Aspects of phase retrieval in x ray crystallography
Full text
Figure
![Figure 1.1Reconstituted diffraction pattern showing spots (reflections) of differentintensities [Rypniewski et al., 1993].](https://thumb-us.123doks.com/thumbv2/123dok_us/9969995.497864/17.595.211.441.114.347/figure-reconstituted-diraction-pattern-showing-reections-dierentintensities-rypniewski.webp)
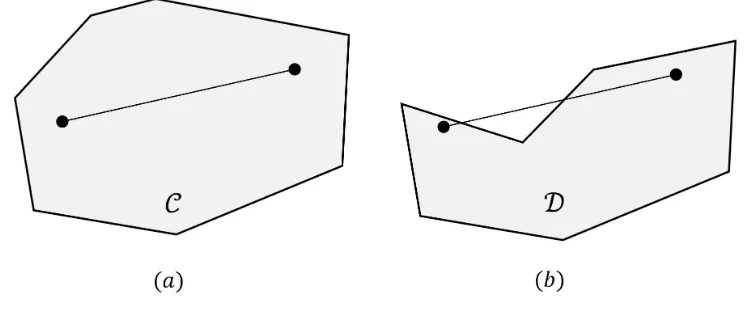
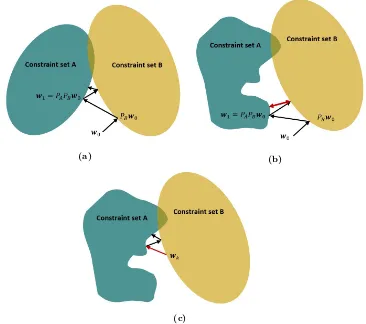
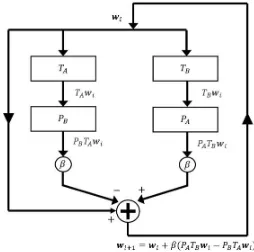
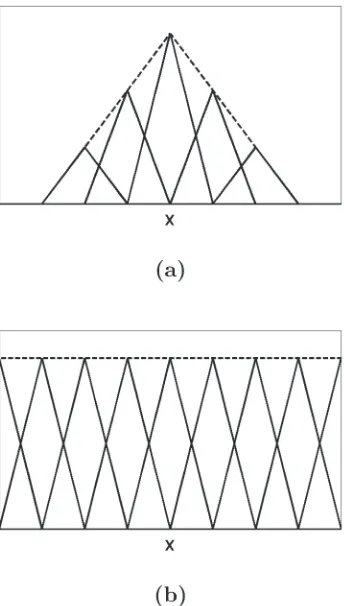

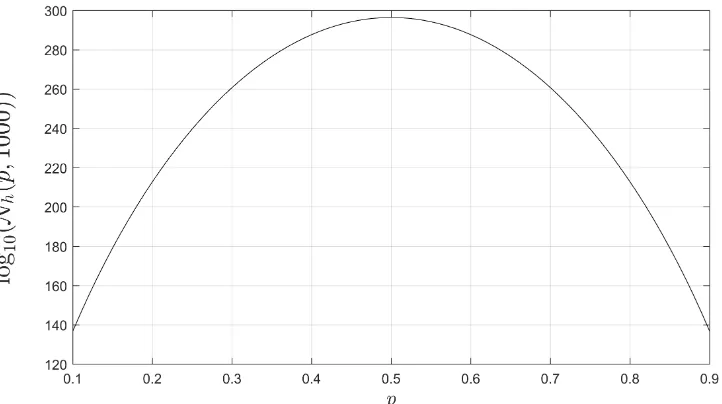
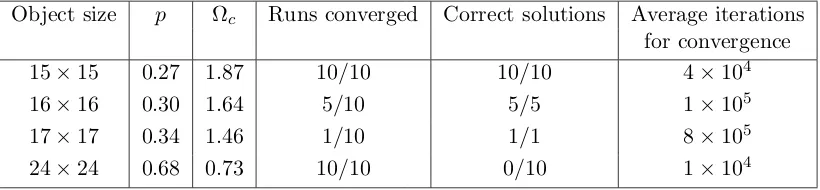

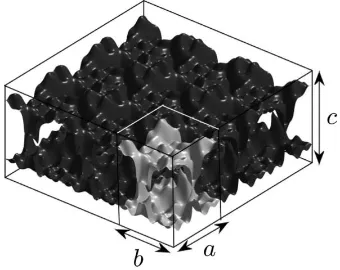
Related documents
When the recently introduced contextual embeddings – ELMo (Peters et al., 2018), BERT (Devlin et al., 2018) and Flair (Akbik et al., 2018) – are added to the architecture, we
the most prominent example of economic studies that use econometric findings. Key words : asset, financial market, modeling, investor. В современной экономике на рынке
ELISA results showed signifi - cantly decreased sH2a contents in peripheral blood samples from liver cirrhosis patients by 52.8% compared to healthy individuals (P<0.05,..
malignant tissues, that epigenetic subtypes exist within human breast cancers, and in particular, that a unique epigenetic gene signature has stronger prognostic value than
Detection of edge is a terminology in image processing and computer vision particularly in the areas of feature detection and extraction to refer to the algorithms which aims
Cumulative infiltration in a silt profile for the VGA parameterization (see Table 1) with conductivity functions according to Mualem (1976) and Alexander and Skaggs (1986) (a) and
METHODOLOGY Open Access Development of one step SYBR Green real time RT PCR for quantifying bovine viral diarrhea virus type 1 and its comparison with conventional RT PCR Ni
The residual concentrations of HCHs, TC, DDTs, THCs and PCBs were measured in Sparus auratus collected from the eastern and western sectors of the Egyptian Mediterranean Sea,
