0095-1137/06/$08.00⫹0 doi:10.1128/JCM.00054-06
Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Sequence-Based Methods for Identifying Epidemiologically Linked
Herpes Simplex Virus Type 2 Strains
Emily Toth Martin,
1David M. Koelle,
2,3,4,5Benjamin Byrd,
3Meei-Li Huang,
2Jeffrey Vieira,
5Lawrence Corey,
2,3,5and Anna Wald
1,2,3,5*
Department of Epidemiology1and Department of Pathobiology,4School of Public Health and Community Medicine, and
Department of Medicine3and Department of Laboratory Medicine,5School of Medicine, University of Washington,
and Program in Infectious Diseases, Fred Hutchinson Cancer Research Center,2Seattle, Washington
Received 10 January 2006/Returned for modification 15 March 2006/Accepted 1 May 2006
Traditional methods for confirming the identity of herpes simplex virus (HSV) isolates use restriction fragment length polymorphism (RFLP). However, RFLP is less amenable to high-throughput analyses of many samples, and the extent to which small differences in RFLP patterns distinguish between different viral strains remains unclear. Viral HSV type 2 (HSV-2) DNA isolates from 14 persons experiencing a primary HSV-2 infection and from their sexual partners were analyzed by RFLP and heteroduplex mobility assays. We also compared the HSV-2 sequences from seven regions, including noncoding regions between UL19 and UL20, UL24 and UL25, UL37 and UL38, and UL41 and UL42 and coding segments of the gC, gB, and gG genes. Although the resulting RFLP patterns of the couples were almost identical, minor banding differences existed between the source and susceptible partners in five couples. Heteroduplex mobility assays were unable to distinguish between unrelated strains. Overall, 22 sites of sequence variation were found in 1,482 bp of analyzed sequence. The DNA sequences differentiated between all unrelated infections, and epidemiologically related isolates had identical sequences in all but two pairs. Our results suggest that a multilocus assay based on several DNA sequences has the potential to be an informative tool for identifying epidemiologically related HSV-2 strains.
Epidemiologic investigations of human herpes simplex virus type 2 (HSV-2) have relied on the use of restriction fragment length polymorphisms (RFLP) to demonstrate superinfection and distinguish between unrelated strains (5, 15, 19). In the past several years, molecular techniques, including heterodu-plex mobility assays (HMA) and sequencing, have superseded RFLP for distinguishing other viruses such as hepatitis C virus and human immunodeficiency virus (1, 3, 22, 23). Recently, Norb-erg et al. (16) showed the utility of HSV-1 DNA sequencing for molecular epidemiology at a population and an individ-ual level through sequencing the near-adjacent coding re-gions of HSV-1 glycoprotein G (gG), gE, and gI. For HSV-1, sequencing of hypervariable regions ReIV and ReVII has been shown to be useful for discriminating between isolates (14). Targeted DNA sequencing of HSV-1 hypervariable regions can also be used to detect a second exogenous HSV-1 infection (17). However, the presence of hypervariable regions in the HSV-2 genome has not been definitively established.
To develop sequence-based methods for distinguishing be-tween HSV-2 strains, we performed RFLP, HMA, and DNA sequence analysis of selected regions of the HSV-2 genome. We attempted to maximize the degree of sequence variability through our selection of genome regions for study. Noncoding genome regions were chosen for sequencing because we hy-pothesized a lack of purifying selection in these regions, and glycoprotein-coding regions were chosen based on previous
sequence variation data in analogous segments in the HSV-1 genome (4, 13, 16). Our goals were to distinguish between strains from unrelated individuals and to identify epidemiolog-ically linked HSV-2 strains.
(Some of the data in this work were presented at the Inter-national Herpesvirus Workshop, 25 to 30 July 2004 [abstr. 4.25].)
MATERIALS AND METHODS
Sample collection.Swabs from genital herpes lesions were collected as part of an ongoing study of HSV-2 transmission conducted at the University of Wash-ington Virology Research Clinic in Seattle. Informed consent was obtained from patients, and human experimentation guidelines of the University of Washington were followed in the conduct of research with human subjects. Transmission events were identified by clinical presentation and documented by viral culture and seroconversion to HSV-2 by Western blotting (2). The HSV-2 samples were isolated on HDF (human tonsil) cells and then passed once in Vero cells, and DNA from the cytoplasm fraction was isolated using phenol-chloroform extrac-tion as previously described (8). Virus was purified from the cytoplasm fracextrac-tion to reduce the amount of human genomic DNA background. Whole-genome amplification was used to increase stocks of purified genomic HSV-2 DNA. This procedure was performed by multiple displacement amplification (MDA) (6) using a Repli-q kit including high-fidelity DNA polymerase and random exonu-clease-resistant primers (Molecular Staging, New Haven, CT).
RFLP.Approximately 750 ng of HSV-2 DNA from each individual was di-gested to completion with SalI and separately with XhoI (Invitrogen, Carlsbad, CA) by methods similar to those reported previously (12). Digests were then electrophoresed on a 0.6% agarose gel overnight. DNA fragments were detected with SYBR green.
PCR.Seven segments of the HSV-2 genome (Table 1) were amplified by PCR from each isolate. Segments gC, noncoding region 1 (NC1), and NC3 were amplified using the Stratagene (La Jolla, CA)PfuUltra enzyme. Segments from gB, gG, NC2, and NC4 were amplified using the Applied Biosystems (Foster City, CA) Amplitaq enzyme when they failed to amplify efficiently with the PfuUltra enzyme. PCRs included 8% glycerol to increase reaction efficiency, 0.3 * Corresponding author. Mailing address: Virology Research Clinic,
University of Washington, 600 Broadway, Suite 400, Seattle, WA 98122. Phone: (206) 720-4340. Fax: (206) 720-4371. E-mail: annawald @u.washington.edu.
2541
on May 16, 2020 by guest
http://jcm.asm.org/
M of each primer (Table 1), and 104to 108copies of DNA template based on
standard quantitative real-time TaqMan PCR (Applied Biosystems, Foster City, CA) (18). Because of high GC content in the regions amplified, 5% dimethyl sulfoxide was added to the PCRs for all regions except NC1. SYBR green in dimethyl sulfoxide and ROX (Invitrogen, Carlsbad, CA) were added to enable real-time detection of amplicons. Three-step PCR cycling conditions included denaturing at 94°C for 20 s, annealing at 68°C for 30 s for NC1 and NC3 and at 60°C for 30 s for gG, gB, gC, NC2, and NC4, and extension at 72°C for 1 min for 35 cycles. Amplicons were then purified using DNA Clean & Concentrator (Zymo Research, Orange, CA).
HMA.PCR amplicons of the NC1 region for each individual HSV-2 sample were hybridized to a radiolabeled probe by denaturing at 95°C for 5 min and incubating at 50°C for 2 h in the presence of the32P-end-labeled probe. The
amplified NC1 region of HSV-2 strain 333 (11) was used as the probe. The samples were then electrophoresed on an MDE gel (Cambrex, East Rutherford, NJ) at 350 V overnight. The gel was dried at 80°C and then exposed to Kodak film at⫺70°C with an enhancer for 2 days.
DNA sequencing.All PCR amplicons were routinely sequenced bidirection-ally, except for that in region NC3, using BigDye Terminator (Applied Biosys-tems, Foster City, CA). Second-strand sequencing of NC3 was done only if polymorphisms were suspected on unidirectional sequencing. Sequencing prim-ers were identical to those used for PCR (Table 1). The sequences were analyzed using Sequencher, version 4.1.4 (Gene Codes, Ann Arbor, MI). HSV-2 strain HG52 (GenBank accession no. NC001798) was used as the reference sequence. Visual genotypes were generated with polymorphism data using VG2 software (20). If the sequences from related pairs did not match, a second confirmatory PCR and sequence for the questionable individual and region were obtained using the original DNA product from culture. No unmatched polymorphisms were found to be caused by MDA or PCR artifacts through the study.
To assess the stability of the genomic HSV-2 regions during limited passage in culture, we compared DNA prepared as published elsewhere (18) directly from swabs of genital lesions to DNA prepared from HSV-2 cultured from the same lesions. The cultures were initially isolated on mink lung cells and were passed once on mink lung cells and then once in Vero cells prior to DNA isolation (QIAGEN [Valencia, CA] blood kit). The samples used for this stability analysis were isolated on mink lung cells instead of HDF cells because mink lung cells are currently used in our laboratory for this procedure, whereas HDF cells were used for isolation before our era of PCR specimen analysis. DNA was amplified and bidirectional sequencing performed as described for the other specimens in this report. For this analysis of stability in culture, genital HSV-2 lesion DNA spec-imens from four additional individuals before and after culture were aligned, and the original chromatograms were examined at any candidate loci of discrepancy.
Nucleotide sequence accession numbers.Sequences from this study have been submitted to GenBank under accession no. AY827201 to AY827401 and DQ236133 to DQ236152. The similarity of these sequences to the HSV-2 ge-nome has been confirmed by comparison to the GenBank database by using megaBLAST (December 2005).
RESULTS
Fourteen pairs of sexual partners were included in the anal-ysis. All of the pairs were heterosexual couples, and 5 of the 28
individuals belonged to racial minorities. Each pair included a person who was thought to transmit HSV-2 (partner A) and that person’s sexual partner, who was diagnosed with a labo-ratory-documented newly acquired HSV-2 infection (partner B) thought to be acquired from partner A. Acquisition of initial HSV-2 infection was diagnosed by clinical presentation with genital lesions, HSV-2 isolation from the genital tract, and seroconversion to HSV-2 by Western blotting (2); all three criteria were present for all 14 partner B’s.
[image:2.585.42.546.82.209.2]RFLP.Nine couples had identical RFLP patterns after di-gestion with each enzyme (Table 2; Fig. 1). Minor banding differences were observed between partners in five couples with SalI and between partners in three couples with XhoI. Related individuals showed no more than a 2-band difference by use of either enzyme. However, comparisons of unrelated individuals showed that no particular number of band differ-ences could be used as a threshold for determining relatedness with one enzyme. For example, between pairs 2, 4, 7, 8, 13, and 14, the mean number of band differences between unrelated individuals was 3.9, with some unrelated individuals differing by only 1 band (Fig. 1). However, each individual was more closely related to his or her partner than to any unrelated individual.
TABLE 1. Regions of HSV-2 genome amplified by PCR
Region ORFa HSV-2 genome
locationb
Size
(bp) Left primer Right primer
NC1 Between UL19 and UL20
40560–40908 348 5⬘CTTGAAGGCACCCGTCGCGTTCT 3⬘ 5⬘CAGGAGCGGCCATTGGGTTC 3⬘ NC2 Between UL41
and UL42
93273–93464 192 5⬘CATTCCGGCACGATTGTAG 3⬘ 5⬘CACCAATCCGCACGTAGAT 3⬘ NC3 Between UL24
and UL25
48706–49047 342 5⬘CCGGTGGCCGCCAAGAGCAGA 3⬘ 5⬘CGTCCAGCGCGTCGAAAGGGTAGT 3⬘ NC4 Between UL37
and UL38
84732–84981 250 5⬘TATACGGCGCGTTATGTCC 3⬘ 5⬘ACACGCCACCAGAAAACAC 3⬘ gB UL27 54093–54319 227 5⬘CAGCATGGTGATGTTCAGGT 3⬘ 5⬘GCTTTCGGTACGAAGACCAG 3⬘ gC UL44 97570–97797 228 5⬘CTCATCATCGAAGAGCTGACC 3⬘ 5⬘GTCCTCGAACCAGACGAACTC 3⬘ gG US4 138209–138376 168 5⬘TGACGTACTACCGGCTCACC 3⬘ 5⬘GCAGGGAAGCATTTACGAGA 3⬘
a
ORF, open reading frame.
b
HSV-2 strain HG52 (GenBank accession no. NC001798) coordinates.
TABLE 2. Number of band differences within pairs, by restriction enzyme
Pair IDa Differences
with SalI
Differences with XhoI
Time (no. of days) between collection dates
1 0 0 Same day
2 0 0 Same day
3 0 0 56
4 2 bands 0 3
5 0 0 Same day
6 2 bands No data Same day
7 0 0 83
8 0 0 31
9 0 0 43
10 2 bands 2 bands 17
11 2 bands 2 bands 13
12 0 0 51
13 2 bands 1 bands 12
14 0 0 2
aID, identification number.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:2.585.303.542.558.716.2]HMA. NC1 amplicons were chosen for HMA analysis be-cause the high yield of the PCR provided ample product for multiple analysis methods. Slight mobility shifts were observed for pairs 2 and 8 and for partner B from pair 1 and partner A from pair 3 (Fig. 2). All other samples had indistinguishable migration. Concurrent DNA sequencing showed the variation between the test strains and the HSV-2 strain 333 probe to be lower than the 1% to 2% sequence divergence previously found to be necessary for measurable differences in heterodu-plex mobility between samples (3). The slight band differences found in pairs with identical sequences (pairs 1 and 3) may be due to a low level of variation from the probe strain, resulting in ambiguous HMA profiles (3). HMA was not used to analyze the other regions due to the lack of migration seen using the NC1 region.
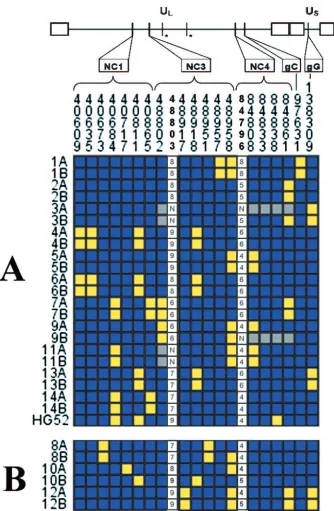
DNA sequencing. Eight regions of HSV-2 sequence were amplified from each isolate except 3A and 9B. These two isolates did not have amplification at region NC4. For pairs 3 and 11, genotypes for the first two polymorphic sites of NC3 could not be determined due to inefficient sequencing across the poly(C) region of the amplicon.
Noncoding regions showed the most variation. Among all 28 samples, one or more strains showed variation at a total of
22.44 nucleotides/1,000, 30.04 nucleotides/1,000, and 33.33 nu-cleotides/1,000 for NC1, NC3, and NC4, respectively. No vari-ation was found in 190 bp of NC2. In coding regions, one single-nucleotide polymorphism each was found in 152 bp of gG and 228 bp of gC, and no variation was present in 217 bp of gB. Overall, 21 sites of sequence variation were found in 1,482 bp of analyzed sequence (Table 3). Five of those sites consisted of variable-length repeats of one or two nucleotides. Two of these repeat mutation sites were in NC1, two were in NC2, and one was in NC4 (Table 3). The remaining sites were single-nucleotide polymorphisms (Fig. 3). In addition to the variations listed, one individual had the sequence GGAGAG GGAGAGGGA in place of a variable-length G repeat located near position 84794 in the NC4 region.
The HG52 sequence differed from the consensus sequence of our samples at two sites in NC1 and one site in NC4 (Fig. 3). In regions with no variation (NC2 and gB), the HG52 sequence (7) was consistent with all other samples.
Taking into account all sites of variation, it was possible to differentiate between all unrelated infections (Fig. 3). All se-quences were also distinguishable from the published HG52 sequence. Eleven of the 14 pairs had sequences that were identical between the related partners at all sites. Pair 8 dif-fered at site 48958, a variable-length C repeat in a noncoding region (Table 3). Pair 12 differed at site 84796, a
[image:3.585.70.263.67.309.2]variable-FIG. 1. Results of RFLP for six selected pairs. HSV-2 genomic DNA was digested using the SalI enzyme and electrophoresed on a 0.6% agarose gel overnight with a 1-kb molecular weight marker. RFLP pattern differences between partners can be seen with pairs 13 and 4.
[image:3.585.301.541.79.301.2]FIG. 2. HMA results for HSV-2 region NC1 amplicons by pair and partner. A radiolabeled amplicon from HSV-2 strain 333 was hybridized to each sample and then electrophoresed on a 0.6% polyacrylamide gel overnight. Slight mobility shifts are visible for pair 2, pair 8, partner B of pair 1, and partner A of pair 3.
TABLE 3. HSV-2 sequence polymorphisms
Region Genome site Major allele Minor allele Third allele
NC1 40609 G A
40635 G A
40673 C T
40684 5A 4A
40717 C A
40735 3A 2A
40811 T C
40865 7C 6C
NC3 48802 C T
48803 6C–9Ca 6C–9Ca
48917 T C
48918 C A
48951 T C
48957 C T
48958 6C 5C 4Cb
NC4 84796 4GT–8GTc 4GT–8GTc
84808 G A
84833 T C
84838 T C
84861 C T
gCd 97631 T C
gGd 138309 G C
aSite 48803 is a C repeat that ranges from 6 to 9 iterations. bThis allele was found only in pair 11.
cSite 84796 is a GT repeat that ranges from 4 to 8 iterations. dCoding region.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:3.585.113.469.658.698.2]length GT repeat in a noncoding region. Pair 10 differed at six sites. A clinical chart review revealed multiple concurrent sex-ual partners for partner B from pair 10. This sexsex-ual history, in combination with the sequence data, indicated that partner A is an unlikely source of partner B’s genital herpes infection.
To assess the stability of the sequences during tissue culture, we evaluated DNA samples from HSV-2 lesions from four
[image:4.585.125.459.64.575.2]individuals before and after culture. All genomic regions were evaluated for specimens from all but one individual. We found no differences in sequence between DNA amplified directly from lesions and DNA obtained after virus was cultured from the same clinical lesions and passaged twice. During this se-quencing of additional strains, occasional 3-nucleotide poly-morphisms were uncovered, two in the noncoding region NC1
FIG. 3. Individual genotypes at each polymorphism. The consensus alleles (blue) and minor alleles (yellow) determined by the population studied are listed for each individual at each locus where a sequence polymorphism was observed by DNA sequence analysis. Missing data are indicated by gray squares or the letter N. The number of C repeats at locus 48802 and the number of GT repeats at locus 84796 are listed for each individual. Asterisks indicate regions where no sequence variation was found. (A) Related pairs with matching sequences; (B) related pairs with differing sequences.
on May 16, 2020 by guest
http://jcm.asm.org/
and one in the noncoding region NC4. These can be accessed from the GenBank submissions accompanying this report. These data are consistent with stability in HSV-2 genomic DNA during a limited number of in vitro passages in these loci and the identification of bona fide polymorphisms in our study.
DISCUSSION
We found that sequencing short segments of both coding and noncoding regions of HSV-2 allowed us to correctly iden-tify epidemiologically linked strains of HSV-2 as well as to differentiate between all of the unrelated strains. These results were confirmed by RFLP, which has historically been used to demonstrate strain variation (5, 15, 19). As a result of the low rate of mutations overall, HMA was found to be of limited utility for differentiating HSV-2 strains. A few differences be-tween related strains were observed by both RFLP and DNA sequencing. Among the 13 pairs that are assumed to be epi-demiologically related, 3 pairs were found to have DNA se-quence differences and 5 pairs were found to have RFLP differences. These results indicate that a minor level of varia-tion can be expected even between individuals related by a transmission event, and therefore, care needs to be taken in the interpretation of minor differences between individuals. Similar minor variations have been reported in strains recov-ered from consecutive recurrences from individuals over time (5, 12, 15, 21). However, the RFLP method requires visual interpretation to determine the relatedness of samples. DNA sequencing is more suitable for efficient analyses of large numbers of samples than RFLP and may be more amenable to standardization. Therefore, we believe that it is better suited for analysis involving large numbers of HSV-2 strains to assess epidemiological relationships. DNA poly-morphisms can be objectively identified and labeled. Spec-ifying the most appropriate set of polymorphisms to use for comparison of HSV-2 isolates can lead to a more rigorous standard for determining relatedness that is more easily communicated than RFLP findings.
Past HSV sequencing efforts have focused on coding re-gions, particularly glycoprotein sequences (4, 13, 16). Two pub-lications have reported that large sections of HSV-1 sequence, including complete coding regions, could be used for examin-ing strain differences between populations (4) and individuals (16). However, a small segment (179 bp) of the HSV-2 DNA polymerase gene was found to have no variability between two samples and a standard strain (13). Nevertheless, we found that similarly small segments could be of use if they were generated from intergenic spacers that do not code for a product.
Five of the 21 polymorphisms observed were variable-length repeats of one or two bases. Similar variations in the length of GT or C repeats in HSV sequences due to DNA polymerase error have been reported elsewhere (9, 10). These polymerase errors are possible at several points in the pathogenesis of HSV-2. Mutations may have occurred during viral replication in the recurrent lesion of partner A or the primary lesion of partner B. Alternatively, a variant strain may have colonized the ganglia of partner A in the intervening time between sam-ple collection from partner A and partner B, which was 31 days for pair 8 and 51 days for pair 12.
We find it unlikely that HSV-2 sequence variation was in-troduced by the methods used in our study. First, even though mutations in DNA sequences can be introduced by the poly-merases used in PCR and sequencing, all polymorphisms were confirmed by second-strand sequencing, and nucleotide differ-ences between partners were confirmed by a second PCR and sequencing using the DNA harvested from culture. Second, we found the sequences to be stable when DNA samples taken directly from the lesion swab were compared to samples ob-tained after 2 passages in cell culture. There are no data to suggest that genetic drift in 2 passages in mink lung cells would be any more or less likely than drift in passages in HDF cells. Sequence drift during culture has been reported in noncoding hypervariable regions of HSV-1 (14); however, we found no such drift in our study.
We expected to observe increased variability between indi-viduals in noncoding regions due to our hypothesized absence of purifying selection in these regions. Our study confirmed that noncoding regions exhibit a greater level of variability than similar-length segments of glycoprotein-coding regions. It is possible that, during HSV-2 replication, DNA polymerase errors in noncoding regions may not decrease the fitness of the virus. This is in contrast to the coding regions, which evolve with functional restraints and therefore are expected to be more conserved. The increased variability of noncoding re-gions facilitates the detection of variation with a much smaller, and thus more easily obtainable, amplicon. Overall, noncoding regions may be preferable for distinguishing HSV-2 strains at an individual level.
While we believe our major conclusions are firm, we do recognize that we sampled a small portion of the HSV-2 ge-nome. While our source partners did not acquire HSV-2 ex-clusively in Seattle, we did analyze only a limited number of persons. It is possible that sequencing of HSV strains from a larger geographical area may show greater variability. How-ever, it is noteworthy that HG52 differed from the consensus sequence only at three sites, even though its origin is geograph-ically distant from that of our samples (7). In addition, the amplified segments of gC, gB, and gG were chosen by ease of primer selection and amplification. These factors may have influenced the selection of a more conserved region. Conse-quently, the variation we observed in those segments may not be indicative of the level of variation present across each entire coding region, and further research is needed to determine to what extent these polymorphisms, as well as others, can be used to define and compare HSV-2 strains across individuals and populations. Specific attention must be given to sequenc-ing efforts that analyze large numbers of individuals to refine the use of HSV-2 sequencing for molecular epidemiology. Fur-thermore, DNA sequencing of consecutive isolates from an individual can confirm whether an HSV-2 infection in a single individual is limited to a single strain or whether more than one strain has colonized the ganglia. While existing literature suggests that most infections occur with a single strain, infec-tion with more than a single strain has been described using RFLP (5, 19, 21).
In conclusion, we found that DNA sequencing was more powerful than both RFLP and HMA in distinguishing HSV-2 strains. The sequences obtained from noncoding regions pro-vided the most information on differences between unrelated
on May 16, 2020 by guest
http://jcm.asm.org/
samples. Our results suggest that a multilocus assay based on several DNA sequences has the potential to be an informative tool for identifying specific strains in an HSV-2 transmission event.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI-30731.
We acknowledge Lizette Embuscado for her technical assistance with the RFLP analysis.
REFERENCES
1.Arens, M.1999. Methods for subtyping and molecular comparison of human viral genomes. Clin. Microbiol. Rev.12:612–626.
2.Ashley, R. L., J. Militoni, F. Lee, A. Nahmias, and L. Corey.1988. Compar-ison of Western blot (immunoblot) and glycoprotein G-specific immunodot enzyme assay for detecting antibodies to herpes simplex virus types 1 and 2 in human sera. J. Clin. Microbiol.26:662–667.
3.Barlow, K. L., J. Green, and J. P. Clewley.2000. Viral genome characteri-sation by the heteroduplex mobility and heteroduplex tracking assays. Rev. Med. Virol.10:321–335.
4.Bowden, R., H. Sakaoka, P. Donnelly, and R. Ward.2004. High recombina-tion rate in herpes simplex virus type 1 natural popularecombina-tions suggests signif-icant co-infection. Infect. Genet. Evol.4:115–123.
5.Buchman, T. G., B. Roizman, and A. J. Nahmias.1979. Demonstration of exogenous genital reinfection with herpes simplex virus type 2 by restriction endonuclease fingerprinting of viral DNA. J. Infect. Dis.140:295–304. 6.Dean, F. B., S. Hosono, L. Fang, X. Wu, A. F. Faruqi, P. Bray-Ward, Z. Sun,
Q. Zong, Y. Du, J. Du, M. Driscoll, W. Song, S. F. Kingsmore, M. Egholm, and R. S. Lasken.2002. Comprehensive human genome amplification using multiple displacement amplification. Proc. Natl. Acad. Sci. USA99:5261– 5266.
7.Dolan, A., F. E. Jamieson, C. Cunningham, B. C. Barnett, and D. J. McGeoch.
1998. The genome sequence of herpes simplex virus type 2. J. Virol.72:2010– 2021.
8.Dworkin, M. S., P. C. Shoemaker, C. Spitters, A. Cent, A. C. Hobson, J. Vieira, L. Corey, and L. R. Frumkin.1999. Endemic spread of herpes sim-plex virus type 1 among adolescent wrestlers and their coaches. Pediatr. Infect. Dis. J.18:1108–1109.
9.Eckert, K. A., and S. E. Hile.1998. Alkylation-induced frameshift mutagen-esis during in vitro DNA synthmutagen-esis by DNA polymerases alpha and beta. Mutat. Res.422:255–269.
10.Eckert, K. A., A. Mowery, and S. E. Hile.2002. Misalignment-mediated DNA
polymerase beta mutations: comparison of microsatellite and frame-shift error rates using a forward mutation assay. Biochemistry41:10490–10498. 11.Kit, S., M. Kit, H. Qavi, D. Trkula, and H. Otsuka.1983. Nucleotide
se-quence of the herpes simplex virus type 2 (HSV-2) thymidine kinase gene and predicted amino acid sequence of thymidine kinase polypeptide and its comparison with the HSV-1 thymidine kinase gene. Biochim. Biophys. Acta
741:158–170.
12.Lonsdale, D. M.1979. A rapid technique for distinguishing herpes-simplex virus type 1 from type 2 by restriction-enzyme technology. Lanceti:849–852. 13.Madhavan, H. N., K. Priya, and R. Bagyalakshmi.2003. Phenotypic and genotypic methods for the detection of herpes simplex virus serotypes. J. Virol. Methods108:97–102.
14.Maertzdorf, J., L. Remeijer, A. Van Der Lelij, J. Buitenwerf, H. G. Niesters, A. D. Osterhaus, and G. M. Verjans. 1999. Amplification of reiterated sequences of herpes simplex virus type 1 (HSV-1) genome to discriminate between clinical HSV-1 isolates. J. Clin. Microbiol.37:3518–3523. 15.Maitland, N. J., I. W. Smith, J. F. Peutherer, D. H. Robertson, and K. W.
Jones.1982. Restriction endonuclease analysis of DNA from genital isolates of herpes simplex virus type 2. Infect. Immun.38:834–842.
16.Norberg, P., T. Bergstrom, E. Rekabdar, M. Lindh, and J. A. Liljeqvist.2004. Phylogenetic analysis of clinical herpes simplex virus type 1 isolates identified three genetic groups and recombinant viruses. J. Virol.78:10755–10764. 17.Roest, R. W., W. F. Carman, J. Maertzdorf, A. Scoular, J. Harvey, M. Kant,
W. I. Van Der Meijden, G. M. Verjans, and A. D. Osterhaus.2004. Genotypic analysis of sequential genital herpes simplex virus type 1 (HSV-1) isolates of patients with recurrent HSV-1 associated genital herpes. J. Med. Virol.
73:601–604.
18.Ryncarz, A. J., J. Goddard, A. Wald, M. L. Huang, B. Roizman, and L. Corey.
1999. Development of a high-throughput quantitative assay for detecting herpes simplex virus DNA in clinical samples. J. Clin. Microbiol.37:1941– 1947.
19.Schmidt, O. W., K. H. Fife, and L. Corey.1984. Reinfection is an uncommon occurrence in patients with symptomatic recurrent genital herpes. J. Infect. Dis.149:645–646.
20.SeattleSNPs.November 2004, date accessed. NHLBI Program for Genomic Applications, UW-FHCRC. [Online.] http://pga.gs.washington.edu/. 21.Sucato, G., A. Wald, E. Wakabayashi, J. Vieira, and L. Corey.1998. Evidence
of latency and reactivation of both herpes simplex virus (HSV)-1 and HSV-2 in the genital region. J. Infect. Dis.177:1069–1072.
22.Sullivan, D. G., S. S. Kim, J. J. Wilson, C. Stehman-Breen, and D. R. Gretch.
2001. Investigating hepatitis C virus heterogeneity in a high prevalence setting using heteroduplex tracking analysis. J. Virol. Methods96:5–16. 23.Truong, H. M., M. M. Berrey, T. Shea, K. Diem, and L. Corey.2002.
Concordance between HIV source partner identification and molecular con-firmation in acute retroviral syndrome. J. Acquir. Immune Defic. Syndr.
29:232–243.