antigen; the effect of coding and non-coding sequences
Sorena Kiani-AIikhan
A thesis subm itted to the University of London to fulfil the requirem ents
for the degree of Doctor of Philosophy
Tum our Im munology Unit
Im perial Cancer Research Fund
91 Riding House Street
London W IP 8BT
D epartm ent of Oncology,
University College London
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U643726
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
This project was conducted m ainly at the T um our Im m unology U nit
(TIU) of Imperial Cancer Research Fund (ICRF) based at the University College
London (UCL) and partly at the departm ent of Clinical Oncology at the Royal
Free Hospital (RFH), London.
I am grateful to my supervisor Prof. Peter Beverley for his continuous
support, guidance and warmth. I w ould like to thank Dr. Kerry Chester for her
supervision, encouragement as well as friendship. Dr. Jonothan Lederm ann and
Prof. Robert H aw kins for their input and interest in the project an d Prof.
Richard Begent and Dr. Allison Jones for their interest and support.
I am indebted to Lynda Robson and Jeetandra Bhatia of RFH for their
generosity and technical support and Drs. Kathrine King and Delin Z hu of
Southam pton University Hospital for their collaborative work.
I w ould like to thank all the people at UCL departm ent of Oncology for
housing me tow ards the end the project and especially M aurine Cohen for her
friendship. Dr. Berend Tolner for his guidance through the band shift assays
and John Bingham for his help with the computers. I w ould also like to express
m y gratitude to UCL departm ent of Medicine for giving me the opportunity to
carry out this project.
The TIU was a friendly and dynamic environm ent, thanks to exceptional
people such as M arcus Timon, Mala Maini, Peter Nobel, Pip O'Brian, Judith
W alter, Christine H uges, Ann Benfield and m y special friends Lindsey Goff,
H arry White, Hal Drakesmith and Dan Ornadel.
Last b u t not least, m y sisters, Brenda, Suzie, Claire, Caroline, Farzin,
Ronen, Lukas and m any other people w ho in one w ay or another helped me
at birth with a term, or a sentence, stating precisely the duration of
one's presence here: the way it is done in prison camps. That this
doesn't happen suggests that the affair is not entirely human; that
something we've got no idea or control of interferes. That there is
an agency which is not subject to our chronology or, for that
matter, our sense of virtue. Hence all these attempts to foretell or
figure out one's future, hence one's reliance on physicians and
gypsies, which intensifies once we are ill or in trouble, and which is
but an attempt at domesticating -or demonizing- the divine. The
same applies to our sentiment for beauty, natural and man made
alike, since the infinite can be appreciated only by the fin ite ...."
Joseph Brodsky
Chapter 1 ____________________________________________________ 17
General Introduction__________________________________________ 17
I n tr o d u c tio n____________________________________________________________18
T u m ou r im m u n o lo g y____________________________________________________18
Im m u n e su rv e illa n c e...18
A n ti- tu m o u r im m u n e responses...21
Identification of Tumour antigens 22
T cells specific for tumour antigens 24
Inefficiency of anti-tumour responses 26
E xp e rim e n ta lly induced a n ti-tu m o u r resp o n ses...31
D N A im m u n is a tio n_____________________________________________________ 32
B a c k g ro u n d...32
Mechanism of the immune respons 34
Advantages of D N A immunisation 35
Safety of D N A immunisation 35
The type o f immune response 38
In d u c tio n o f im m u n e responses a fter D N A im m u n is a tio n...39
D N A im m u n isa tio n results in b o th T fii and Th2 resp o n ses...42
Route or method of immunisation 44
A ntigen presentation and structure 47
Conditioning o f the immune system 48
Changes o fT helper phenotypes with time 48
Im m u n o s tim u la to r y sequence-m otifs in D N A...50
ISS are adjuvants that induce cytokines synthesis 52
Innate im m unity and T k l cytokine synthesis 53
ISS m y act as adjuvants by augm enting costimulation 55
C a rcin o e m b ryo n ic a n tig e n______________________________________________ 56
B a c k g ro u n d...56
hCEA Functions...61
Carcinoembryonic antigen as a target tumour a n tig en...61
Chapter 2 ____________________________________________________ 64
Material and M ethods_________________________________________ 64
P l a s t i c s_________________________________________________________________ 65
M i c e____________________________________________________________________ 65
R e a g e n t s________________________________________________________________ 65
R a d io a c tiv e I s o to p e s____________________________________________________ 65
P e p tid e s a n d P r o t e i n s___________________________________________________ 66
M e d ia___________________________________________________________________ 67
M edia used fo r mam malian cell cultures... 67
M edia used fo r bacterial cell cultures...68
Sera _____________________________________________________________________68
B u ffe r s__________________________________________________________________ 69
A n tib o d ie s_______________________________________________________________70
C ell l i n e s________________________________________________________________ 73
C ell c o u n tin g____________________________________________________________ 73
Cryopreservation and retrieval o f cells...74
T ra n sfectio n o f M a m m a lia n c e l l s________________________________________74
E lectroporation... 74
L ipofection... 75
Im m u n oflu orescen ce s ta in in g o f c e ll su rface m a r k e r s_____________________ 76
I n tr a c e llu la r s ta in in g a n d co n fo ca l m i c r o s c o p y__________________________76
M ounting cells onto slid es... 77
Confocal m icroscopy... 77
E nrich in g d e n d ritic cells fr o m s p le e n_____________________________________ 79
E nriching ly m p h o c y te s fr o m s p le e n_______________________________________80
P r o life r a tio n a s s a y_____________________________________________________ 81
CTL a s s a y_______________________________________________________________81
Target ce lls...82
Effector cells...82
R e s tr ic tio n e n zy m e d ig e s ts_______________________________________________83
P u r ific a tio n o f D N A fr o m a g a ro se g e ls___________________________________83
E s tim a tio n o f nucleic a c id c o n c e n tr a tio n_________________________________83
L ig a tio n o f r e s tr ic tio n fra g m e n ts in to p l a s m id s__________________________84
T ra n sfo rm a tio n o fE .c o li w ith p la s m id D N A_____________________________84
P la s m id p r e p a r a tio n____________________________________________________85
D N A a m p l if ic a t io n_____________________________________________________ 86
O ligonucleotide prim ers...86
Polym erase Chain Reaction (PCR)...86
cDNA syn th esis...87
RNA extraction 87
Reverse Transcription Reaction 88
G el ele c tro p h o re sis_______________________________________________________89
Agarose Gel Electrophoresis...89
Polyacrylam ide Gel Electrophoresis (PAGE)...90
D N A S equ en cing_________________________________________________________90
Single stranded DNA preparation...90
Sequencing reaction...91
Chapter 3 ___________________________________________________ 93
M e th o d s_________________________________________________________________ 96
Vaccination con structs...96
Im m unisation schedule...99
M easurement o f anti-hCEA an tibody le v e ls... 99
Lym phoproliferative a ssa y...100
C ytotoxic T Lym phocyte a s s a y...100
R e s u lts_________________________________________________________________102 Transfection o f mammalian cell lines w ith vaccination vecto rs...102
In itial im munisations w ith pVAC/hCEA and pVAC/hCEADo...105
Humoral responses 105 Cellular responses 108 V ariability o f response in larger groups o f m ic e...113
Immunisation w ith hCEA genes coding fo r different form s o fh C E A...116
Anti-hCEA responses after im m unisation w ith hCEADoTag... 119
Does the nucleotide or protein sequence o f ta g increase hCEADo immunogenicity?...121
A ntibody responses after protein b o o s t... 122
D is c u s s io n_____________________________________________________________ 126 Chapter 4 __________________________________________________ 135 The effect of LPS and tetanus toxoid fragm ent-c on D N A immunisation against hCEA ________________________________________ 135 I n tr o d u c tio n___________________________________________________________ 136 M e th o d s________________________________________________________________140 Vaccination con structs...140
Immunisation schedule...141
Measurement o f an tibody leve ls... 141
R esu lts ____________________________________________________________ 143 LPS as an adjuvant fo r DNA vaccin ation... 143
A n ti Fragment c antibody responses...150
Does fusion o f hCEADo to Fr-c change the typ e o f effector respon se...152
The importance of fusion o f Fragment-c to hCE A D o...155
CTL responses against Fr-c...156
D is c u s s io n_____________________________________________________________159 Chapter 5 __________________________________________________ 164 The effect of CpG DNA on antigen presentation________________ 164 I n tr o d u c tio n___________________________________________________________165 M e th o d s_______________________________________________________________ 167 Splenocyte cultures...167
MLR cultures...167
CTL cultures...167
Bone m arrow derived dendritic c e lls...168
Dendritic-cell stim ulation o f lym p h o cytes...168
Proliferation and cyto to xicity a ssa ys...169
Staining fo r cell surface an tigen s...169
Detection o flL -1 2...169
R e s u lts_________________________________________________________________170 CG oligodeoxynucleotides m ay interact w ith A P C s...170
Do CG oligodeoxynucleotides interact w ith DCs ?...175
Effect of CG ODN on antigen presentation by mature BM DC...176
Effect of CG ODN on antigen presentation o f im mature BM DC...178
Phenotype of BMDC treated w ith CG O D N...179
Cells stim ulated by CG O D N -treated D C s...183
D is c u s s io n_____________________________________________________________186
Chapter 6 __________________________________________________ 191
The target molecules of CpG D N A _____________________________ 191
Proliferation of splenocytes in response to b io tin yla ted oligodeoxynucleotides
...195
Staining CBl cells w ith oligodeoxynucleotides...195
Preparation of the cytoplasm ic fraction o f C Bl c e lls...196
M o b ility shift DNA-binding (band shift) a s s a y...197
Isolation of proteins bound to oligodeoxynucleotides...197
Western b lo t a n a lysis...198
R e s u lts_________________________________________________________________199 The binding site o f CBl cells fo r C G -O D N...199
A nalysis of CG ODN interaction w ith cytoplasm ic m olecules...204
CG O D N induces appearance o f a high afin ity target in cytop la sm ic extract..204
CG O D N m ay interact w ith NF-kB signalling p a th w a y s...208
D is c u s s io n_____________________________________________________________ 213
Chapter 7 __________________________________________________ 217
Final D iscu ssion _____________________________________________ 217
D N A im m u n is a tio n____________________________________________________218
I m m u n o s tim u la to r y D N A Sequences ___________________________________ 223
S u m m a r y______________________________________________________________ 2 2 7
Figure 1.1: Some reasons fo r failure of effective... 27
Figure 1.2: D N A immunisation can result in both... 45
Figure 1.3: Im m unostim ulatory sequences (IS S )... 54
Figure 1.4: Diagramatic representation o fh C E A ... 60
Figure 3.1: Diagramatic representation of the vectors... 98
Figure 3.2: Staining o f a R M A /hC E A single cell colony... 101
Figure 3.3: Verification o f expression o fh C E A ... 103
Figure 3.4: Staining of cells, transfected w ith ... 104
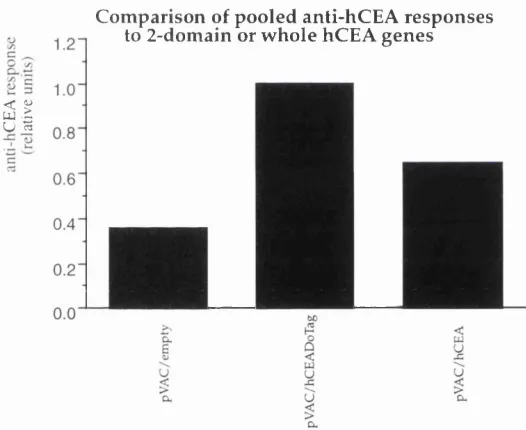
Figure 3.5: The relative levels ofanti-hC EA responses... 105
Figure 3.6: Titration of the anti-hCEA responses... 107
Figure 3.7: Individual anti-hCEA responses... 107
Figure 3.8: In vitro proliferation o fT cell enriched... 109
Figure 3.9: Percentage cytotoxicity o f splenocytes... I l l
Figure 3.10: Percentage cytotoxicity of splenocytes... 112
Figure 3.11: a) The relative levels ofanti-hC EA antibody... 115
Figure 3.12: A nti-hC E A responses o f fo u r groups... 118
Figure 3.13: A nti-hC E A responses o f fo u r groups o f 10 m ice... 120
Figure 3.14: A nti-hC E A antibody responses of fiv e groups... 123
Figure 3.15: A nti-hC E A antibody response of m ice... 124
Figure 3.16: A nti-hC E A response o f mice after i.p ... 125
Figure 4.1: A n ti hCEA antibody levels as a result o f... 144
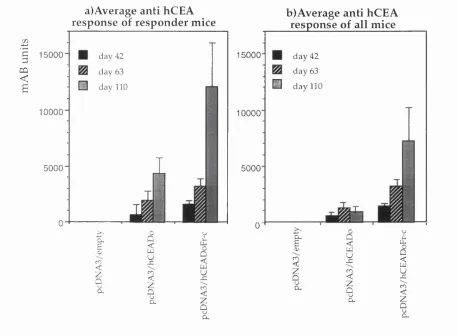
Figure 4.2: M ean m Ab units o f the responding m ice... 145
Figure 4.3: A n ti hCEA responses o f groups of eight m ice... 147
Figure 4.4: A n ti hCEA responses o f groups o f twelve m ice... 148
Figure 4.5: Average anti hCEA antibody level in the m ice... 149
Figure 4.6: Average anti hCEA antibody level in ... 150
Figure 4.7: Average ofanti-Fr-c antibody responses... 151
Figure 4.8: Comparison o f anti hCEADo with anti Fr-c... 152
Figure 4.9: Different isotypes produced against h C E A ... 154
Figure 4.10: Different isotypes produced against Fr-c... 155
Figure 4.11: A n ti hCEA and anti Fr-c responses... 156
Figure 5.2: Proliferation o f splenocytes in response... 171
Figure 5.3: A nti-H -2^ cytotoxicity ofBALBIc splenocytes... 174
Figure 5.4: Proliferation o f BALBIc or C57BLI6 splenocytes... 177
Figure 5.5: Proliferation of BALB/c or C57BLI5 splenocytes... 179
Figure 5.6: A higher percentage o f bone marrow derived dendritic... 181
Figure 5.7: A higher mean fluorescent intensity (M F I)... 181
Figure 5.8: Proliferation of 10^ syngeneic T or B cell enriched... 184
Figure 6.1: Time course o f proliferation of non-adherent... 199
Figure 6.2: Cell surface staining o f C B l cells w ith ... 201
Figure 6.3: Intracellular staining o f C B l dendritic cells... 202
Figure 6.4: Confocal micrographs o f C B l cells stained... 203
Figure 6.5: M obility shift D N A -binding analysis o f the cytoplasmic... 205
Figure 6.6: M obility shift D N A -binding analysis o f the cytoplasmic... 207
Figure 6.7: SD S-PAG E electrophoresis of the cytoplasmic extract... 210
Figure 6.8: SD S-PAG E electrophoresis of material bound to C G ... 211
Figure 6.9: Western blot analysis of material bound to CG O D N ... 212
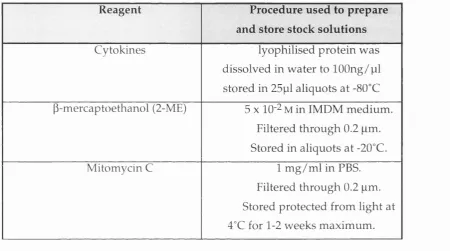
Table 2.1: Storage procedures for certain reagents... 66
Table 2.2: Details of peptides used in these studies... 67
Table 2.3: Primary (1st layer) antibodies used in cell staining... 70-71
Table 2.4: Secondary antibodies and Streptavidin-Cychrome C ... 72
Table 2.5: Cell lines used as targets in CTL assays... 73
Table 3.1: Dose and combinations o f plasmid used fo r im m unisation... 121
Table 4.1: Percentage o f responding mice producing... 153
Table 5.1: Percentages o f cells stained with antibodies against B ... 172
Table 5.2: The number of live cells/well in CG O D N treated... 173
Table 5.3: Percentages of cells stained with antibodies against... 173
Table 5.4: Percentage o f cells stained for C D llb , C D llc o r... 182
Abbreviations
EU Endotoxin Units
BMDC Bone Marrow drived Dendritic Cells
BSA Bovine Serum Albumin
CD66e Cluster of Differentiation 66e; hCEA
cDNA complementarey DNA
CFA Complete Freund's Adjuvant
Con-A Concavalin-A
CS Circumsporozoite
CTL Cytotoxic T Lymphocyte
CTLp Cytotoxic T Lymphocyte precursors
DC Dendritic Cell
ddNTPs 2' ,3 '-dideoxynucleoside 5 ' -triphophates
dNTPs deoxynucleoside triphosphates
D IT dithiothreitol
ELISA Enzyme Linked Im m unosorbant Assay
Fr-c Tetanus toxoid fragment-c
g-8' gene gun
CM -CSF Granulocyte Macrophage-Colony Stimulating Factor
gP glycoprotein
hAAT H um an-a-l-antitrypsin
hCEA Human Carcinoembryonic A ntigen
hCEADo N-terminal & A1 subdom ain of hCEA
hCEADoTM hCEADo w ith a transm em brane dom ain
HEL Hen Egg Lysozyme
hC H Hum an G row th H orm one
hig Hum an Im m unoglobulin
HIV Hum an Immunodeficiency Virus
HSA Heat Stable Antigen
HSV Herpes Simplex Virus
i.n. intranasal
IFN-T Interferon-T
Ig
ImmunoglobulinISS Immunostimulatory
mAb Monoclonal Antibody
MEM Minimal Essential M edium
MHC Major Histocompatibility Complex
m ig membrane Ig
MLR Mixed Lymphocyte Reaction
MMC Mitomycin-c
NK Natural Killer Cell
NP nucleoprotein
ODN Oligodeoxynucleotide
OVA Ovalbumin
PBMCs Peripheral Blood Mononuclear Cells
RSV Rous Sarcoma Virus
rt-PCR reverse transcriptase Polymerase Chain Reaction
scFv single chain Fv
SCID Severe Combined Immunodficiency
SEE Systemic Lupus Erythromatosis
TOR T Cell Receptor
Th Helper T Cell
TcH Helper T Cell Hybridom as
Chapter 1
Introduction
The idea th at the im m une system m ay be u sed as the m eans of
com bating tum our cells has been around for at least a h u n d red years. To
achieve this, William Coley immunised animals with extracts of Mycobacterium
m ixed w ith killed tu m o u r cells as early as 1905. Such id e a s an d
experim entations set the foundations of the discipline of tum our imm unology.
In this chapter current concepts of tum our imm unology will be discussed and a
review of the literature on DNA immunisation and interaction of DNA w ith the
im m une system will be presented. The final section w ill describe h u m an
carcinoem bryonic antigen that was used in our studies as a target tu m o u r
antigen.
Tumour immunology
The im m une system is known to interact w ith tum our cells resulting in tu m o u r regression or potentiation of grow th. The aim of this section is to discuss the evidence for natural anti-tumour imm une responses and the reasons for the inefficiency of such responses against tum our growth.
Im m une surveillance
The concept of im m une surveillance, that a function of the im m une
system is to seek out transformed cells and eliminate them , is now considered
to apply m ost effectively only to lim ited tu m o u r types. These are m ainly
virally-induced tum ours, although, there is also some evidence for possible
existence of surveillance for a few other tum ours (see below). In 1970, Burnet
proposed that the imm une system developed prim arily to prevent transm ission
show ed evidence of somatic m utation (i.e. cancer cells) (Burnet 1970) . He
proposed that the extensive polymorphism in MHC molecules evolved to allow
description of a unique self (Burnet 1973). Thus, the im m une system w ould
detect any alterations in MHC m olecules and consider it as non-self. A
fundam ental criticism of this m odel is that som atic m utations resu ltin g in
carcinogenesis do not have to necessarily occur in the MHC genes. Burnet also
proposed th at T cells w ere "almost solely" responsible for surveillance and
antibody producing cells had a "negligible" role. There is , how ever, evidence
show ing th at other cells such as NK cells are also involved in im m une
surveillance (Kurosawa, H arada et al. 1995). Antibodies have also been show n
to play a role in im m une surveillance of feline leukaem ia (Essex, Sliski et al.
1975).
D espite the criticisms of Burnet's m odel, different lines of evidence
suggest that im m une surveillance does operate but m ainly for virally induced
tum ours (Melief, Vasmel et al. 1989). Epidemiological evidence suggests that
there is a higher risk of virally-induced tum ours, such as Burkitt's lym phom a,
K aposi sarcom a and nasopharyngeal carcinom a, in im m une su p p re ssed
patients (Penn 1988). Im m une surveillance against virally-induced tu m o u rs
could exist as a result of the presence of CTL against viral epitopes that are
expressed by the tum our precursors. Therefore, the increased prevalence of
virally-induced tum ours in im m unosuppressed individuals and the observed
deficiency of im m une surveillance may reflect defective anti-viral responses in
such patients.
The risk of cancers, some of which are not virally induced, is higher in
transplantation patients than normal population. In a tw enty year follow -up
stu d y of 5,692 renal tran sp lan t p atien ts in the N ordic co u n tries, after
b lad d er cancers, and in men the risk of cancers of prostate and testis, and in
w om en cancers of cervix and vulva or vagina. These studies do not, of course,
directly link im m une suppression w ith increased risk of cancer since in renal
tran sp lan t patients, apart from im m une suppression, there are m any other
physiological anomalies compared to the general population and som e of the
dru g s used for im m unosuppression can have m utagenic effects. H ow ever,
im m une surveillance against non-viral cancer cells should not be com pletely
ruled out since there are still some data that m ay be explained in this way. For
instance, in a study of 34 hum an lung cancer lines, in HLA A 0201 positive cell
lines the p53 m utations did not occur in the regions of p53 th a t could
potentially bind HLA A 0201 (W iedenfeld, Fernandez-V ina et al. 1994). In
addition, in some cell lines from HLA A 0201 patients m utations d id occur in
HLA A 0201-restricted regions of p53 , however, those tum ours had lost A 0201
expression. A clinical observation that also supports the presence of im m une
surveillance is that some tum ours undergo spontaneous regressions associated
w ith detectable imm une activity (Rosenberg 1991).
et al. 1993). In this study, SCID and SCID-beige mice w ere used as syngeneic tu m o u r recipients. In contrast to the rapid grow th of the IL-2 transfected fibrosarcoma in SCID-beige mice, these cells did not grow in the mice that lack functional B and T cells (SCID mice). In addition, the inhibition of tu m o u r grow th in SCID mice was reversed by treatm ent w ith antibodies against natural killer (NK) cells dem onstrating that the innate imm une system m ay u n d er some circumstances m ount anti-tum our responses.
NK cells have been show n to express heterogeneous M HC class I receptors, NK inhibitory receptors (NKiR), that inhibit killing of target cells expressing native class I antigens (Farrell, Vally et al. 1997). This finding su p p o rts the notion of self recognition th ro u g h MHC m olecules th at w as p ro p o sed by Burnet, how ever, it is the NK cells th at have the ability to recognise self rather than T cells of the adaptive im m une system. Non-specific im m unity has also been dem onstrated against acute T cell leukaem ias in a TCRV'yl.ljY4Cy4 transgenic m ouse (Penninger, W en et al. 1995). Protection against haematopoeitic tum ours in this model was conferred by yô+ T cells. The role played by the innate im m une system especially in surveillance of early neoplastic cells, deserves more attention, as com ponents of the innate im m une system such as NK cells are capable of responding im m ediately, w hereas CTL m ay need a lag period before being able to lyse their target cells.
A nti-tum our im m une responses
im m une system are not all neo-antigens and m ay also be derived from norm al cell constituents or non-self antigens such as viral products.
Identification of Tumour antigens
The initial w ork on identification of tum our antigens w as based on the assum ption that tum our responses were best m ediated by antibodies. Some of the approaches used will be described here.
Sera from cancer patients were analysed for antibodies against tum our an tig en s using the 'autologous typing' m ethod. Three classes of tu m o u r associated antigens w ere described by testing the sera for reactivity w ith surface antigens of cultured autologous or allogeneic tu m o u r cells and non- m alignant cells. Sera that reacted to autologous tum ours exclusively w ere considered to detect class 1 antigens. Class 2 antigens w ere identified by sera th at w ere reactive w ith autologous and allogeneic tu m our cells and class 3 antigens were detected by sera w ith reactivity against tum our and norm al cells (Ueda, Shiku et al. 1979; Pfreundschuh, Shiku et al. 1980).
A nother approach that led to the discovery of antigens that w ere over expressed in tum our cells but had a low level of expression in norm al cells, was im m unisation of anim als w ith tum our extracts. One such antigen is hu m an carcinoembryonic antigen (hCEA) that was discovered by injecting rabbits w ith pooled tum our extracts (Gold and Freedm an 1965). TTie resulting antisera were purified by absorption w ith excess norm al colon extract. The antisera w ere further absorbed w ith tum our plasma fibrin and killed gut bacteria, and then gave a precipitin band w ith colonic tum our extracts in gel diffusion assays but not w ith extracts of norm al colon.
deglycosylated form of this antigen show ed differential reactivity w ith the m ucin produced by norm al and m alignant breast epithelium (Girling, Bartkova et al. 1989). SM-3 binds a region of the core protein that is not glycosylated in
m alignant breast carcinoma.
Currently, it is widely accepted that tum our im m unity is best m ediated by T cells of particularly CDS phenotype. This is based on evidence from anim al studies in which CDS T cells, alone or in com bination w ith CD4 cells, have been either transferred to naive anim als or deleted from im m unised anim als resulting in effective anti-tum our im m unity or loss of an ti-tu m o u r responses, respectively. In addition, in both anim al and h u m an studies it has been repeatedly observed that tum our progression is associated w ith dow n regulation of class I MHC, suggesting outgrow th of escape m utants u n d er CDS m ediated pressure. Accordingly, in the past decade various approaches have been used to identify tum our specific T-cell epitopes.
In one approach, to identify target epitopes, the MHC class I associated peptides on tum our cells are extracted, fractionated and used for coating target cells (Cox, Skipper et al. 1994). Using this m ethod, a p ep tid e fragm ent of gpl00/M eI17, a norm al protein expressed by melanocytes, was identified as the epitope recognised by m elanom a specific CTL derived from five different patients. Alternatively, CTL epitopes are identified by transfection of MHC m atched tum our cells that do not express the antigen (i.e. are not killed by the CTL) w ith a cDNA library of the tumour. Sequencing of the plasm ids from the cells that become susceptible to CTL lysis after transfection identifies the gene coding for CTL epitopes expressed by the original tum our cells (Brichard, Van
Pel et al. 1993). This technique has been used successfully to identify epitopes
T cells specific for tumour antigens
The T-cell repertoire that reacts against tu m o u r antigens w o u ld be shaped by the process of thymic selection. T cells that are capable of reacting against non-self or neoantigens, regardless of their affinity, w ould be positively selected as these antigens are not expressed in the thymus. Virally transform ed tum ours m ay express antigens derived from viral transform ing proteins (e.g. hum an papillom a virus E6 or E7) or structural proteins. There is good evidence th at virally induced tum our cells dow n regulate expression of the m ost im m unogenic viral proteins such as envelope proteins (Klein and Boon 1993). This m ay represent anti-tum our responses resulting in selection of tum our cells th at dow n regulate immunogenic epitopes. A ltered cellular proteins such as m utated p53 products, w ould also be recognised by T cells th at have been allowed to leave the thymus as the antigen is not coded by the genome during T-cell ontogeny.
costim ulation. Alternatively, naive autoreactive T cells m ay rem ain in lym ph nodes before they encounter their antigen.
The an ti-tu m o u r T cells th at recognise self antigen, therefore, are probably anergic cells that are activated in vitro under conditions that results in triggering these cells above activation threshold. These conditions include in vitro overexpression of peptide epitopes, for instance w hen transfected cells are
used for stim ulation of T cells, or addition of cytokines such as IL-2 that are norm ally secreted by helper T cells or provision of costim ulation by accessory cells presenting the tum our antigens.
Inefficiency of anti-tumour responses
The evidence above argues that there are tum our antigens that can be recognised by CTL. The same argum ents m ay apply to antigens recognised by other effector elements of the adaptive im m une system such as B cells and h elp er T cells. A dditionally, the presence of anergic h elp er T cells m ay contribute to B-cell and CTL anergy. An im portant question that arises is w hy tum our-specific im m une cells are anergic and do not m o u n t an effective response leading to eradication of tumours. In particular, cells specific for viral antigens or m utated self antigens would be expected to become activated as the target antigens is non-self. As m entioned before for virally induced tum ours, the cells that express imm unogenic antigens m ay be selected against b u t the cells that dow n regulate these antigens grow progressively. This m ight also be the case for the tum our cells expressing im m unogenic m utated proteins. An effective response is also absent against the rem aining tum our cells that m ay express antigens seen by anergic cells even w hen the antigen is overexpressed (e.g. MART or tyrosinase) and w ould be expected to efficiently trigger the responding T cells. The reasons for lack of efficient responses could be divided into factors associated w ith the effector phase and those associated w ith the induction of response.
suppression of both innate and adaptive anti-tumour responses (Chouaib, Asselin-Paturel et al. 1997). In principle, the ability of tumour cells to divide rapidly and the instability of the genome allows tumours to evolve under the pressure of the immune response and become poorer targets. The rate of tumour growth in relation to the magnitude of the immune response would also effect the outcome of the response such that rapidly growing tumours may become too great a burden for the immune response to manage.
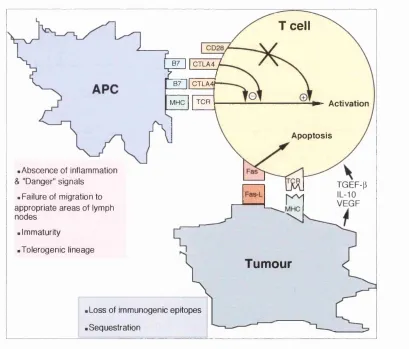
.Abscence of inflammation & "Danger" signals
. Failure of migration to appropriate areas of lymph nodes
• Immaturity
•Tolerogenic lineage
8 7
II
CTLA4 /^ ~ l
B7 GTLA4f—
MHC TOR
.Loss of immunogenic epitopes .Sequestration
T cell
- Activation
Apoptosis
TGEF-(i
T u m o u r
Figure 1.1: Some reasons for failure of effective anti-tumour immune responses. At low levels of B7 expression CTLA4 would blocks all the binding sites for CD28. MHC molecules on tumour cells mutate and are not recognised by specific activated T cells. Tumour cells secrete immunosuppressive cytokines.
The absence of costimulatory signals on potential target cells has been shown to allow maintenance of peripheral tolerance (Guerder, Meyerhoff et al.
1994). The mechanism of maintenance of anergic T cells and failure of induction of effective anti-tum our responses, has been proposed to be the absence of costim ulatory molecules on target cells (figure 12.1). This assum es that only tum our cells, that are not professional antigen presenting cells (APCs), present tum our antigens. This assum ption was m ade on the basis of the paradigm that class I presentation exclusively operates for endogenous antigens. However, the description of the phenom enon of cross-prim ing of CTL against exogenous antigens and its possible physiological significance, w hich probably has been underestim ated (discussed elsewhere), indicates that in an anti-tum our im m une response, professional APCs could potentially p resent tu m o u r antigens in association w ith class I and II molecules.
P resen tatio n of tu m o u r antigen by pro fessio n al APCs has been experim entally proven by induction of CD8+CTL in mice after im m unisation w ith GM-CSF-transfected B6 melanoma cells that lacked expression of MHC class I molecules (Huang, Golumbek et ah 1994). In another study, H uang et ah p ro v id ed direct evidence that even if a tu m o u r cell is transfected w ith a costim ulatory molecule such as B7-1, tum our antigens are prim arily presented by bone m arrow derived APCs (Huang, Bruce et ah 1996). In this m odel H- 2^—> H -2^^ bone m arrow chimeras were im m unised w ith H-2‘^ tum ours that w ere transfected w ith a model antigen and B7-1. The CTL against the m odel antigen were restricted to the bone m arrow haplotype (H-2^). After repeated rounds of im m unisation, however, CTL specific for the tum our haplotype also developed indicating that the tum our was also contributing to induction of the T cells b u t to a lesser degree.
required for T cell tolerance to that antigen (Adler, M arsh et al. 1998). They studied T cell tolerance to an epitope of haem aglutinin (HA) in HA transgenic mice of H-2^(^ haplotype. These animals were reconstituted w ith bone m arrow from non-transgenic animals of either H-2^ or H-2‘^ haplotypes. N aive T cells expressing a transgenic TCR specific for a I-E^^-restricted epitope of H A from H -2bd anim als were transferred to the bone m arrow reconstituted animals. In these experiments, only HA-specific T cells from mice reconstituted w ith H-2‘^ bone m arrow became tolerant to HA antigen. In addition, these T cells had a p h en o ty p e consistent w ith antigen recognition indicating th at the T cell tolerance w as an active process. A part from the fact that this transgenic TCR m ay have a high affinity for its target, this m odel is similar to the encounter of tum our specific cells w ith tum our cells that are not professional APCs. The interesting question here is w hat makes such a presentation tolerogenic.
The experim ent of A dler et al. described above show th at antigen presenting cells derived from bone m arrow can be tolerogenic. This could occur as a result of presentation of antigen by a population of APCs different from professional APCs th at are im m unogenic (figure 1.1). L ym phoid derived dendritic cells have been proposed to be tolerogenic (de St G roth 1998) and m ight be the cells that present tum our antigen. O n the other hand, the same professional APCs m ight present tum our antigen b u t have become tolerogenic as a resu lt of either not receiving activation or m a tu ratio n signals, or responding to signals that render them tolerogenic (figure 1.1). The chemokines released from the tum our site may also be different to those released from inflam m atory sites, resulting in m igration of either tolerogenic APCs, if they exist, or of APCs potentially capable of inducing either tolerance or activation. If the latter w as the case then these APCs m ay become m ature and capable of antigen presentation in response to other signals that m ay be absent at the tum our site.
"danger" signals received by professional APCs result in their m atu ratio n to im m unogenic APCs. In tum ours, "danger" signals could be the m olecules released from cells only w hen they undergo necrotic rather th an apoptotic dam age (M atzinger 1994). On the other hand, Janeway et al. propose th at the innate im m une system has inherited a set of prim itive "pattern recognition receptors"(PRR) th at recognise m olecular p attern s th at are u n iq u e to and essential for specific groups of organism s (M edzhitov and Janew ay 1997; M edzhitov, Preston-H urlburt et al. 1997). These receptors detect the presence of foreign organism s and transduce signals th a t resu lt in a ctiv atio n and m a tu ra tio n of APCs. The signals th at cause m a tu ra tio n of A PC s to im m unogenic ones or result in their m igration to the appropriate areas of the lym ph nodes, m ay be absent in tum our sites or there m ay exist other signals that counteract them. Finally, it is possible that the APCs that take u p tum our antigens do not migrate to the T cell areas of the draining lym ph nodes w ithout the presence of appropriate signals (De Smedt, Pajak et al. 1996).
A stu d y perform ed by Leach et al. m ay provide som e clues into the signals im portant in tolerance induction to tum our cells. In this stu d y , it was show n that blockade of cytotoxic T lym phocyte antigen-4 (CTLA-4) result in tum our regression both in challenge and therapy experiments (Leach, Krum m el et al. 1996). CTLA-4 is expressed by T cells and binds B7-1 and B7-2 w ith a
signalling events. It has been show n th at in renal cell cancer, intracellular pathw ays leading to T cell activation are im paired as a result of an unknow n product secreted by the tum our cells (Kolenko, W ang et al. 1997).
Experimentally induced anti-tum our responses
A new m ethod of eliciting CTL responses is DNA im m unisation that has been used for tum our vaccination. This m ethod has the advantage of allowing im m unisation w ith the gene for a protein that may contain m ultiple CTL and B cell epitopes resulting in a broad cellular as w ell as hum oral response in anim als of different haplotypes. Moreover, there is no need to identify CTL epitopes before im m unisation and the possibility of alteration in the epitopes d uring synthesis (for instance glycosylation patterns) is circum vented as the antigen is produced in its native form by m am m alian cells, in vivo. A nother advantage of DNA immunisation is that im m unom odulatory molecules such as cytokines or costim ulatory molecules can be adm inistered at the same time as the antigen. As m entioned earlier tolerant self reactive T cells m ay be the cells that recognise tum our antigens. It has been show n that DNA vaccination is capable of breaking self tolerance against a transgenic self antigen (Mancini, H adchouel et al. 1996). This may be because in DNA im m unisation the antigen is synthesised under the control of a strong viral prom oter, such epitopes may be presented at high concentrations if antigen presenting cells are transfected w ith the vector.
DNA im m unisation
B ackground
antibodies against the viral proteins (A tanasiu, O rth et al. 1962). As these experiments were not entirely clear due to the possibility of contam ination with viral particles, their significance was ignored u n til 1992 w h en T ang et al. rep o rted in N a ture that it was possible to raise antibodies against h u m an grow th horm one (hGH) and hum an-a-1-antitrypsin (hAAT) by introducing the gene encoding these proteins directly into m ouse cells, in vivo. (Tang, De Vit et al. 1992). This w as achieved by in tro d u c tio n of D N A -co ated gold
microprojectile particles into mouse ear skin. In 1993, Ulmer et al. dem onstrated that i.m. injection of BALB/c mice w ith DNA encoding nucleoprotein (NP) resulted in developm ent of antigen specific IgG antibodies and CTL responses th at protected the anim als against lethal challenge w ith the v irus (Ulmer, Donnelly et al. 1993). After a short period of silence there w as an explosion of reports on DNA vaccination which has continued ever since an d grow n by geometric progression. Most of these reports, however, dem onstrate that DNA im m unisation can be used to elicit antibody a n d /o r CTL responses against antigens from a variety of organisms in different m am m als. O nly a sm all m inority of reports have dem onstrated novel uses of DNA im m unisation or shed light on the physiology of immune response w hen antigen is encountered in this form.
Mechanism of the immune response
Advantages of D N A immunisation
DNA im m unisation as a m ethod of vaccination has m any theoretical advantages over conventional forms of vaccination. These include the ease of preparation and the stability of DNA molecules at am bient tem peratures that w ould low er the cost of vaccination; the absence of dem onstrable responses against the vector allowing its repeated use for vaccination ag ain st other pathogens; the possibility of administration of m ultiple vaccines in one vector; the absence of the risk of reactivation en co u n tered in a tte n u a te d viral vaccination and administration of the antigen in its native form. H ow ever, since the discovery of DNA imm unisation very few studies have been published on the theoretical disadvantages and indeed dangers of using this m ethod as a m eans of vaccinating healthy populations against pathogenic organism s. The safety issues that need to be addressed in DNA vaccination include the effect of chronic expression of the antigen; the risk of integration of DNA into the genome and the possibility of autoim m unity directed against the host DN A or proteins.
Safety of D N A immunisation
150,000 nuclei and looked for such events in m uscle and m ost other tissues (Nichols, Ledwith et al. 1995). In these experiments integration of plasm id DNA w as not detected in any tissues from the tw enty mice studied. O n the basis of these studies, Nichols et al. estimate that the chance of plasm id DNA integration is a third of the chance for spontaneous mutations. However, to be able to make any comm ents about such an im portant issue clearly m ore studies need to be carried out.
accelerated by imm unisation with ds-E.Coli DNA conjugated to BSA, however, this paradoxically protects the animals against developing the disease. Gilkeson et al. have show n that immunisation of pre-autoim m une N Z B /W mice w ith ds-
E.Coli DN A/BSA results in a reduction in proteinuria and prolongs survival
com pared to animals who spontaneously develop the disease or are induced to develop early disease by other m ethods (Gilkeson, Ruiz et al. 1996). The anti- DNA antibodies induced in the protected animals have the same specificity and affinity for m am m alian DNA and glom erular antigens as in control anim als (Gilkeson, Ruiz et al. 1996) however protection w as associated w ith a shift in cytokine profiles. The splenocytes of anim als im m unised w ith bacterial DNA secreted m ore interleukin (IL)-12 and INF-Y and less IL-4 than control animals (Gilkeson, Conover et al. 1998).
The type of immune response
An im portant point to consider in DNA im m unisation is the type of im m une response that is elicited. The effector arm of the adaptive im m une response is thought to be directed by helper T-cells to develop appropriate responses so that different pathogens are counteracted according to the nature and location of their interaction w ith the internal environm ent. D uring the 1980's w ork on classification of CD4+ T cells lead to categorisation of these as T helper 1 (Thl) and 2 (Th2) cells. In 1982, MacDonald et al. reported clones of
alloreactive T-cells that were obtained by lim iting d ilu tio n w hich w ere heterogeneous in their cytokine production (M acDonald, G lasebrook et al. 1982). In 1986, the current terminology of T h l/T h 2 responses w as first defined
by M osm ann et al. (Mosmann, Cherwinski et al. 1986). Thus, m ouse helper T- cells have been divided into the Thl sub-type that secretes cytokines such as IL- 2, IFN-T and IL-12, and the Th2 sub-type that produces cytokines such as IL-4,
IL-5 and IL-10. The work of Mosmann et al. and subsequent studies show ed that these distinct patterns of cytokine secretion are associated w ith provision of help for two types of immune responses. Stevens et al. dem onstrated that T h l an d Th2 cells induce antigen specific B-cells to secrete IgG2a and IgG l,
respectively (Stevens, Bossie et al. 1988). A similar classification has been used for hum an T cells, however, the divisions are not as strict. It is im portant to em phasis here that these classifications, that w ere initially based on in vitro observations, have become dogma leading some w orkers to assum e that these responses are m utually exclusive. In DNA im m unisation over fifty papers on the type of helper responses have been published. The initial w ork show ed some evidence for the presence of T h l-ty p e cytokines and IgG2a. This w as
follow ed by a series of publications that reinforced the idea th at DNA im m unisation results only in T hl responses. There are, how ever, some very good examples that show the presence of Th2 cytokines and IgG l antibodies or
of these responses in altering disease processes are review ed later in this chapter.
Studies on the interaction of bacterial DNA w ith the im m une system in late 1980s lead to the discovery that specific bacterial DNA m otifs stim ulate splenocytes to proliferate and secrete cytokines some of w hich are capable of inducing T hl responses. Therefore, these im m unostim ulatory (IBS) m otifs are th o u g h t to be responsible for developm ent of T hl responses after DNA
im m unisation. Studies of the interaction of bacterial DNA w ith the im m une system are reviewed in chapter 5, however, a m ore detailed discussion of these studies and their relevance to DNA im m unisation is found later in this chapter.
Induction o f immune responses after DNA im m unisation
Wolff et al. dem onstrated that direct injection of a plasm id encoding
firefly luciferase into the skeletal muscle of mice resulted in expression of the
reporter gene (Wolff, Malone et al. 1990). A lthough, the m axim um level of
reporter gene expression was at the site of injection in the skeletal m uscle cells,
low levels of expression were also observed in liver, spleen, skin, lungs, brain
and blood cells. After intraderm al injection of a plasm id, expression has been
observed in the derm is and epiderm is in fibroblasts, kératinocytes an d cells
w ith dendritic m orphology (Raz, Carson et al. 1994). C ondon et al. im m unised
mice using the epiderm al gene gun technique and fortuitously found dendrtic
cells containing gold particles in the draining lym ph nodes (Condon, W atkins et
aZ. 1996).
After DNA injection, therefore, antigen can be synthesised potentially by
m any cells in the body. APCs could acquire the antigen synthesised by any cell
by direct uptake if it is a secreted antigen or by taking up dead-cell debris or
apoptotic blebs that contain the antigen. This route of antigen uptake has been
for p resen tatio n of antigen in association w ith class II M HC antigens.
Presentation of antigen in association w ith the class I MHC molecule, however,
h a s been classically th o u g h t to h a p p en w h en a n tig en is sy n th esised
intracellularly. The exogenous pathw ay of antigen presentation w as show n in
1993 to also result in presentation of antigen in association w ith class I MHC
antigens (Kovacsovics-Bankowski and Rock 1995). This p ath w ay of antigen
p resen tatio n , how ever, was thought to be inefficient an d not to have a
significant physiologic role (Reis e Sousa and Germain 1995).
A possible explanation for induction of CTL is direct transfection of
APCs and intracellular synthesis of antigen. As m entioned above, Wolff et a l,
d em onstrated that i.m. injection of a plasm id encoding a reporter gene can
resu lt in expression of the product in m any tissues distant from the site of
injection (Wolff, M alone et al. 1990). Therefore, APCs could theoretically be
transfected after DNA administration. However, Ulmer et al. dem onstrated that
tran sp lan tatio n of m yoblasts that had been previously transfected w ith a
plasm id was sufficient to induce both antibody and CTL responses against the
encoded antigen (Ulmer, Deck et al. 1996). In these experiments, DNA could not
be transferred from myoblasts to other cells because it w as integrated in the
genome. In addition, for induction of CTL and antibody responses the antigen
had to be synthesised by viable transplanted myoblasts. Therefore, induction of
CTL can occur w ithout transfection of APCs. The question is w hether muscle
cells themselves present the antigen or w hether professional APCs are required.
Corr. et al. dem onstrated that in i.m. DNA im m unisation CTL prim ing is
by bone m arrow -derived cells (Corr, Lee et al. 1996). They reconstituted lethally
irradiated mice of H-2^-^ haplotype with bone m arrow of either H-2^ or H-2^
N P antigen of influenza virus resulted in prim ing CTL that w ere restricted only
to the haplotype of the donor bone m arrow.
It follows that, in the myoblast transplantation experiments (Ulmer, Deck
et al. 1996), for CTL induction, antigen synthesised by the m yoblasts w ould
have to be transferred to bone m arrow derived APCs. This w ould m ean that the
process of "cross prim ing" is responsible for CTL in d u ctio n after DNA
imm unisation, unless there is another yet unknow n process by w hich antigen is
transferred from muscle to the class I pathw ay. Recent observations show that
cross prim ing m ay be a major physiologic mechanism of CTL induction. Sigal et
al. have constructed a transgenic mouse m odel in which only non-haem opoetic
cells can be infected by a virus and dem onstrated that efficient CTL prim ing
against the viral epitopes does occur (Sigal, Crotty et al. 1999). They argue that
this could be an im portant m echanism for CTL induction against m u tan t
viruses that avoid infection of professional APCs.
Professional bone m arro w -d eriv ed APCs resp o n sib le for an tig en
presentation after DNA immunisation could be m acrophages, dendritic cells or
B cells. In an in vitro model. Rouse et al. dem onstrated th at m acrophages
stim ulated a CTL response by taking up naked DNA w hereas dendritic cells
had to be transfected to be able to do so (Rouse, N air et al. 1994). This finding
m ay be a result of using m ature dendritic cells that w ere incapable of DNA
uptake. In these experiments, splenic dendritic cells were used. The experience
in our laboratory w ith splenic dendritic cells is that w ithin a few hours after
isolation they acquire a m ature p h enotype and are no longer capable of
endocytosis.
In an in vivo model, dendritic cells and not B cells have been show n to
present antigen after DNA imm unisation (Casares, Inaba et al. 1997). Casares et
cell epitope (Vh-Tb)- Three, 7 or 11 days after i.m. injection of mice w ith this
plasm id, B cells and dendritic cells were sorted using antibodies against B220
and C D llc, respectively. The sorted cells were used to stim ulate specific helper
T cell hybridom as (TcH). In this study only the dendritic cells could stim ulate
the TcH. Furtherm ore, in the presence of the VH-TB p ep tid e b o th sorted
populations could stimulate the TcH, dem onstrating that the B-cell fraction was
also able to present the peptide, however, only the dendritic cells had acquired
that ability in vivo.
In DNA immunisation bone m arrow -derived antigen presenting cells are
responsible for induction of the immune response. Dendritic cells are the m ost
p otent know n APCs but a role for m acrophages cannot be ruled out. A ntigen
can be synthesised in APCs or in other cells and then be transferred to APCs for
p resen tatio n . In addition, skin-derived d en d ritic cells could d irectly be
transfected using the particle bom bardm ent technique.
D NA im m unisation results in both Thl Th2 responses
The im m une system has evolved to have at its disposal different effector
m echanism s for combating invading pathogens. A com bination of the m ost
appropriate effector mechanisms ought to develop for an optim um im m une
response. The choice of effectors w ould, therefore, be influenced by the
invading p ath o g en /an tig en ; where in the body it is encountered an d other
m olecules from the external environm ent th at are in tro d u ced w ith the
pathogen/antigen. In response to these stimuli a variety of signals are initiated
by the im m une system and other cells in the body. In tu rn the com bination of
these signals results in selection of the most appropriate effector response. The
pathogens have also evolved ways of evading the im m une response by for
system of an individual is based on an inherited genetic fram ew ork an d is
conditioned throughout life by unique environm ental factors. Therefore, in
resp o n se to the sam e pathogen, alth o u g h in tw o in d iv id u a ls sim ilar
com bination of effectors may develop, they w ould not be identical and m ay
vary for instance in intensity of different components. As m entioned earlier the
m ouse im m une response has been observed to polarise to T h l and Th2 types,
how ever, such polarisation is not absolute. It is im p o rtan t to b ear these
argum ents in m ind w hen discussing imm une responses to DNA im m unisation
on the basis of detection of individual cytokines. Considering the com plexity of
this system and the multifactorial nature of the outcom e, caution sh o u ld be
used in extrapolating from one experimental system to another. The rest of this
section will use examples in the literature to illustrate the points m entioned
above.
Raz et al. directly compared the type of responses elicited by DNA and
protein immunisation. They demonstrated that intraderm al (i.d.) im m unisation of mice w ith (3-galactosidase (P-gal) in saline or alum induced IgG l and IgE in association w ith IL-4 and 5 whereas IgC2a in association w ith IFN-T w ere induced w hen plasm id DNA encoding p-gal was injected i.d.(Raz, Tighe et al. 1996). There are other reports which also conclude that DNA im m unisation results in T hl-type of immunity. Shiver et al. im m unised mice against h u m an
im m unodeficiency virus (HIV)-l glycoproteins (gp) 120 or 160 and detected 100-fold more IFN-T than IL-4 in the supernatants of splenocytes restim ulated w ith the recom binant protein (Shiver, Perry et al. 1995). On this basis they concluded that this was a T hl response. M anickan et al. detected IgC2a and
im m unisation, how ever, there are also som e reports that d em o n strate the opposite and many others that show a m ixture of the two.
Route or method of immunisation
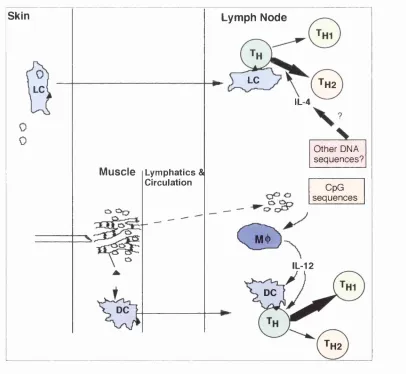
One of the factors that has been thought to result in developm ent of Th2
responses after DNA immunisation is the use of intraderm al or gene gun (g.g.) routes for delivery of im m unogen (figure 1.2). Boyle et al. co m p ared the im m une responses w hen different routes w ere used to im m unise mice. They used vectors encoding ovalbumin (OVA), hen egg lysozyme (HEL) and hum an im m unoglobulin (hig) (Boyle, Silva et a l 1997). In this study i.m. im m unisation resu lted in m arginally higher levels of IgG2a against OVA an d hIg in com parison to i.d. immunisation. This experiment, how ever, w as not reported for HEL. In the case of OVA, i.d. immunisation resulted in secretion of both IL-4 an d IFN-T by restim ulated splenocytes w h ereas splenocytes from i.m. im m unised anim als only secreted IFN-T after restim ulation. M oreover, both IgG l and IgG2a were produced in response to all three antigens regardless of the route of imm unisation. This paper has, rather misleadingly, been quoted by m any subsequent publications to support the statem ent that i.m. im m unisation induces T hl w hereas i.d. immunisation results in Th2 responses. O ther reports
The amount of vector has been equalised in another experiment where the intranasal route of immunisation was used. Intranasal (i.n.) administration of HSV gp-B has been reported to induce IL-5 synthesis and more IgGl than 2a whereas i.m. administration of the same vector results in no IL-5 and more IgC2a than 1 (Kuklin, Daheshia et al 1997). Both methods also resulted in secretion of IFN-Y which supports a skewing role for i.n. immunisation rather than an all or none switching function.
Skin Lymph Node
LC TH2
LC
IL-4
Other DNA sequences? Muscle Lymphatics &
Circulation
CpG sequences
m
IL-12
DC
DC
TH2
Figure 1.2: DNA immunisation can result in both Thl and Th2 responses. Intradermal or gene gun immunisation may result in direct transfection of Langerhan's cells. In intramuscular immunisation a larger amount of DNA is used that potentially could reach lymph nodes and influence the immune response by stimulating cytokine secretion.
Different ports of entry for antigen, therefore, seems to result in different
effector responses. How ever, the route of im m unisation as the reason for
differences in outcome has been disputed by Felquate et al. (Feltquate, H eaney
et al. 1997). They claim that instead of routes of im m unisation different m ethods
are responsible for the differences such that g.g. im m unisation into m uscle or
derm is results in Th2"type whereas needle injection i.m. or i.d. results in T hl-
type responses. Felquate et al. found that the Ig G l/Ig G 2 a ratios w ere higher
w h en DNA encoding influenza haem agglutinin w as adm inistered by g.g.,
w hereas IFN-T/IL-4 ratios were higher after needle injection regardless of the
location of DNA administration. In these studies the DNA dose w as taken into
account by using as little as 1 |ig of DNA in i.m. im m unisation. These results,
however, are in contrast to other work that shows Th2"type responses after i.d.
im m unisation w ith a needle (Raz, Tighe et al. 1996; Boyle, Silva et al. 1997; Li,
Sambhara et al. 1998). This could be due to the fact that different antigens have
been used in different studies.
An im portant signal for development of T h l responses is IL-12 (Manetti,
Parronchi et al. 1993). Hexameric motif sequences in DNA have been show n to
induce IL-12 secretion and T hl responses (Chu, Targoni et al. 1997). These
motifs will be discussed later in this chapter, however, they are m entioned here
to point out that the interaction of DNA molecule w ith the im m une system m ay
be altered by the m ethod or route of im m unisation. In the g.g. m ethod very
little DNA m ay be found extracellularly and the only cells stim ulated by the
DNA molecule w ould be the transfected cells, w hereas in the other m ethods a
large quantity of DNA is normally injected in to the extracellular space that