organic papers
o2426
Wanet al. C8H13NO2S2 doi:10.1107/S1600536805020891 Acta Cryst.(2005). E61, o2426–o2427
Acta Crystallographica Section E Structure Reports Online
ISSN 1600-5368
Acetonyl morpholine-4-carbodithioate
Jun Wan, Chun-Li Li, Xue-Mei Li and Shu-Sheng Zhang*
College of Chemistry and Molecular
Engineering, Qingdao University of Science and Technology, 266042 Qingdao, Shandong, People’s Republic of China
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 293 K
Mean(C–C) = 0.004 A˚
Rfactor = 0.034
wRfactor = 0.099
Data-to-parameter ratio = 17.4
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography
Printed in Great Britain – all rights reserved
In the title molecule, C8H13NO2S2, the morpholine ring adopts
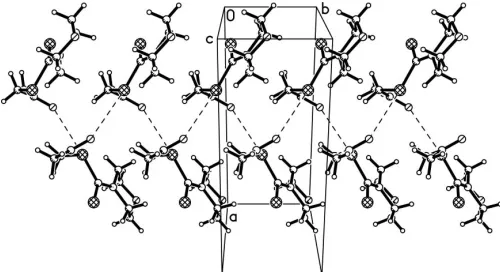
a chair conformation. In the crystal structure, a short intermolecular O S interaction [3.214 (2) A˚ ] links the mol-ecules into spiral chains along thebaxis.
Comment
Morpholine derivatives, as an important type of fungicides, have attracted much interest because of their inward absor-bent and broad-spectrum activities (Badioli et al., 2002). Dialkyl-substituted dithiocarbamate salts have shown inter-esting biological effects as broad-range fungicides. In order to search for new morpholine compounds with high bioactivity, the title compound, (I), was synthesized. We present here its crystal structure.
The bond lengths and angles in (I) (Table 1) are within normal ranges (Allenet al., 1987) and comparable with those in a related compound (Ko¨ysalet al., 2004). The morpholine ring adopts a chair conformation (Fig. 1), with atoms O1 and N1 deviating by 0.66 (2) and 0.57 (1) A˚ , respectively, from the mean plane through atoms C1–C4. There are three intramolecular C—H S interactions (Table 2), each forming a five-membered ring. In the crystal structure, short inter-molecular contacts O2 S1i[3.214 (2) A˚ ; symmetry code: (i)
1 x, 1
2+y, 1
2z] link the molecules into spiral chains
along thebaxis (Fig. 2).
Experimental
1-Bromoacetone was prepared by the reaction of acetone (3.7 ml, 0.05 mol) and bromine (8.0 g, 2.6 ml) in anhydrous diethyl ether according to Xu et al. (2002). (4-Morpholinylcarbothioyl)-sulfanylamine was prepared by reacting morpholine (3.9 ml, 0.05 mol) with carbon disulfide (3.0 ml, 0.05 mol) in ammonia (25– 28%, 10 ml). The title compound was synthesized by reacting 1-bromoacetone and (4-morpholinylcarbothioyl)sulfanylamine in acetone at room temperature for 2 h. The solution was filtered and purified by flash chromatography (silica gel, petroleum ether–ethyl acetate 6:1 (v/v). Single crystals suitable for X-ray diffraction study were obtained by slow evaporation of an ethyl acetate–petroleum ether (1:1 (v/v) solution over a period of two weeks.
Crystal data
C8H13NO2S2
Mr= 219.31 Monoclinic,P21=c
a= 15.043 (2) A˚ b= 4.9119 (7) A˚ c= 17.1097 (17) A˚
= 123.848 (8) V= 1050.0 (2) A˚3 Z= 4
Dx= 1.387 Mg m3 MoKradiation Cell parameters from 2917
reflections
= 2.9–26.0
= 0.48 mm1
T= 293 (2) K Block, colourless 0.500.220.07 mm
Data collection
Siemens SMART 1000 CCD area-detector diffractometer
!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996) Tmin= 0.797,Tmax= 0.968
5540 measured reflections
2048 independent reflections 1794 reflections withI> 2(I) Rint= 0.013
max= 26.1
h=18!17 k=5!6 l=11!21
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.034 wR(F2) = 0.099
S= 1.07 2048 reflections 118 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0575P)2
+ 0.1984P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.20 e A˚
3
min=0.19 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
S1—C5 1.7754 (16)
S1—C6 1.7831 (18)
S2—C5 1.6604 (16)
O1—C2 1.405 (3)
O1—C3 1.421 (3)
N1—C5 1.330 (2)
N1—C4 1.471 (2)
N1—C1 1.473 (2)
C5—S1—C6 102.24 (8)
N1—C5—S2 124.69 (12)
N1—C5—S1 113.31 (12)
[image:2.610.314.564.224.360.2]S2—C5—S1 121.99 (10)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C1—H1A S1 0.97 2.37 2.901 (3) 114
C4—H4B S2 0.97 2.57 3.048 (2) 111
C6—H6A S2 0.97 2.69 3.057 (2) 103
All H atoms were located in difference Fourier maps and constrained to ride on their parent atoms, with C—H distances in the range 0.93–0.97 A˚ and withUiso(H) = 1.2Ueq(C).
Data collection:SMART(Siemens, 1996); cell refinement:SAINT
(Siemens, 1996); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 1997); program(s) used to refine structure:SHELXTL; molecular graphics:SHELXTL; software used
to prepare material for publication:SHELXTL,PARST(Nardelli, 1995) andPLATON(Spek, 2003).
This project was supported by the Program for New Century Excellent Talents in University (No. NCET-04–0649), and the Project of Educational Administration of Shandong Province (No. J04B12).
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Badioli, M., Ballini, R., Bartolacci, M., Bosica, G., Torregiani, E. & Marcantoni, E. (2002).J. Org. Chem.67, 8938–8942.
Ko¨ysal, Y., Isik, S., Septioglu, E. & Calis, U. (2004).Acta Cryst.C60, o757– o758.
Nardelli, M. (1995).J. Appl. Cryst.28, 659.
Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (1997).SHELXTL. Version 5.1. Bruker AXS Inc., Madison,
Wisconsin, USA.
Siemens (1996).SMARTandSAINT. Siemens Analytical X-ray Instruments Inc., Madison, Wisconsin, USA.
Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
Xu, L. Z., Jiao, K., Zhang, S. S. & Kuang, S. P. (2002).Bull. Kor. Chem. Soc.23, 1699–1701.
Figure 2
The crystal packing, viewed approximately down thecaxis, showing the short intermolecular O S interactions (dashed lines).
Figure 1
[image:2.610.44.295.518.565.2]supporting information
sup-1 Acta Cryst. (2005). E61, o2426–o2427
supporting information
Acta Cryst. (2005). E61, o2426–o2427 [https://doi.org/10.1107/S1600536805020891]
Acetonyl morpholine-4-carbodithioate
Jun Wan, Chun-Li Li, Xue-Mei Li and Shu-Sheng Zhang
Acetonyl morpholine-4-carbodithioate
Crystal data
C8H13NO2S2
Mr = 219.31
Monoclinic, P21/c
a = 15.043 (2) Å b = 4.9119 (7) Å c = 17.1097 (17) Å β = 123.848 (8)° V = 1050.0 (2) Å3
Z = 4
F(000) = 464 Dx = 1.387 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2917 reflections θ = 2.9–26.0°
µ = 0.48 mm−1
T = 293 K Block, colourless 0.50 × 0.22 × 0.07 mm
Data collection
Siemens SMART 1000 CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
Detector resolution: 8.33 pixels mm-1
ω scans
Absorption correction: empirical (using intensity measurements)
(SADABS; Sheldrick, 1996)
Tmin = 0.797, Tmax = 0.968
5540 measured reflections 2048 independent reflections 1794 reflections with I > 2σ(I) Rint = 0.013
θmax = 26.1°, θmin = 1.6°
h = −18→17 k = −5→6 l = −11→21
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.034
wR(F2) = 0.099
S = 1.07 2048 reflections 118 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0575P)2 + 0.1984P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.20 e Å−3
Δρmin = −0.19 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
S1 0.34990 (3) −0.40882 (10) −0.28135 (3) 0.05330 (17)
S2 0.16710 (4) −0.56402 (10) −0.47601 (3) 0.05674 (17)
O1 0.09755 (14) −0.9550 (4) −0.22966 (12) 0.0856 (5)
N1 0.19435 (11) −0.7322 (3) −0.31665 (9) 0.0516 (4)
C7 0.40725 (14) −0.3467 (4) −0.41015 (13) 0.0521 (4)
O2 0.45425 (12) −0.5599 (3) −0.38385 (11) 0.0721 (4)
C1 0.25426 (15) −0.7714 (5) −0.21403 (12) 0.0618 (5)
H1A 0.3080 −0.6300 −0.1825 0.074*
H1B 0.2905 −0.9460 −0.1974 0.074*
C2 0.17967 (17) −0.7611 (5) −0.18258 (14) 0.0679 (5)
H2A 0.2195 −0.7944 −0.1154 0.081*
H2B 0.1484 −0.5807 −0.1944 0.081*
C3 0.03533 (17) −0.8969 (5) −0.32760 (16) 0.0739 (6)
H3A 0.0043 −0.7168 −0.3376 0.089*
H3B −0.0229 −1.0270 −0.3596 0.089*
C4 0.09967 (16) −0.9088 (4) −0.36932 (14) 0.0608 (5)
H4A 0.1222 −1.0948 −0.3679 0.073*
H4B 0.0560 −0.8505 −0.4345 0.073*
C5 0.22899 (12) −0.5862 (3) −0.36005 (11) 0.0429 (4)
C6 0.36472 (14) −0.2036 (4) −0.35938 (13) 0.0529 (4)
H6A 0.2955 −0.1261 −0.4061 0.063*
H6B 0.4124 −0.0539 −0.3237 0.063*
C8 0.3897 (2) −0.1986 (5) −0.49329 (18) 0.0850 (7)
H8A 0.4188 −0.3027 −0.5215 0.128*
H8B 0.4247 −0.0247 −0.4738 0.128*
H8C 0.3144 −0.1726 −0.5383 0.128*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
S1 0.0451 (3) 0.0659 (3) 0.0473 (3) −0.00952 (18) 0.0247 (2) −0.00917 (19)
S2 0.0552 (3) 0.0718 (3) 0.0395 (3) −0.0034 (2) 0.0241 (2) −0.00283 (19)
O1 0.0931 (11) 0.1046 (13) 0.0788 (10) −0.0303 (9) 0.0600 (9) 0.0004 (9)
N1 0.0474 (8) 0.0688 (9) 0.0431 (7) −0.0112 (7) 0.0280 (6) −0.0096 (7)
C7 0.0504 (9) 0.0485 (9) 0.0594 (10) −0.0033 (7) 0.0320 (8) −0.0009 (8)
O2 0.0791 (10) 0.0654 (9) 0.0858 (10) 0.0227 (7) 0.0546 (9) 0.0123 (7)
C1 0.0593 (11) 0.0831 (13) 0.0460 (9) −0.0043 (10) 0.0312 (9) −0.0021 (9)
C2 0.0812 (13) 0.0818 (14) 0.0574 (11) −0.0074 (11) 0.0491 (10) −0.0008 (10)
C3 0.0652 (13) 0.0954 (17) 0.0723 (14) −0.0220 (11) 0.0451 (11) −0.0121 (11)
supporting information
sup-3 Acta Cryst. (2005). E61, o2426–o2427
C5 0.0401 (8) 0.0479 (9) 0.0425 (8) 0.0015 (6) 0.0241 (7) −0.0075 (6)
C6 0.0522 (9) 0.0451 (9) 0.0635 (11) −0.0039 (7) 0.0337 (8) −0.0045 (8)
C8 0.121 (2) 0.0705 (14) 0.0863 (16) 0.0011 (14) 0.0721 (16) 0.0111 (12)
Geometric parameters (Å, º)
S1—C5 1.7754 (16) C1—H1B 0.9700
S1—C6 1.7831 (18) C2—H2A 0.9700
S2—C5 1.6604 (16) C2—H2B 0.9700
O1—C2 1.405 (3) C3—C4 1.490 (3)
O1—C3 1.421 (3) C3—H3A 0.9700
N1—C5 1.330 (2) C3—H3B 0.9700
N1—C4 1.471 (2) C4—H4A 0.9700
N1—C1 1.473 (2) C4—H4B 0.9700
C7—O2 1.202 (2) C6—H6A 0.9700
C7—C8 1.485 (3) C6—H6B 0.9700
C7—C6 1.509 (2) C8—H8A 0.9600
C1—C2 1.491 (2) C8—H8B 0.9600
C1—H1A 0.9700 C8—H8C 0.9600
C5—S1—C6 102.24 (8) C4—C3—H3B 109.1
C2—O1—C3 109.37 (16) H3A—C3—H3B 107.9
C5—N1—C4 121.52 (14) N1—C4—C3 110.45 (16)
C5—N1—C1 124.48 (14) N1—C4—H4A 109.6
C4—N1—C1 113.22 (15) C3—C4—H4A 109.6
O2—C7—C8 122.63 (19) N1—C4—H4B 109.6
O2—C7—C6 122.67 (17) C3—C4—H4B 109.6
C8—C7—C6 114.66 (17) H4A—C4—H4B 108.1
N1—C1—C2 110.00 (16) N1—C5—S2 124.69 (12)
N1—C1—H1A 109.7 N1—C5—S1 113.31 (12)
C2—C1—H1A 109.7 S2—C5—S1 121.99 (10)
N1—C1—H1B 109.7 C7—C6—S1 115.96 (13)
C2—C1—H1B 109.7 C7—C6—H6A 108.3
H1A—C1—H1B 108.2 S1—C6—H6A 108.3
O1—C2—C1 111.60 (17) C7—C6—H6B 108.3
O1—C2—H2A 109.3 S1—C6—H6B 108.3
C1—C2—H2A 109.3 H6A—C6—H6B 107.4
O1—C2—H2B 109.3 C7—C8—H8A 109.5
C1—C2—H2B 109.3 C7—C8—H8B 109.5
H2A—C2—H2B 108.0 H8A—C8—H8B 109.5
O1—C3—C4 112.35 (18) C7—C8—H8C 109.5
O1—C3—H3A 109.1 H8A—C8—H8C 109.5
C4—C3—H3A 109.1 H8B—C8—H8C 109.5
O1—C3—H3B 109.1
C5—N1—C1—C2 140.45 (18) C1—N1—C5—S2 174.18 (14)
C4—N1—C1—C2 −49.6 (2) C4—N1—C5—S1 −175.64 (13)
N1—C1—C2—O1 56.7 (2) C6—S1—C5—N1 −172.76 (12)
C2—O1—C3—C4 61.0 (3) C6—S1—C5—S2 6.65 (12)
C5—N1—C4—C3 −141.50 (19) O2—C7—C6—S1 −18.8 (2)
C1—N1—C4—C3 48.2 (2) C8—C7—C6—S1 163.18 (16)
O1—C3—C4—N1 −53.6 (3) C5—S1—C6—C7 −78.56 (14)
C4—N1—C5—S2 5.0 (2)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C1—H1A···S1 0.97 2.37 2.901 (3) 114
C4—H4B···S2 0.97 2.57 3.048 (2) 111