organic papers
o4358
Han and Zhen C18H18O4 doi:10.1107/S160053680503905X Acta Cryst.(2005). E61, o4358–o4359 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
4-[4-(4-Formylphenoxy)butoxy]benzaldehyde
Jian-Rong Han* and Xiao-Li Zhen
College of Sciences, Hebei University of Science & Technology, Shijiazhuang 050018, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 294 K
Mean(C–C) = 0.002 A˚ Rfactor = 0.039 wRfactor = 0.118
Data-to-parameter ratio = 15.3
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The molecule of the title compound, C18H18O4, lies on crystallographic center of symmetry. The benzaldehyde group makes a dihedral angle of 62.82 (16)with the four C atoms in
the central chain. The two aromatic ring are parallel to each other by symmetry.
Comment
The pioneering work of Pedersen (1967) on the synthesis of macrocyclic crown ethers was a milestone in this field of chemistry. These compounds are capable of forming stable and selective complexes with metal cations, halide anions and small organic molecules. Consequently, these species have been used to study their molecular recognition for special guest molecules and cations (Habata et al., 1996; Zhang & Buchwald, 2000). As part of our interest in the molecular and ionic recognition properties of crown ethers, we investigated the title compound, (I), used as a precursor in the preparation of crown ethers.
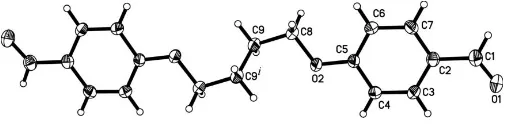
In (I) (Fig. 1), a crystallographic center of symmetry is located at the mid-point of the central C9—C9i bond [symmetry code: (i) x, y, z]. The benzaldehyde group (C1–C7/O1/O2) is planar, with an r..m.s. deviation for fitted atoms of 0.0353 A˚ . The chain of atoms C8—C9—C9i—C8i linking the two aromatic systems is exactly planar by symmetry. The results are similar to that reported recently for the closely related species 4-[6-(4-formyl-2-methoxyphen-oxy)hexyloxy]-3-methoxybenzaldehyde (Diaoet al., 2005) and 4-[4-(4-formyl-2-methoxyphenoxy)butoxy]-3-methoxybenz-aldehyde (Duan & Zhang, 2005). However, the dihedral angle between the bridge plane (C8/C9/C9i/C8i) and the benzalde-hyde group plane is 62.82 (16), in comparison with 3.0 (3)
and 55.80 (17) in
4-[6-(4-formyl-2-methoxyphenoxy)hexyl-oxy]-3-methoxybenzaldehyde and
[image:1.610.206.459.658.720.2]4-[4-(4-formyl-2-methoxy-Received 21 November 2005 Accepted 24 November 2005 Online 30 November 2005
Figure 1
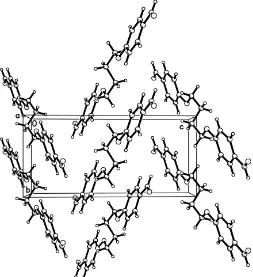
phenoxy)butoxy]-3-methoxybenzaldehyde, respectively. All bond lengths and angles (Table 1) are within normal ranges (Allenet al., 1987). The crystal packing arrangement, a zigzag pattern, is shown in Fig. 2.
Experimental
To a solution of 4-hydroxybenzaldehyde (12.2 g, 100 mmol) and potassium carbonate (13.8 g, 100 mmol) in acetonitrile (500 ml), 1,4-dibromobutane (10.8 g, 50 mmol) was added dropwise over a period of 30 min, and the mixture refluxed for 24 h under nitrogen. The solvent was removed and the resultant mixture poured into ice–water (500 ml). The white precipitate was isolated and recrystallized from ethanol to give the pure compound in 46% yield. Colorless single crystals of (I) suitable for X-ray analysis were obtained by slow evaporation of an acetonitrile solution.
Crystal data
C18H18O4 Mr= 298.32
Monoclinic,P21=c a= 7.988 (2) A˚
b= 6.6635 (16) A˚
c= 14.260 (4) A˚ = 96.354 (4) V= 754.4 (3) A˚3 Z= 2
Dx= 1.313 Mg m
3
MoKradiation Cell parameters from 1332
reflections = 2.9–26.5
= 0.09 mm1 T= 294 (2) K Block, colorless 0.300.260.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer ’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.960,Tmax= 0.984
4120 measured reflections
1547 independent reflections 1034 reflections withI> 2(I)
Rint= 0.027
max= 26.5 h=6!10
k=8!8
l=17!16
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.039 wR(F2) = 0.118 S= 1.00 1547 reflections 101 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.0611P)2
+ 0.0823P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.16 e A˚
3
min=0.14 e A˚
3
Extinction correction:SHELXL97
[image:2.610.314.567.72.349.2]Extinction coefficient: 0.053 (7)
Table 1
Selected geometric parameters (A˚ ,).
O1—C1 1.2110 (19) O2—C5 1.3582 (17)
O2—C8 1.4346 (17)
C5—O2—C8 118.84 (11) O1—C1—C2 125.16 (17) O2—C5—C6 124.70 (13)
O2—C5—C4 115.31 (13) O2—C8—C9 107.58 (12)
The H atoms were included in calculated positions and refined
= 1.2Ueq(C) for aromatic CH; C—H = 0.97 A˚ andUiso(H) = 1.2Ueq(C) for methylene CH2.
Data collection:SMART(Bruker, 1999); cell refinement:SAINT (Bruker, 1999); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997a); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997a); molecular graphics: SHELXTL(Sheldrick, 1997b); software used to prepare material for publication:SHELXTL.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L. Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans 2, pp. S1–19.
Bruker (1999).SMART(Version 5.0) andSAINT(Version 4.0) for Windows NT. Bruker AXS Inc., Madison, Wisconsin, USA.
Diao, C.-H., Guo, M.-J., Yu, M., Chen, X. & Jing, Z.-L. (2005).Acta Cryst.E61, o3670–o3671.
Duan, Z.-Y. & Zhang, W.-J. (2005).Acta Cryst.E61, o3355–o3356.
Habata, Y., Bradshaw, J. S., Young, J. J., Castle, S. L., Huszthy, P., Pyo, T., Lee, M. L. & Izatt, R. (1996).J. Org. Chem.61, 8391–8396.
Pedersen, C. J. (1967).J. Am. Chem. Soc.89, 7017–7036.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997a). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
Sheldrick, G. M. (1997b).SHELXTL97. Version 5.10 for Windows NT. Bruker AXS Inc., Madison, Wisconsin, USA.
Figure 2
supporting information
sup-1 Acta Cryst. (2005). E61, o4358–o4359
supporting information
Acta Cryst. (2005). E61, o4358–o4359 [https://doi.org/10.1107/S160053680503905X]
4-[4-(4-Formylphenoxy)butoxy]benzaldehyde
Jian-Rong Han and Xiao-Li Zhen
4-[4-(4-Formylphenoxy)butoxy]benzaldehyde
Crystal data C18H18O4
Mr = 298.32 Monoclinic, P21/c
Hall symbol: -P 2ybc a = 7.988 (2) Å b = 6.6635 (16) Å c = 14.260 (4) Å β = 96.354 (4)° V = 754.4 (3) Å3
Z = 2
F(000) = 316 Dx = 1.313 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 1332 reflections θ = 2.9–26.5°
µ = 0.09 mm−1
T = 294 K Block, colorless 0.30 × 0.26 × 0.18 mm
Data collection
Bruker SMART APEX CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin = 0.960, Tmax = 0.984
4120 measured reflections 1547 independent reflections 1034 reflections with I > 2σ(I) Rint = 0.027
θmax = 26.5°, θmin = 2.6°
h = −6→10 k = −8→8 l = −17→16
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.039
wR(F2) = 0.118
S = 1.00 1547 reflections 101 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0611P)2 + 0.0823P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.16 e Å−3
Δρmin = −0.14 e Å−3
Extinction correction: SHELXL97, Fc*=3DkFc[1+0.001xFc2λ3/sin(2θ)]-1/4
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is
used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 0.96911 (15) 0.5594 (2) 0.22503 (9) 0.0679 (4)
O2 0.28261 (12) 0.17200 (15) 0.04352 (8) 0.0479 (4)
C1 0.8247 (2) 0.6180 (3) 0.21598 (11) 0.0494 (4)
H1 0.8044 0.7464 0.2377 0.059*
C2 0.68015 (19) 0.5032 (2) 0.17366 (10) 0.0395 (4)
C3 0.69980 (19) 0.3054 (2) 0.14486 (11) 0.0417 (4)
H3 0.8049 0.2443 0.1551 0.050*
C4 0.56526 (18) 0.2003 (2) 0.10155 (11) 0.0414 (4)
H4 0.5798 0.0690 0.0818 0.050*
C5 0.40675 (18) 0.2895 (2) 0.08698 (10) 0.0369 (4)
C6 0.38422 (19) 0.4844 (2) 0.11758 (11) 0.0423 (4)
H6 0.2782 0.5437 0.1097 0.051*
C7 0.52146 (18) 0.5890 (2) 0.15986 (11) 0.0427 (4)
H7 0.5070 0.7203 0.1796 0.051*
C8 0.11617 (17) 0.2537 (2) 0.02442 (11) 0.0409 (4)
H8A 0.1185 0.3741 −0.0137 0.049*
H8B 0.0719 0.2883 0.0830 0.049*
C9 0.00722 (19) 0.0962 (2) −0.02776 (11) 0.0440 (4)
H9A 0.0533 0.0645 −0.0861 0.053*
H9B −0.1047 0.1511 −0.0440 0.053*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0444 (8) 0.0688 (9) 0.0861 (10) −0.0105 (6) −0.0123 (7) −0.0053 (7)
O2 0.0301 (6) 0.0414 (7) 0.0706 (8) 0.0003 (5) −0.0016 (5) −0.0114 (5)
C1 0.0499 (10) 0.0511 (10) 0.0460 (9) −0.0085 (8) 0.0001 (8) −0.0038 (8)
C2 0.0394 (9) 0.0439 (9) 0.0351 (8) −0.0052 (7) 0.0040 (7) −0.0009 (7)
C3 0.0294 (7) 0.0474 (9) 0.0478 (9) 0.0022 (7) 0.0021 (7) 0.0001 (7)
supporting information
sup-3 Acta Cryst. (2005). E61, o4358–o4359
Geometric parameters (Å, º)
O1—C1 1.2110 (19) C5—C6 1.388 (2)
O2—C5 1.3582 (17) C6—C7 1.380 (2)
O2—C8 1.4346 (17) C6—H6 0.9300
C1—C2 1.459 (2) C7—H7 0.9300
C1—H1 0.9300 C8—C9 1.506 (2)
C2—C7 1.385 (2) C8—H8A 0.9700
C2—C3 1.395 (2) C8—H8B 0.9700
C3—C4 1.371 (2) C9—C9i 1.518 (3)
C3—H3 0.9300 C9—H9A 0.9700
C4—C5 1.393 (2) C9—H9B 0.9700
C4—H4 0.9300
C5—O2—C8 118.84 (11) C7—C6—H6 120.5
O1—C1—C2 125.16 (17) C5—C6—H6 120.5
O1—C1—H1 117.4 C6—C7—C2 121.60 (15)
C2—C1—H1 117.4 C6—C7—H7 119.2
C7—C2—C3 118.65 (14) C2—C7—H7 119.2
C7—C2—C1 120.83 (15) O2—C8—C9 107.58 (12)
C3—C2—C1 120.51 (15) O2—C8—H8A 110.2
C4—C3—C2 120.49 (14) C9—C8—H8A 110.2
C4—C3—H3 119.8 O2—C8—H8B 110.2
C2—C3—H3 119.8 C9—C8—H8B 110.2
C3—C4—C5 120.20 (14) H8A—C8—H8B 108.5
C3—C4—H4 119.9 C8—C9—C9i 113.82 (16)
C5—C4—H4 119.9 C8—C9—H9A 108.8
O2—C5—C6 124.70 (13) C9i—C9—H9A 108.8
O2—C5—C4 115.31 (13) C8—C9—H9B 108.8
C6—C5—C4 119.98 (14) C9i—C9—H9B 108.8
C7—C6—C5 119.04 (14) H9A—C9—H9B 107.7
O1—C1—C2—C7 −174.99 (16) C3—C4—C5—C6 −0.9 (2)
O1—C1—C2—C3 4.0 (2) O2—C5—C6—C7 −179.22 (14)
C7—C2—C3—C4 1.7 (2) C4—C5—C6—C7 1.8 (2)
C1—C2—C3—C4 −177.30 (14) C5—C6—C7—C2 −1.0 (2)
C2—C3—C4—C5 −0.8 (2) C3—C2—C7—C6 −0.8 (2)
C8—O2—C5—C6 2.0 (2) C1—C2—C7—C6 178.20 (14)
C8—O2—C5—C4 −178.96 (12) C5—O2—C8—C9 177.31 (12)
C3—C4—C5—O2 180.00 (13) O2—C8—C9—C9i 61.3 (2)