫾1.9 DY3923 17.9⫾1.9
SWI5 cbk1 DY5794 15.9⫾0.6 DY5796 15.2⫾0.6
swi5 CBK1 DY3925 32.7⫾0.6 DY4653 6.2⫾1.1
swi5 cbk1 DY5798 6.0⫾1.2 DY5800 5.0⫾1.0
on September 8, 2020 by guest
http://ec.asm.org/
S288c strains display axial budding in haploids and bipolar budding in diploids, W303 cells show bipolar bud site selection in haploids as well as diploids. Low-copy plasmids bearing
BUD4derived from S288c genomic DNA convert the W303
haploid pattern from bipolar to axial, and genetic crosses with
a markedBUD4⫹allele shows complete 2:2 segregation with
respect to the W303 bipolar haploid phenotype. W303ace2
strains converted toBUD4⫹now show a smooth colony
mor-phology similar to S288cace2cells. Thus, combining a
muta-tion that reduces cell separamuta-tion, such asace2orcbk1, with a
mutation that causes bipolar budding, such as bud4or axl1,
causes a change in colony morphology. Importantly,ace2/ace2
a/␣diploids show rough colonies in either strain background,
even though allace2strains still show the same cell separation
defect regardless of cell type orBUD4dosage. This indicates
that the rough colony appearance is caused by the combination of two effects: a bipolar budding pattern and cells remaining attached postmitotically. We suggest that this combination re-sults in more extended assemblages of cells than for unsepa-rated cells budding axially, thus affecting the colony morphol-ogy.
Mutations in genes of the RAM network are lethal in certain genetic backgrounds, including the S288c-derived strain used by the Yeast Genome Deletion Consortium (41). However, these gene disruptions are viable in the W303 background. This strain-dependent difference in lethality is due to
polymor-phism at theSSD1locus, with S288c possessing the dominant
SSD1-v allele and W303 bearing the recessive ssd1-d allele
encoding a truncated protein (20). Additionally, S288c strains
tolerate RAM network gene disruptions if anssd1mutation is
introduced (16, 20). Thus, most studies of RAM network genes
have necessarily been carried out in the ssd1 mutant W303
background. Several lines of evidence point to a physical in-teraction of Ssd1 with members of the RAM network (19, 28), and the molecular role of Ssd1 is hypothesized to be as a regulator of protein dephosphorylation through protein phos-phatases (34).
The fact that viable RAM network mutations were isolated
primarily in W303, which also bore abud4mutation, has made
analysis of the effects of RAM network genes on bud site selection, cell separation, and colony morphology problematic. Microscopic observation is especially challenging, considering the large clusters of cells due to defects in cell separation. We
have constructed W303 strains with restored BUD4function
such that both diploids and haploids now display the normal
bipolar and axial patterns, respectively. W303 haploidBUD4⫹
strains with a cbk1 mutation are still fully viable and show
completely normal nonrandom axial budding. WhileBUD4⫹
CBK1⫹ diploids show the normal bipolar budding pattern,
BUD4⫹cbk1⫺diploids bud randomly. This is reflected in
col-ony morphology and sedimentation rate due to the cell
sepa-ration defect and occurs because theBUD4⫹(axial) function is
dispensable in diploids. Thus the cytoskeletal cell polarity cues programmed by Cbk1 and other RAM factors are necessary only for the bipolar pattern, in which budding machinery is deposited at alternating, antipolar termini of the long axis of the cell in each successive cycle, but not for the axial pattern in which localized tags must only be retained proximal to the site of previous budding (8). Without the polarity axis dependent
on Cbk1, undirected budding machinery randomly locates to medial sites in the absence of the axial program.
Budding pattern and cell separation effect colony morphol-ogy and sedimentation.Mutations in genes of the RAM
net-work, includingCBK1,MOB2,KIC1, andHYM1, as well as a
mutation in the downstream transcription factorACE2, cause
a rough colony morphology in the W303 genetic background
with abud4mutation. This defect consists of scalloped rather
than smooth colony margins and a surface with many branched channels and invaginations resulting in an eroded appearance. At the microscopic level, these rough colony mutants exhibit incomplete scission of the mother-daughter cell wall septum after cytokinesis. Since all other aspects of the cell cycle in-cluding cytokinesis continue in an apparently unaffected way, this loss of cell separation leads to the formation of multicel-lular aggregates in liquid medium, called clusters or clumps. The failure of cell separation is distinct from flocculent phe-notypes caused by aggregation due to expression of cell surface agglutinins (36). Unlike flocculent strains, the clustered
phe-notype of RAM network andace2mutants cannot be restored
to unicellularity by chelation of Ca2⫹from the medium and
thus is a stable morphogenetic state of the cell wall.
Impor-tantly, a multicopy plasmid withACE2suppresses the
unsepa-rated state of RAM mutants, demonstrating that the decreased
expression ofACE2target genes is responsible for the
pheno-type.
The sedimentation rate assay was developed as a way to quantify phenotypes which were normally observed in only a qualitative way as colony morphology defects. Several factors probably contribute to the end measurement. The obvious one is the average size of cell clusters brought about by incomplete scission after cytokinesis. Such clumping will lead to a lower aggregate frontal surface area for a given number of cells and thus less overall resistance to movement through a viscous fluid. Additionally, changes in the overall compactness of clus-ters of a given size would affect the sedimentation rate. For instance, bipolar bud site selection should result in a more extended filament-like structure than an axial program, in which daughter buds are formed in close apposition to previ-ous daughters. Random bud site selection, in which a large number of medial buds form, might result in an intermediate density for a clump of a given cell number. In contrast, axially budding clumpy cells would exhibit the densest clusters. How-ever, our observations indicate that clumpy cells with a bipolar or random pattern actually sediment more rapidly than those with an axial pattern (Fig. 4), and yet there is no evidence for differences in cell number per cluster due to bud site selection pattern differences (Fig. 2) or in expression of cell separation
genes in response toBUD4genotype (Fig. 5). To explain the
more rapid sedimentation rates of bipolar clumpy cells relative to axial clumpy cells, we suggest that the bipolar clusters have longer, narrower, and more rod-like clusters, leading to more rapid sedimentation. In contrast, axially budded clusters, al-though not larger in any one dimension than bipolar clusters of the same cell number, are more uniform in all dimensions and so are unable to adopt any orientation with a low frontal surface area that would speed sedimentation.
One phenotype ofcbk1mutants is that the cells themselves
are rounder, having a smaller length-to-width ratio than wild-type cells (28). Rounder cells may form a more compact clump
on September 8, 2020 by guest
http://ec.asm.org/
than the typical ellipsoid cells, since any resulting chain-like structures of a given number of cells would be shorter in contour length and thus manifested in changes in the sedimen-tation assay. Coupled with microscopic observations of bud-ding patterns and cell shape, the sedimentation assay allows measurements of relatively subtle differences in phenotype in a quantifiable way.
Additive genetic effects. Strong additive effects are seen
when abud4mutation is combined with eitherace2orcbk1,
resulting in altered colony morphology and more rapid sedi-mentation. Slight additive effects are also seen in the
sedimen-tation assay whenace2andcbk1mutations are combined (Fig.
4). One interpretation of thisace2 cbk1additivity is that the
Cbk1 kinase also directs cell separation, independently of Ace2, by modifying other transcriptional activators for cell separation enzymes or specific cell wall components. Previous quantification of the extent of cell separation defects indicated
a small increase in the average clump size inace2/ace2diploids
relative to cbk1/cbk1 diploids, irrespective of cell shape or
budding pattern differences (3). Additionally, although
expres-sion ofACE2target genes is more reduced in anace2mutant
than in acbk1mutant,ace2is epistatic tocbk1, since there is
not an additive effect in theace2 cbk1double mutant (Fig. 5)
(data not shown).Thus, the similar sedimentation rates inace2
bud4andcbk1 bud4strains could be a product of the slightly
reduced clumpiness ofcbk1cells in combination with the
ran-domized budding pattern. The round cell phenotype seen in
cbk1mutants could also contribute to a more compact average
cluster with reduced frontal area and less resistance to
sedi-mentation relative to the total cell number. Whilecbk1 BUD4
haploid cells bud in the wild-type axial manner, these cells still possess a shortened long axis due to overall polarity defects. This may also lead to a more compact cluster due to shorter chains of unseparated cells, which is again quantitatively
indis-tinguishable from ellipsoidal ace2 strains in sedimentation
measurements, due to their slightly larger clump size. The
slight additive effect inace2 cbk1strains could be due to the
slightly increased clump size caused byace2, coupled with the
round cell phenotype and/or budding pattern change caused by
thecbk1mutation, depending on the allele ofBUD4present.
Most of the target genes identified to date that require Ace2 for activation are cell wall-degrading enzymes or putative cell wall components (11, 12, 14). This is consistent with the
in-complete cell wall scission phenotypes oface2mutants, as well
as for the phenotypes of mutations inCBK1, necessary for full
Ace2 activity. However, some genes which are directly acti-vated by Ace2 can also be actiacti-vated by Swi5, a transcription
factor paralogous to Ace2 (14, 24).EGT2, which encodes an
endoglucanase, is activated by either Ace2 or Swi5 (Fig. 5), and
egt2-null mutants show defects in cell separation (21). Addi-tionally, some cell separation genes which are predominantly
activated by Ace2 are expressed at a low level in an ace2
mutant, but this residual expression is completely abolished in anace2 swi5double mutant. Our sedimentation assays show a
strong additive effect in combiningcbk1 and swi5 mutations
(Table 7), leading to the question of whether acbk1mutation
affects Swi5 localization. However, the pattern of nuclear lo-calization of an Swi5-green fluorescent protein fusion protein
is the same in bothCBK1 and cbk1 cells (data not shown).
Instead, we believe thecbk1 swi5additivity in the
sedimenta-tion assays is consistent with the idea that activasedimenta-tion of some genes requires either Ace2 or Swi5. Thus, the sedimentation assays are able to detect relatively subtle changes in expression
of cell separation genes such asEGT2.
TheACE2gene itself is expressed in the late G2and early M
phase of the cell cycle (12), as are other genes required for
cytokinesis, which are part of theCLB2cell cycle group (10,
33). This timing leads to protein production at the time needed
for cell division.BUD4is part of thisCLB2group, and
tem-porally the Bud4 protein localizes to the bud neck region similarly to Cbk1, along with many other morphogenetic
fac-tors (30). BUD9 encodes one of the localized tags for the
bipolar pattern (31), andbud9mutant diploids adopt a
unipo-lar budding pattern (43).BUD9expression is reduced in acbk1
mutant (25) and also in anace2mutant (W. P. Voth., Y. Yu,
and D. J. Stillman, unpublished observations). Most laboratory strains of yeast grow in a predominantly unicellular manner, but yeasts freshly isolated from their wild habitats are naturally clumpy (42). This difference has been attributed to the fact that
laboratory strains of yeasts are mutated for theAMN1gene,
while the wild isolates bear a functional version of this gene (42). Genetically, Amn1 behaves as a repressor of Ace2 func-tion (42). Further work is needed to understand this relafunc-tion-
relation-ship, as AMN1is reported to be expressed only in daughter
cells (11), butAMN1expression can be activated by either Swi5
or Ace2 (14). Thus, it is tempting to speculate that facultative modulation of Cbk1 and other RAM gene activity could cause alterations in bud site selection, cell polarity, and cell separa-tion, which could be a normal part of coordinated regulation of morphogenetic transitions due to the influence of environmen-tal factors.
ACKNOWLEDGMENTS
We thank Emily Parnell for determining the effect of acbk1 muta-tion on localizamuta-tion of the Swi5-green fluorescent protein fusion pro-tein.
This work was supported by a grant from the National Institutes of Health awarded to D.J.S.
REFERENCES
1.Baudin, A., O. Ozier-Kalogeropoulos, A. Denouel, F. Lacroute, and C. Cul-lin.1993. A simple and efficient method for direct gene deletion in Saccha-romyces cerevisiae. Nucleic Acids Res.21:3329–3330.
2.Bhoite, L. T., and D. J. Stillman.1998. Residues in the Swi5 zinc finger protein that mediate cooperative DNA binding with the Pho2 homeodomain protein. Mol. Cell. Biol.18:6436–6446.
3.Bidlingmaier, S., E. L. Weiss, C. Seidel, D. G. Drubin, and M. Snyder.2001. The Cbk1p pathway is important for polarized cell growth and cell separa-tion inSaccharomyces cerevisiae. Mol. Cell. Biol.21:2449–2462.
4.Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter, and J. D. Boeke.1998. Designer deletions strains derived fromSaccharomyces cerevi-siaeS288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast14:115–132.
5.Breeden, L., and K. Nasmyth.1987. Cell cycle control of the yeastHOgene: cis- and trans-acting regulators. Cell48:389–397.
6.Butler, G., and D. J. Thiele.1991.ACE2, an activator of yeast metallothio-nein expression which is homologous toSWI5. Mol. Cell. Biol.11:476–485. 7.Casamayor, A., and M. Snyder.2002. Bud-site selection and cell polarity in
budding yeast. Curr. Opin. Microbiol.5:179–186.
8.Chant, J.1999. Cell polarity in yeast. Annu. Rev. Cell Dev. Biol.15:365–391. 9.Chant, J., and I. Herskowitz.1991. Genetic control of bud site selection in yeast by a set of gene products that constitute a morphogenetic pathway. Cell
65:1203–1212.
10.Cho, R. J., M. J. Campbell, E. A. Winzeler, L. Steinmetz, A. Conway, L. Wodicka, T. G. Wolfsberg, A. E. Gabrielian, D. Landsman, D. J. Lockhart, and R. W. Davis.1998. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell2:65–73.
11.Colman-Lerner, A., T. E. Chin, and R. Brent.2001. Yeast Cbk1 and Mob2
on September 8, 2020 by guest
http://ec.asm.org/
activate daughter-specific genetic programs to induce asymmetric cell fates. Cell107:739–750.
12.Dohrmann, P. R., G. Butler, K. Tamai, S. Dorland, J. R. Greene, D. J. Thiele, and D. J. Stillman.1992. Parallel pathways of gene regulation: the homol-ogous regulatorsSWI5andACE2differentially control transcription ofHO
and chitinase. Genes Dev.6:93–104.
13.Dohrmann, P. R., W. P. Voth, and D. J. Stillman.1996. Role of negative regulation in promoter specificity of the homologous transcriptional activa-tors Ace2p and Swi5p. Mol. Cell. Biol.16:1746–1758.
14.Doolin, M. T., A. L. Johnson, L. H. Johnston, and G. Butler.2001. Over-lapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol.40:422–432.
15.Dorland, S., M. L. Deegenaars, and D. J. Stillman.2000. Roles for the
Saccharomyces cerevisiae SDS3, CBK1 andHYM1genes in transcriptional repression bySIN3. Genetics154:573–586.
16.Du, L. L., and P. Novick.2002. Pag1p, a novel protein associated with protein kinase Cbk1p, is required for cell morphogenesis and proliferation in Sac-charomyces cerevisiae. Mol. Biol. Cell13:503–514.
17.Fujita, A., M. Lord, T. Hiroko, F. Hiroko, T. Chen, C. Oka, Y. Misumi, and J. Chant.2004. Rax1, a protein required for the establishment of the bipolar budding pattern in yeast. Gene327:161–169.
18.Fujita, A., C. Oka, Y. Arikawa, T. Katagai, A. Tonouchi, S. Kuhara, and Y. Misumi.1994. A yeast gene necessary for bud-site selection encodes a protein similar to insulin-degrading enzymes. Nature372:567–570. 19.Ho, Y., A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A.
Millar, P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donald-son, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. Hogue, D. Figeys, and M. Tyers.
2002. Systematic identification of protein complexes inSaccharomyces cer-evisiaeby mass spectrometry. Nature415:180–183.
20.Jorgensen, P., B. Nelson, M. D. Robinson, Y. Chen, B. Andrews, M. Tyers, and C. Boone.2002. High-resolution genetic mapping with ordered arrays of
Saccharomyces cerevisiaedeletion mutants. Genetics162:1091–1099. 21.Kovacech, B., K. Nasmyth, and T. Schuster.1996.EGT2gene transcription
is induced predominantly by Swi5 in early G1. Mol. Cell. Biol.16:3264–3274.
22.Lord, M., T. Chen, A. Fujita, and J. Chant.2002. Analysis of budding patterns. Methods Enzymol.350:131–141.
23.Madden, K., and M. Snyder. 1998. Cell polarity and morphogenesis in budding yeast. Annu. Rev. Microbiol.52:687–744.
24.McBride, H. J., Y. Yu, and D. J. Stillman.1999. Distinct regions of the Swi5 and Ace2 transcription factors are required for specific gene activation. J. Biol. Chem.274:21029–21036.
25.Nelson, B., C. Kurischko, J. Horecka, M. Mody, P. Nair, L. Pratt, A. Zoug-man, L. D. McBroom, T. R. Hughes, C. Boone, and F. C. Luca.2003. RAM: a conserved signaling network that regulates Ace2p transcriptional activity and polarized morphogenesis. Mol. Biol. Cell14:3782–3803.
26.Ni, L., and M. Snyder. 2001. A genomic study of the bipolar bud site selection pattern inSaccharomyces cerevisiae. Mol. Biol. Cell12:2147–2170.
27.Pruyne, D., and A. Bretscher.2000. Polarization of cell growth in yeast. J. Cell Sci.113:571–585.
28.Racki, W. J., A. M. Becam, F. Nasr, and C. J. Herbert.2000. Cbk1p, a protein similar to the human myotonic dystrophy kinase, is essential for normal morphogenesis inSaccharomyces cerevisiae. EMBO J.19:4524–4532. 29.Rothstein, R.1991. Targeting, disruption, replacement, and allele rescue:
integrative DNA transformation in yeast. Methods Enzymol.194:281–302. 30.Sanders, S. L., and I. Herskowitz.1996. TheBUD4protein of yeast, required
for axial budding, is localized to the mother/BUD neck in a cell cycle-dependent manner. J. Cell Biol.134:413–427.
31.Schenkman, L. R., C. Caruso, N. Page, and J. R. Pringle.2002. The role of cell cycle-regulated expression in the localization of spatial landmark pro-teins in yeast. J. Cell Biol.156:829–841.
32.Sherman, F.1991. Getting started with yeast. Methods Enzymol.194:1–21. 33.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeastSaccharomyces cer-evisiaeby microarray hybridization. Mol. Biol. Cell9:3273–3297. 34.Sutton, A., D. Immanuel, and K. T. Arndt.1991. The SIT4 protein
phos-phatase functions in late G1for progression into S phase. Mol. Cell. Biol.
11:2133–2148.
35.Tamaskovic, R., S. J. Bichsel, and B. A. Hemmings.2003. NDR family of AGC kinases—essential regulators of the cell cycle and morphogenesis. FEBS Lett.546:73–80.
36.Teunissen, A. W., and H. Y. Steensma.1995. Review: the dominant floccu-lation genes of Saccharomyces cerevisiae constitute a new subtelomeric gene family. Yeast11:1001–1013.
37.Thomas, B. J., and R. Rothstein.1989. Elevated recombination rates in transcriptionally active DNA. Cell56:619–630.
38.Vieira, J., and J. Messing.1991. New pUC-derived cloning vectors with different selectable markers and DNA replication origins. Gene100:189– 194.
39.Voth, W. P., Y. W. Jiang, and D. J. Stillman.2003. New ‘marker swap’ plasmids for converting selectable markers on budding yeast gene disrup-tions and plasmids. Yeast20:985–993.
40.Weiss, E. L., C. Kurischko, C. Zhang, K. Shokat, D. G. Drubin, and F. C. Luca.2002. TheSaccharomyces cerevisiaeMob2p-Cbk1p kinase complex promotes polarized growth and acts with the mitotic exit network to facilitate daughter cell-specific localization of Ace2p transcription factor. J. Cell Biol.
158:885–900.
41.Winzeler, E. A., D. D. Shoemaker, A. Astromoff, H. Liang, K. Anderson, B. Andre, R. Bangham, R. Benito, J. D. Boeke, H. Bussey, A. M. Chu, C. Connelly, K. Davis, F. Dietrich, S. W. Dow, M. El Bakkoury, F. Foury, S. H. Friend, E. Gentalen, G. Giaever, J. H. Hegemann, T. Jones, M. Laub, H. Liao, R. W. Davis et al.1999. Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science285:901–906. 42.Yvert, G., R. B. Brem, J. Whittle, J. M. Akey, E. Foss, E. N. Smith, R.
Mackelprang, and L. Kruglyak.2003. Trans-acting regulatory variation in
Saccharomyces cerevisiaeand the role of transcription factors. Nat. Genet.
35:57–64.
43.Zahner, J. E., H. A. Harkins, and J. R. Pringle.1996. Genetic analysis of the bipolar pattern of bud site selection in the yeastSaccharomyces cerevisiae. Mol. Cell. Biol.16:1857–1870.