organic papers
o1148
Anthonysamyet al. C22H17N3O doi:10.1107/S1600536807003674 Acta Cryst.(2007). E63, o1148–o1150 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
4
000-(4-Methoxyphenyl)-2,2
000:6
000,2
000000-terpyridine
Arockiam Anthonysamy,a Sengottuvelan
Balasubramanian,a K. Chinnakalib‡ and Hoong-Kun Func*
aDepartment of Inorganic Chemistry, School of
Chemical Sciences, University of Madras, Guindy Campus, Chennai 600 025, India,
b
Department of Physics, Anna University, Chennai 600 025, India, andcX-ray Crystallography Unit, School of Physics, Universiti Sains Malaysia, 11800 USM, Penang, Malaysia
‡ Additional correspondence author, email: [email protected]
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 100 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.056
wRfactor = 0.173
Data-to-parameter ratio = 29.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 28 December 2006 Accepted 23 January 2007
#2007 International Union of Crystallography
All rights reserved
In the title molecule, C22H17N3O, the three pyridyl rings are
almost coplanar. The molecule exhibits atrans–trans arrange-ment of the pyridine rings about the interannular C—C bonds. The methoxyphenyl substituent makes a dihedral angle of 6.17 (7) with the central pyridine ring.
Comment
2,20:60,200-Terpyridines and their 40-substituted derivatives
form metal complexes with a wide range of transition metal ions (Sauvageet al., 1994; Duprezet al., 2005), leading to the construction of linear, rod-like complexes with potential applications in the fields of macromolecular chemistry, nanoscience and photophysics. Biochemical applications include potential use of terpyridine complexes as sensors in tumor research and as DNA/RNA binding agents (Zhang et al., 2002). In particular, 2,20:60,200-terpyridine and its
deriv-atives have been used as key building blocks in supra-molecular architectures, such as double helicates and dendrimers, and its ruthenium(II) polypyridyl complexes have been exploited in nanocrystalline TiO2-based solar cells
(Schubert & Eschbaumer, 2002; Reagen & Gratzel, 1991). We report here the crystal structure of the title compound, (I) (Fig. 1).
The molecule exhibits a trans–trans arrangement of pyri-dine rings about the interannular C—C bonds. The three pyridine rings are approximately coplanar. The two terminal pyridine rings, N1/C6–C10 and N3/C11–C15, make dihedral angles of 1.66 (7) and 3.30 (7), respectively, with the central pyridine ring (N1/C1–C5). The methoxyphenyl substituent makes a dihedral angle of 6.17 (7)with the N1/C1–C5 ring.
Molecules of the title compound associate via C22— H22A N3iiinteractions, forming a zigzag chain along theb
H13 N2i interactions, leading to the formation of a
two-dimensional network (Fig. 3); see Table 1 for symmetry codes.
Experimental
2-Acetylpyridine (10.0 g, 8.25 mmol) was added to a suspension of crushed NaOH (3.3 g, 8.25 mmol) in PEG300 (70 ml), and stirred at 273 K for 10 min.p-Methoxybenzaldehyde (5.61 g, 4.12 mmol) was added through a syringe and the suspension was kept at 273 K for 2 h. The suspension was stirred manually with a spatula every 15 min as the viscosity became too high for adequate mixing with a magnetic stirrer. After 2 h, NH4OAc (20 g) was added in excess and the
suspension was heated at 373 K for 2 h. During this time, the colour
of the mixture changed from red to dark green, accompanied by the formation of a fine precipitate. Millipore water (200 ml) was added and the precipitate of the substituted terpyridine was isolated by filtration, washed with water (100 ml) and cold ethanol (20 ml), and dried under vacuum. Compound (I) was dissolved in acetonitrile and allowed to evaporate slowly to give golden yellow needle-shaped crystals (yield 65%; m.p. 434–436 K).
Crystal data
C22H17N3O
Mr= 339.39
Monoclinic,P21=c
a= 18.9035 (4) A˚
b= 5.1670 (1) A˚
c= 17.1558 (3) A˚
= 90.069 (1)
V= 1675.68 (6) A˚3
Z= 4
Dx= 1.345 Mg m
3
MoKradiation
= 0.09 mm1
T= 100.0 (1) K Needle, yellow 0.470.220.09 mm
Data collection
Bruker SMART APEX2 CCD diffractometer
!scans
Absorption correction: multi-scan (SADABS; Bruker, 2005)
Tmin= 0.945,Tmax= 0.993
21868 measured reflections 6903 independent reflections 4606 reflections withI> 2(I)
Rint= 0.035
max= 34.2
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.056
wR(F2) = 0.173
S= 1.07 6903 reflections 236 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0934P)2
+ 0.0299P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.42 e A˚
3
min=0.24 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C13—H13 N2i 0.95 2.54 3.2922 (14) 136
C22—H22A N3ii
0.98 2.59 3.4617 (15) 149
Symmetry codes: (i)x;yþ1 2;z
1
2; (ii)xþ1;y 1 2;zþ
1 2.
H atoms were positioned geometrically and were refined as riding on their parent C atoms, with C—H = 0.95–0.98 A˚ andUiso(H) =
organic papers
Acta Cryst.(2007). E63, o1148–o1150 Anthonysamyet al. C
[image:2.610.62.287.71.325.2]22H17N3O
o1149
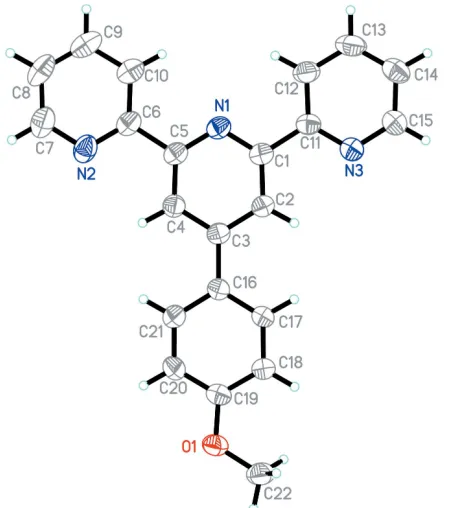
Figure 1 [image:2.610.313.564.74.202.2]The molecular structure of (I), showing 80% probability displacement ellipsoids and the atomic numbering scheme.
Figure 2
A view of a hydrogen-bonded (dashed lines) chain in (I). H atoms not involved in hydrogen bonding have been omitted.
Figure 3
[image:2.610.46.292.368.552.2]1.2Ueq(C) or 1.5Ueq(methyl C). A rotating-group model was used for
the methyl group.
Data collection:APEX2(Bruker, 2005); cell refinement:APEX2; data reduction: SAINT (Bruker, 2005); program(s) used to solve structure: SHELXTL (Sheldrick, 1998); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTLandPLATON(Spek, 2003).
HKF thanks the Malaysian Government and Universiti Sains Malaysia for Scientific Advancement Grant Allocation (SAGA) grant No. 304/PFIZIK/653003/A118 and USM short-term grant No. 304/PFIZIK/635028.
References
Bruker (2005).APEX2(Version 1.27),SAINT(Version 7.12A) andSADABS
(Version 2004/1). Bruker AXS Inc., Madison, Wisconsin, USA.
Duprez, V., Biancardo, M., Spanggaard, H. & Krebs, F. C. (2005).
Macromolecules,38, 10436–10448.
Reagen, B. O. & Gratzel, M. (1991).Nature (London),353, 737–740. Sauvage, J. P., Collin, J. P., Chambron, J. C., Guillerez, S., Coudret, C., Balzani,
V., Barigelletti, F., De Cola, L. & Flamigni, L. (1994).Chem. Rev.94, 993– 1019.
Schubert, U. S. & Eschbaumer, C. (2002).Angew. Chem. Int. Ed.41, 2892– 2926.
Sheldrick, G. M. (1998).SHELXTL. Version 5.1. Bruker AXS Inc., Madison, Wisconsin, USA.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Zhang, Y., Murphy, C. B. & Jones, W. E. (2002).Macromolecules,35, 630–636.
organic papers
o1150
Anthonysamyet al. Csupporting information
sup-1
Acta Cryst. (2007). E63, o1148–o1150
supporting information
Acta Cryst. (2007). E63, o1148–o1150 [https://doi.org/10.1107/S1600536807003674]
4
′
-(4-Methoxyphenyl)-2,2
′
:6
′
,2
′′
-terpyridine
Arockiam Anthonysamy, Sengottuvelan Balasubramanian, K. Chinnakali and Hoong-Kun Fun
4′-(4-Methoxyphenyl)-2,2′:6′,2′′-terpyridine
Crystal data
C22H17N3O Mr = 339.39 Monoclinic, P21/c
Hall symbol: -P 2ybc a = 18.9035 (4) Å b = 5.1670 (1) Å c = 17.1558 (3) Å β = 90.069 (1)° V = 1675.68 (6) Å3 Z = 4
F(000) = 712 Dx = 1.345 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 4472 reflections θ = 2.6–34.0°
µ = 0.09 mm−1 T = 100 K Needle, yellow 0.47 × 0.22 × 0.09 mm
Data collection
Bruker SMART APEX2 CCD diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.33 pixels mm-1 ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2005) Tmin = 0.945, Tmax = 0.993
21868 measured reflections 6903 independent reflections 4606 reflections with I > 2σ(I) Rint = 0.035
θmax = 34.2°, θmin = 1.1° h = −29→29
k = −8→8 l = −27→26
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.056 wR(F2) = 0.173 S = 1.07 6903 reflections 236 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0934P)2 + 0.0299P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.42 e Å−3
Δρmin = −0.24 e Å−3
Special details
supporting information
sup-2
Acta Cryst. (2007). E63, o1148–o1150
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
supporting information
sup-3
Acta Cryst. (2007). E63, o1148–o1150
H22A 0.5977 0.4501 0.4597 0.042* H22B 0.5378 0.6464 0.4305 0.042* H22C 0.5845 0.4911 0.3683 0.042*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0212 (3) 0.0328 (5) 0.0219 (3) −0.0021 (3) −0.0056 (3) 0.0047 (3) N1 0.0181 (3) 0.0226 (5) 0.0184 (4) −0.0014 (3) 0.0006 (3) −0.0032 (3) N2 0.0289 (4) 0.0271 (5) 0.0212 (4) −0.0090 (4) 0.0037 (3) −0.0042 (4) N3 0.0229 (4) 0.0220 (5) 0.0207 (4) −0.0006 (4) −0.0017 (3) 0.0012 (3) C1 0.0174 (4) 0.0210 (5) 0.0178 (4) 0.0010 (4) 0.0007 (3) −0.0030 (4) C2 0.0171 (4) 0.0195 (5) 0.0199 (4) −0.0005 (4) −0.0008 (3) −0.0007 (4) C3 0.0179 (4) 0.0178 (5) 0.0175 (4) 0.0006 (4) −0.0002 (3) −0.0032 (4) C4 0.0208 (4) 0.0198 (5) 0.0177 (4) −0.0015 (4) 0.0002 (3) −0.0012 (4) C5 0.0185 (4) 0.0202 (5) 0.0183 (4) −0.0012 (4) 0.0011 (3) −0.0046 (4) C6 0.0203 (4) 0.0234 (5) 0.0198 (4) −0.0030 (4) 0.0027 (3) −0.0055 (4) C7 0.0313 (5) 0.0286 (6) 0.0269 (5) −0.0092 (5) 0.0072 (4) −0.0047 (5) C8 0.0249 (5) 0.0298 (6) 0.0388 (6) −0.0107 (5) 0.0062 (4) −0.0090 (5) C9 0.0218 (5) 0.0361 (7) 0.0404 (6) −0.0065 (5) −0.0036 (4) −0.0072 (5) C10 0.0224 (5) 0.0289 (6) 0.0298 (5) −0.0040 (5) −0.0027 (4) −0.0022 (5) C11 0.0165 (4) 0.0227 (5) 0.0174 (4) 0.0021 (4) −0.0001 (3) −0.0022 (4) C12 0.0189 (4) 0.0372 (7) 0.0219 (4) −0.0037 (4) −0.0030 (3) 0.0027 (4) C13 0.0207 (4) 0.0462 (8) 0.0208 (4) 0.0017 (5) −0.0037 (3) 0.0039 (5) C14 0.0245 (5) 0.0325 (6) 0.0194 (4) 0.0059 (5) 0.0011 (3) 0.0040 (4) C15 0.0258 (5) 0.0246 (6) 0.0225 (4) −0.0001 (4) −0.0008 (4) 0.0021 (4) C16 0.0180 (4) 0.0182 (5) 0.0170 (4) 0.0010 (4) 0.0000 (3) −0.0007 (4) C17 0.0269 (5) 0.0244 (6) 0.0206 (4) −0.0052 (4) −0.0069 (3) 0.0054 (4) C18 0.0240 (5) 0.0253 (6) 0.0219 (4) −0.0054 (4) −0.0057 (3) 0.0027 (4) C19 0.0191 (4) 0.0244 (5) 0.0166 (4) 0.0030 (4) −0.0010 (3) −0.0009 (4) C20 0.0242 (5) 0.0261 (6) 0.0254 (5) −0.0016 (4) −0.0042 (4) 0.0088 (4) C21 0.0210 (4) 0.0237 (6) 0.0269 (5) −0.0035 (4) −0.0037 (3) 0.0045 (4) C22 0.0222 (4) 0.0362 (7) 0.0246 (5) −0.0038 (5) −0.0046 (4) −0.0006 (5)
Geometric parameters (Å, º)
supporting information
sup-4
Acta Cryst. (2007). E63, o1148–o1150
C3—C4 1.3988 (14) C17—C18 1.3924 (14) C3—C16 1.4888 (13) C17—H17 0.95 C4—C5 1.3969 (13) C18—C19 1.3872 (15) C4—H4 0.95 C18—H18 0.95 C5—C6 1.4906 (14) C19—C20 1.3893 (15) C6—C10 1.3943 (15) C20—C21 1.3853 (14) C7—C8 1.3883 (17) C20—H20 0.95 C7—H7 0.95 C21—H21 0.95 C8—C9 1.3800 (19) C22—H22A 0.98 C8—H8 0.95 C22—H22B 0.98 C9—C10 1.3857 (17) C22—H22C 0.98
supporting information
sup-5
Acta Cryst. (2007). E63, o1148–o1150
C12—C11—C1 120.41 (9)
C5—N1—C1—C2 −0.59 (15) N1—C1—C11—N3 176.99 (8) C5—N1—C1—C11 −179.76 (8) C2—C1—C11—N3 −2.21 (14) N1—C1—C2—C3 −0.18 (15) N1—C1—C11—C12 −2.60 (14) C11—C1—C2—C3 178.95 (9) C2—C1—C11—C12 178.21 (10) C1—C2—C3—C4 0.67 (14) N3—C11—C12—C13 −1.32 (16) C1—C2—C3—C16 −179.71 (9) C1—C11—C12—C13 178.24 (10) C2—C3—C4—C5 −0.43 (14) C11—C12—C13—C14 −0.24 (17) C16—C3—C4—C5 179.96 (9) C12—C13—C14—C15 1.28 (17) C1—N1—C5—C4 0.85 (14) C11—N3—C15—C14 −0.58 (16) C1—N1—C5—C6 −179.08 (8) C13—C14—C15—N3 −0.91 (17) C3—C4—C5—N1 −0.35 (15) C4—C3—C16—C17 −174.00 (9) C3—C4—C5—C6 179.58 (9) C2—C3—C16—C17 6.40 (15) C7—N2—C6—C10 0.59 (16) C4—C3—C16—C21 6.12 (15) C7—N2—C6—C5 −178.66 (9) C2—C3—C16—C21 −173.47 (9) N1—C5—C6—N2 179.47 (9) C21—C16—C17—C18 0.85 (16) C4—C5—C6—N2 −0.46 (14) C3—C16—C17—C18 −179.03 (10) N1—C5—C6—C10 0.21 (15) C16—C17—C18—C19 −0.21 (17) C4—C5—C6—C10 −179.72 (10) C22—O1—C19—C18 3.71 (15) C6—N2—C7—C8 −0.41 (17) C22—O1—C19—C20 −176.86 (9) N2—C7—C8—C9 0.06 (18) C17—C18—C19—O1 178.75 (10) C7—C8—C9—C10 0.10 (18) C17—C18—C19—C20 −0.66 (16) C8—C9—C10—C6 0.07 (18) O1—C19—C20—C21 −178.58 (10) N2—C6—C10—C9 −0.44 (17) C18—C19—C20—C21 0.88 (17) C5—C6—C10—C9 178.78 (10) C19—C20—C21—C16 −0.22 (17) C15—N3—C11—C12 1.71 (15) C17—C16—C21—C20 −0.63 (16) C15—N3—C11—C1 −177.87 (9) C3—C16—C21—C20 179.25 (10)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C13—H13···N2i 0.95 2.54 3.2922 (14) 136
C22—H22A···N3ii 0.98 2.59 3.4617 (15) 149